The Molecular Basis of Human Anophthalmia and Microphthalmia



1
UCL Institute of Ophthalmology, London, EC1V 9EL, UK
2
Moorfields Eye Hospital NHS Foundation Trust, London, EC1V 2PD, UK
3
Great Ormond Street Hospital for Children NHS Foundation Trust, London, WC1N 3JH, UK
*
Author to whom correspondence should be addressed.
J. Dev. Biol. 2019, 7(3), 16; https://0-doi-org.brum.beds.ac.uk/10.3390/jdb7030016
Submission received: 19 June 2019
/
Revised: 8 August 2019
/
Accepted: 8 August 2019
/
Published: 14 August 2019
(This article belongs to the Special Issue Women in Developmental Biology)
Abstract
:Human eye development is coordinated through an extensive network of genetic signalling pathways. Disruption of key regulatory genes in the early stages of eye development can result in aborted eye formation, resulting in an absent eye (anophthalmia) or a small underdeveloped eye (microphthalmia) phenotype. Anophthalmia and microphthalmia (AM) are part of the same clinical spectrum and have high genetic heterogeneity, with >90 identified associated genes. By understanding the roles of these genes in development, including their temporal expression, the phenotypic variation associated with AM can be better understood, improving diagnosis and management. This review describes the genetic and structural basis of eye development, focusing on the function of key genes known to be associated with AM. In addition, we highlight some promising avenues of research involving multiomic approaches and disease modelling with induced pluripotent stem cell (iPSC) technology, which will aid in developing novel therapies.
Keywords:
anophthalmia; microphthalmia; coloboma; eye; genetics; development; induced pluripotent stem cells; SOX2; OTX2; genes1. Introduction
The development of the human eye is a tightly controlled morphogenetic process which requires precise spatial and temporal gene regulation [1,2,3,4,5]. Perturbation of early eye organogenesis, either due to genetic or environmental factors, can result in lack of eye induction or abortion of eye development [6]. Anophthalmia and microphthalmia (AM) are, respectively, the absence or reduced size of the ocular globe compared to the population age-adjusted mean (Figure 1) [7,8]. Both are part of the same clinical spectrum and, therefore, can overlap, as the phenotype can be difficult to delineate in severe cases [9,10]. True anophthalmia occurs when eye development is aborted at the stage of the developing optic vesicle around 3–4 weeks gestation, leading to absence of the eye, optic nerve and chiasm, which can be confirmed through MRI brain and orbit scans [9]. Frequently, a small cystic remnant is detectable termed clinical anophthalmia, which occurs when the optic vesicle has formed but subsequently degenerates, hence a hypoplastic optic nerve, chiasm or tract may be present [10]. In microphthalmia, the eye has a smaller volume, and can be associated with a reduced corneal diameter (microcornea defined as a horizontal diameter <9 mm in a newborn and <10 mm in children >2 years) [11]. An axial length of <19 mm at 1 year of age or <21 mm in an adult using ultrasound B-scan substantiates a diagnosis of microphthalmia, representing ≥2 standard deviations below normal [11]. The estimated prevalence of microphthalmia and anophthalmia is 1 in 7000 and 1 in 30,000 live births, respectively [12]. The effect on vision is dependent on the severity, size of the eye and associated ocular malformations [9,10,11]. AM are considered part of the phenotypic continuum with ocular coloboma, a structural malformation resulting from incomplete fusion of the optic fissure, and are likely to have the same genetic basis [12,13,14,15]. Microphthalmia is reported in 3.2%–11.2% of blind children, but despite this there are no treatment options to improve visual function in microphthalmic patients, with current management focusing on maximising existing vision and enhancing cosmetic appearance [9,11,16].
AM has high clinical heterogeneity, and is rarely an isolated condition, often associated with other ocular abnormalities within the microphthalmic or contralateral eye (complex), such as anterior segment dysgenesis, ocular coloboma, cataract or vitreoretinal dysplasia (Table 1) [11]. Furthermore, 33%–95% of AM occurs alongside additional non-ocular systemic malformations, with 20%–45% of individuals diagnosed with a recognised syndrome [6,7,10,17,18,19]. The phenotypic variation observed with AM may be due to the multitude of genetic variants, including genetic modifiers as significant intrafamilial variability is seen, and/or environmental factors, such as maternal vitamin A deficiency or alcohol consumption [7,20,21,22,23]. The genetic variation is attributable to chromosomal and monogenic causes, with over 90 identified associated genes (Table 1). Genetic diagnosis can be obtained through molecular testing, such as array comparative genomic hybridisation (aCGH) or whole exome/genome sequencing [11]. Identifying the underlying genetic cause can influence appropriate disease management with recruitment of the correct multidisciplinary team to screen for potential syndromic associations, as well as guide genetic counselling and family planning [6,11]. The molecular diagnosis can be established in approximately 70% of bilateral anophthalmia or severe microphthalmia cases, but less than 10% of unilateral cases, which consists of the majority of patients [6,9,11,12]. Through understanding the genetic regulation underlying human eye development, and clarifying the roles of signalling molecules, we can elucidate how aberrant gene expression results in eye developmental disorders. Additionally, it is possible to better understand genotype–phenotype relationships, prognosis and develop new treatments.
2. The Genetic Basis of Human Eye Development
2.1. Eye Field Specification
Human eye development is initiated at three weeks gestation in the anterior neural plate (Figure 2a) [2,24]. OTX2 is required for the specification of the forebrain, and allows the neural plate to become competent for eye development by coordinating expression of eye field initiators SIX3, RAX and PAX6 [2,3,24,25,26,27]. The role of Otx2 in initiating eye development has been shown through animal studies investigating eye development, which have shown Six3 activates Pax6 and Rx2 expression in medaka fish, and is sufficient to induce ectopic eyes in the presence of Otx2 in medaka fish and Xenopus [2,3,24,25,26,27]. OTX2 and SOX2 coregulation of RAX, along with SIX3-mediated repression of WNT allows upregulation of eye field transcription factors (EFTFs), including PAX6, RAX, LHX2, TBX3 and SIX6 (Figure 2a,b) [1,2,25,28,29]. Rax has been shown to be necessary for inducing eye development in mice, as Rax knockouts do not form optic structures [29,30]. In Xenopus, Otx2 and Sox2 have been shown to directly synergistically activate Rax through binding to a non-coding sequence 2 kb upstream of the Rax promoter, which is conserved across vertebrates [28]. Furthermore, Sox2 missense mutations identified in patients with ocular disorders were found to disrupt the interaction of Otx2 and Sox2, as well as Sox2 DNA-binding ability, perturbing the ability of Otx2 and Sox2 to regulate Rax expression [28]. The role of Six3 in early eye development has been shown in mice, where Six3 is required for repression of Wnt1 transcription, which is necessary for regionalisation of the forebrain, as Six3-/- mice show expansion of Wnt1 expression, truncated rostral forebrain and lack eyes [3,31]. EFTFs form a self-regulating network which are sufficient to coordinate the development of the eye through suppression of genes which antagonise eye development, such as WNT, and upregulation of genes required for eye development, such as BMP4, demonstrated in Xenopus by the ability of EFTF RNA cocktails to induce ectopic eye formation [1,2,4,25,32,33]. OTX2 is subsequently downregulated in the eye field [2,24].
2.2. Splitting of the Eye Field
At 22 days gestation, SIX3-regulated SHH establishes the midline and factors from TGFβ, FGF and SHH families secreted from the underlying axial mesoderm stimulate the anterior migration of prospective hypothalamic cells, causing the eye field to split in two (Figure 2c) [1,2,3,24]. The direct regulation of Shh by Six3 has been observed in mice, and loss of Shh signalling in mice leads to defective midline patterning, including cyclopia, also found in humans with mutations in SIX3 and SHH [1,3,34,35]. Experiments using medaka fish show that FGF signalling is required for neural patterning to establish a competence region for Shh signalling, and FGFs interact with Shh to activate downstream targets of Shh required for optic development [2,36]. Ndr2, a member of the TGFβ family, is required for midline signalling in zebrafish, and loss of Ndr2 expression leads to cyclopia [2,3,37]. Tracking the movement of neural cells in zebrafish has shown that the single eye field is split through the anterior migration of cells, and cells in Ndr2 mutant zebrafish fail to migrate, resulting in a single cyclopic eye [2,38]. RAX-positive cells of the eye field proliferate and migrate bilaterally, resulting in evagination of the optic primordia from the forebrain and the generation of bilateral optic pits/grooves, observed through fluorescent tagging of Rx3 in wild-type and mutant medaka fish (Figure 2d,e) [2,4,24,25,30,39,40]. From 22–27 days gestation, further evagination of the optic pits through the cephalic mesenchyme results in the formation of optic vesicles (Figure 2e,f) [2,3].
2.3. Lens Placode Formation
Within the overlying surface ectoderm, expression of SIX3 directly activates PAX6 and SOX2, stimulating the formation of a pre-placodal region from 22–27 days gestation (Figure 3a) [4,41,42,43]. Six3 regulation of the lens pre-placodal region has been demonstrated by conditional deletion of Six3 in mice, leading to downregulation of Pax6 and loss of Sox2 expression, as well as failure of the pre-placodal region to thicken [4,42]. Furthermore, ChIP, EMSA and luciferase reporter assays, show that Six3 activates Pax6 and Sox2 expression in mice [42]. LHX2-regulated BMP4 expression from the evaginating optic vesicle signals the thickening of the lens placode at day 27–28 of gestation (Figure 3b) [1,3,4,41,43,44,45]. The importance of Bmp4 in lens induction has been shown in mouse models, where homozygous mutant mice lack lenses, but the phenotype can be rescued by exogenous BMP4 protein [45]. Lhx2 has been shown to regulate Bmp4 expression in the optic vesicle of mice, as Lhx2-/-mice have downregulated Bmp4 in the optic vesicle, and failure of lens formation [43,46].
2.4. Optic Cup Formation
The developing lens placode releases BMP and retinoic acid which bind to the optic vesicle, stimulating invagination of the optic vesicle from 31–33 days gestation (Figure 3c,d) [1,4,47,48,49,50,51,52]. Simultaneously, retinoic acid signalling from the surrounding periocular mesenchyme aids invagination of the optic vesicle (Figure 3c) [48,51]. Retinoic acid requirement in optic cup formation has been studied in mouse models, where double knockouts of retinoic acid synthesising retinaldehyde dehydrogenases developed optic vesicles that failed to invaginate, which were rescued by maternal dietary retinoic acid administration [1,4,47,48,49,50,51,52]. Experiments in mice show that filopodia extending mostly from the lens ectoderm tether the lens to the presumptive retina, allowing coordinated invagination of the presumptive lens and optic vesicle, and loss of filopodia in mutant mouse models results in reduced lens pit invagination (Figure 3c–e) [4,53]. In mice and chicks, lens invagination has been shown to occur through apical constriction, but the mechanisms driving optic vesicle invagination are not entirely clear, although experiments disrupting the extracellular matrix in chick embryos, alongside computational investigation have shown the extracellular matrix is required for proper formation and invagination of the lens placode and optic vesicle, through spatial restriction of proliferating cells [54,55,56,57].
The invagination of the optic vesicle results in the formation of a bilayered optic cup (Figure 3d,e) [33]. TGFβ signalling from the extraocular mesenchyme induces and maintains MITF and OTX2 expression in the outer layer of the optic cup, which forms the presumptive retinal pigmented epithelium (RPE), demonstrated experimentally through the removal of the extraocular mesenchyme in developing chick eyes resulting in absent RPE, which was restored through addition of the TGFβ family member activin (Figure 3e,f) [4,24,44,58,59,60,61]. MITF and OTX2 in the distil optic vesicle activate RPE differentiation genes, such as QNR71, Tyr, Trp1 and Trp2 in mice [24,62]. Reduction of MITF signalling in a human embryonic stem cell (hESC) model of eye development disrupted RPE maturation [61]. Additionally, Otx1 and Otx2 mutant mouse models show loss of RPE-specific Mitf expression, and expansion of neural retina (NR)/optic stalk regions [63]. FGF signals from the surface ectoderm stimulate the inner layer of the optic cup to form VSX2-expressing presumptive NR, shown in chick development through Fgf8 localisation to the neural retina, and in vivo application of FGF8, inducing presumptive chick RPE to differentiate into ectopic NR through preventing expression of RPE marker Mitf, and inducing NR markers Rx and Sgx1 (Figure 3e,f) [3,4,24,64]. VSX2 in the proximal optic vesicle represses RPE marker MITF, and is required for cell proliferation [24,65,66,67,68]. In Vsx2 mutant mice, Mitf is ectopically expressed in the NR, indicating Vsx2 represses Mitf expression in the NR [24,68]. Furthermore, mice with impaired Vsx2 production have a small eye phenotype due to decreased proliferation of retinal progenitor cells [24,69]. PAX6 expression in the presumptive NR maintains multipotency in retinal progenitor cells, allowing subsequent differentiation into the seven retinal types: retinal ganglion cells, horizontal cells, cone photoreceptors, rod photoreceptors, amacrine cells, bipolar cells and Müller glia [24,70]. Inactivation of Pax6 in mice retinal progenitor cells using conditional gene targeting restricts the cell fate of retinal progenitor cells to amacrine cells [24,70].
The optic cup is patterned through antagonistic expression of PAX2 and PAX6, shown in mouse models as Pax6-/- mutants show expansion of the Pax2 expression domain into the optic stalk, and Pax2-/- mutants show expansion of the Pax6 expression domain into the optic cup, and analysis of the molecular interactions of Pax6 and Pax2 in mice show reciprocal transcriptional repression [24,32,71,72]. SHH-regulated VAX1/VAX2 expression induces PAX2 expression and represses PAX6 expression in the proximal optic vesicle, inducing optic stalk formation, demonstrated experimentally by injection of Shh mRNA into Xenopus embryos, resulting in expansion of the Vax1 and Pax2 domain, at the expense of the Pax6 domain, in addition to abrogation of Vax1/Vax2 in zebrafish, leading to the expansion of retinal tissue into the optic nerve territory [24,32,73,74,75]. The optic fissure forms on the ventral surface of the optic vesicle from four weeks gestation to enable the hyaloid vasculature to enter and supply the developing eye with nutrients [76]. This opening fuses by week seven of gestation through apposition of the epithelial margins around the vasculature, spatial specification along the proximal-distal axis of the fissure and basement membrane breakdown, resulting in the formation of a continuous epithelial layer [77,78,79].
3. The Genetic Basis of Anophthalmia and Microphthalmia
AM has a complex aetiology, with dominant, recessive, autosomal and X-linked inheritance, although most mutations associated with non-syndromic AM arise de novo sporadic [11,12]. Non-penetrance has been observed, where family members carrying the same variant as the AM proband exhibit no ocular phenotype, as well as variable expressivity, where relatives with the same variant exhibit a spectrum of AM and related ocular features [80,81]. Variable expressivity and non-penetrance may be due to genetic modifiers, and/or stochastic environmental factors which influence the levels of gene expression [80]. Some cases of AM have been shown to arise through parental germline mosaicism, where a parent expresses no ocular phenotype as the mutation is not present in the ocular-derived tissue, but is present in gonadal tissue due to postzygotic mutational events [10,80,82,83,84]. This complex pattern of inheritance has implications for providing meaningful genetic counselling [85]. Chromosomal abnormalities, including aneuploidy, triploidy, translocations, deletions and duplications account for 20%–30% of AM [9,10,85,86]. Over 90 individual genes are associated with AM (Table 1), most of which encode transcription factors involved in eye morphogenesis, or components of the retinoic acid signalling pathway [12,87,88]. Pathogenic monogenic mutations include missense, nonsense, deletions, insertions and splice-site variants [12,66].
There is significant inter- and intra-familial phenotypic variability amongst patients and this may be related to specific genotype–phenotype correlations, or yet unidentified genetic modifiers. In PAX6-related ocular disease, haploinsufficiency gives rise to a range of phenotypes; the majority of loss-of-function alleles result in classic aniridia (with full iris and foveal hypoplasia, nystagmus, cataract, glaucoma and corneal keratopathy [211]) and missense mutations in the DNA-binding paired domain result in a milder phenotype, such as dominant nystagmus with foveal hypoplasia and normal irides [212]. Interestingly, a recent report highlights recurrent missense variants, p.S54R and p.N124K, associated with severe bilateral microphthalmia, phenocopying SOX2-associated AM [213]. An electromobility gel-shift assay (EMSA) revealed that for p.S54R, an 85% reduction in binding to the PAX6 protein-responsive element termed the LE9 enhancer was seen compared to wild-type protein, but with no effect on binding to the SIMO element (an autoregulatory PAX6 binding site with enhancer activity, resulting in correct maintenance of PAX6 expression). The p.N124K variant showed a moderate reduction in binding to both DNA enhancer sequences. The precise location of variants can affect protein function, and the lack of established genotype–phenotype relationships highlight the need for more correlation studies in disorders with Mendelian inheritance and phenotypic heterogeneity, such as AM. This will guide clinicians on prognosis and identifying potential therapeutic targets [85]. Furthermore, exploring the genetic pathways frequently disrupted in AM will support the identification of novel causative genes, variants, and regulatory elements which may act as genetic modifiers [85]. The more common genes associated with AM, with evidence from animal and cellular modelling, will be outlined below. The main genes fall into two distinct categories (i) eye field initiating transcription factors, such as SOX2, OTX2, RAX, VSX2 and PAX6 or (ii) retinoic acid signalling pathway components, including STRA6, ALDH1A3 and RARβ [12,87,88].
3.1. Eye Field Initiating Transcription Factors
3.1.1. SOX2
Heterozygous variants of sex-determining region Y-box 2 (SOX2) are the most common known cause of bilateral anophthalmia and severe microphthalmia, accounting for 15%–40% of cases [8,12,19]. SOX2 mutations are associated with isolated unilateral or bilateral AM, complex AM, and Syndromic Microphthalmia 3 (MCOPS3), where extraocular features include brain anomalies, neurocognitive delays, seizures, sensorineural hearing loss, oesophageal atresia, short stature, microcephaly and genital anomalies (Table 1) [8,12,89]. Over 76 disease-associated variants of SOX2 have been identified, incorporating loss-of-function deletion, missense, frameshift and nonsense mutations [7,8,12,19]. The majority of AM pathogenic SOX2 mutations arise de novo (60%), but alleles can also be inherited in an autosomal dominant manner, causing AM due to SOX2 haploinsufficiency [7,8,12,89]. Disease-associated SOX2 variants have been inherited from asymptomatic parents as a result of mosaicism, such as a case of a mother with no ocular features who had a daughter with bilateral anophthalmia along with extraocular features of SOX2 syndrome as a result of germinal mosaicism [83,84]. Consequently, it is important to screen parents for accurate genetic counselling and future family planning [6,9,10,11].
SOX2 is a high-mobility group transcription factor [8,28]. In Xenopus, Sox2 and Otx2 have been shown to function together to coordinate Rax expression in the early eye field [28,29]. Furthermore, introduction of point mutations in Xenopus Sox2 protein (to replicate missense mutations found in human ocular abnormalities) resulted in disrupted Rax transactivation and reduced DNA and Otx2-binding ability of Sox2 [28]. These findings indicate a key role for Sox2 in early eye formation, through coordinated regulation of EFTF Rax in conjunction with Otx2, and consequently, disruption of Sox2 results in disrupted eye development through perturbation of the Rax signalling network [28,29]. The severity of ocular abnormalities observed in humans indicate the role of SOX2 may reflect that of Xenopus, with a conserved key function in transcriptional regulation of key genes in human early eye development.
A genotype–phenotype correlation has been suggested for SOX2 mutations in AM cohorts, as missense mutations, particularly in the DNA-binding or transactivation domains, often result in non-penetrance or a milder ocular phenotype, such as ocular coloboma, and fewer developmental and systemic abnormalities, including postnatal growth retardation [8,10,12]. However, a clear correlation has not been definitively proved [8]. Variable Sox2 ocular phenotypes have been observed in a gene-dosage allelic series of mouse models, which displayed a range of eye phenotypes where the severity correlated to the expression of Sox2, suggesting a relationship between SOX2 genotype and clinical phenotype in humans may exist, due to dosage sensitivity in eye development [10,214].
3.1.2. OTX2
Between 2%–8% of AM cases are estimated to be the result of a pathogenic mutation in OTX2 [8,9,18,90,91]. Combined, SOX2 and OTX2 variants account for at least 60% of bilateral severe cases of AM [12,19]. Variants of OTX2 include indel, missense, frameshift and nonsense mutations, and AM is usually the result of a heterozygous loss-of-function [8,12,19,91]. While 40% of OTX2 mutations arise de novo, 35% are inherited from an unaffected parent, which complicates inheritance patterns [8,12,19,91]. Frequency of non-penetrance is high, with carrier parents displaying no ocular phenotype, as well as variable expressivity, for example, a case where one patient displayed unilateral microphthalmia, and his brother had a right anophthalmic eye and left ocular coloboma [12,18,19,80,91]. This case was believed to be a result of gonosomal mosaicism, as neither parent was found to carry the mutation in blood or buccal cell DNA [18]. Mosaicism has been confirmed in other OTX2 cases, such as a patient with bilateral severe microphthalmia who was the daughter of a phenotypically normal mother shown to have low levels of the variant in blood and buccal DNA, indicating gonosomal mosaicism [12,18,19,80,91]. OTX2 patients encompass extremely variable phenotypes, including additional eye malformations such as anterior segment dysgenesis, retinal dystrophy and hypoplasia or aplasia of the optic nerve and optic chiasm, as well as syndromic features including pituitary abnormalities, hypopituitarism, brain anomalies, seizures and developmental delay (Table 1) [8,91,93,215]. Homozygous mouse knockouts of homeobox gene Otx2 lack eyes, while heterozygous knockout mouse models also show a highly variable phenotype, from acephaly, micrognathia, anophthalmia, microphthalmia to normal development, depending on the genetic background [8,25,63,80,215,216,217,218,219]. A relationship between genotype and clinical phenotype has been observed, as mutations in the second half of OTX2 seem to occur more frequently with abnormal pituitary function, although this correlation is not always seen [8,91,215].
OTX2 is required for neural development, and coordinates the expression of EFTFs SIX3, RAX and PAX6 to initiate eye field development, but is subsequently downregulated in the eye field by EFTFs including RAX [3,25,32,220,221,222]. Otx2 has been shown to be necessary for eye development in mouse and Xenopus, as early EFTFs Six3 and Pax6 only possesses the ability to induce ectopic eyes in the presence of Otx2 expression [25,26,27,63,223]. Furthermore, overexpression of Otx2 in Xenopus results in upregulation of Rax, indicating Otx2 is a positive regulator of Rax in early development [28].
OTX2 also plays a role later in eye development, during the regionalisation of the optic vesicle, where TGFβ signalling from the extraocular mesenchyme induces OTX2 expression in the distil optic vesicle [24]. WNT signalling then induces upregulation of OTX2 in the prospective RPE [24]. OTX2 is required for RPE maintenance and differentiation, including regulation of key RPE marker MITF [24,63,224]. Inactivation of Wnt signalling activator β-catenin in mice at the optic vesicle stage results in a lack of Otx2 and Mitf signalling in the optic cup, and expansion of the NR domain, leading to anophthalmia [24,59].
3.1.3. RAX
Biallelic mutations in the EFTF RAX are estimated to account for 2%–3% of AM [6,7,11,94]. Disease-associated loss-of-function variants of RAX include missense, nonsense, frameshift and splicing mutations, as well as whole gene deletions [6,12]. Heterozygous carriers of disease-associated variants display no ocular phenotype, indicating an autosomal recessive inheritance pattern [6,7,12]. Homozygous or compound heterozygous RAX variants are often associated with bilateral severe AM, alongside neurological features such as intellectual deficiency and autism [7].
RAX has been shown to play a conserved role in establishment/proliferation of the eye field, as expression of ectopic Rax in Xenopus results in formation of ectopic retinal tissue [1,30], and zebrafish/mouse heterozygous knockouts of Rax have an eyeless phenotype [30,225,226,227]. The homeobox gene Rax is expressed early in eye development, and has been identified as necessary for the initiation of eye field development through regulation by Otx2 and EFTFs Six3 and Pax6 in targeted mouse knockout models [228]. Rax has been shown in zebrafish to coordinate the splitting of the eyefield, by downregulating nlcam in the eye field, to decelerate the convergence of retinal progenitor cells to the midline of the neural plate [39,40]. Overexpression of nlcam results in abnormal migration of the eye field, converging further towards the midline, resulting in a small eye phenotype in zebrafish [39]. Furthermore, in chimeric mouse embryos of Rax-/- and wild-type cells, Rax-deficient cells were not incorporated into optic tissue, indicating a role for Rax in cellular segregation in the early developing eye [4,29].
3.1.4. VSX2
Biallelic mutations in VSX2 are thought to account for 2% of isolated microphthalmia, and were identified in 1.5% of all MAC (microphthalmia, anophthalmia and coloboma) cases in a screening of >880 samples [9,12]. Disease-associated variants include missense, nonsense, exon deletions and splice-site variants [66,95]. VSX2 has an autosomal recessive inheritance pattern, and homozygous or compound heterozygous VSX2 variants are usually associated with bilateral AM [12]. VSX2 variants can result in isolated AM, although complex cases often include cataracts, as well as iris abnormalities, retinal dystrophy and colobomas, and individual cases with systemic features have been reported, including developmental delay with behavioural problems, autism, cryptorchidism, ovarian defects, or hearing impairment, although no genotype–phenotype correlations have been identified [12,17].
VSX2 is a homeodomain-containing transcription factor [95]. Inactivation of the VSX2 orthologue Alx1 in zebrafish results in small eyes [95,229]. Vsx2 null mice are microphthalmic, and show reduced proliferation of retinal progenitor cells, as well as transdifferentiation of the neural retina into RPE [69,95,230,231]. Vsx2 has been shown to regulate retinal cell number via CycD1-mediated repression of the negative regulator of proliferation cyclin-dependent kinase inhibitor Kip1, in retinal progenitor cells [231]. Consequently, Vsx2 enables cell proliferation by preventing retinal progenitor cells from exiting the cell cycle [66,231]. This has been shown as in Vsx2 null mice, Kip1 is abnormally present in retinal progenitor cells, resulting in cell number decrease in the retina and a small eye phenotype [231].
In the optic vesicle, VSX2 expression marks the developing neural retina, while MITF is expressed in the developing RPE [32]. Fgf expression activates Vsx2 in mouse neural retina which represses Mitf transcription, and Vsx2 null mice show erroneous expression of Mitf in the neural retina [66,68]. Analysis of the function of VSX2 in human iPSC-derived optic vesicles showed WNT to be a downstream target [232]. Patient-derived iPSCs with a mutation in VSX2 found upregulation of WNT pathway components and RPE markers, demonstrating VSX2-mediated repression of WNT contributes to maintaining neural retina identity [232].
3.1.5. PAX6
PAX6 is a transcription factor most commonly associated with aniridia [92,233]. A study of >500 MAC cases found 1%–2% to be PAX6 related [12]. Heterozygous loss-of-function PAX6 variants have been found in patients with unilateral and bilateral AM, indicating an autosomal dominant inheritance pattern [7,12,92]. Patients with PAX6-associated microphthalmia often have a complex phenotype, with associated ocular abnormalities including aniridia, iris hypoplasia, coloboma and optic nerve hypoplasia (Table 1) [12]. Mosaicism has been reported in PAX6-associated microphthalmia, such as a case with two brothers with bilateral complex microphthalmia with a heterozygous missense mutation, and an unaffected mosaic mother [92].
Missense mutations in the paired-box domain of PAX6 have been more frequently been associated with MAC, which is the domain known to interact with SOX2 [12,213,234]. The complex formed by PAX6 and SOX2 is known to be key for initiation of lens development by activation of crystallin genes [3,234]. Correct formation of the lens is vital to optic cup development, indicating a potential pathway for PAX6 mutations in the paired-box domain to have an increased likelihood to cause AM.
The homeobox gene PAX6 has been shown to be a conserved regulator of eye development [42,235]. Pax6 mutant mouse models display small eyes alongside other ocular phenotypes [92,236,237]. Pax6-/- mice develop eyes, but they fail to be maintained, arresting in development prior to the optic cup stage [2,236,237,238]. PAX6 is one of the first EFTFs to be expressed, and ectopic expression of Pax6 in Xenopus can be sufficient to induce ectopic eye tissue [223,235]. Furthermore, misexpression of Pax6 in Xenopus results in erroneous upregulation of EFTFs including Six3, Rax and Lhx2 [25,223,235]. Pax6 is known to form functional complexes with Lxh2 in mice, which are required for the regulation of Six6 in the optic vesicle [235,238]. These results indicate PAX6 plays a conserved key role in initial eye formation by initiating a cascade of gene expression which regulates eye development [223].
Rax-deficient mice show loss of Pax6 expression in the region of the neural plate which would form the optic primordium, suggesting Rax is an upstream regulator of Pax6 in early eye field formation [32,228]. In Six3 mutant mice, Pax6 is downregulated in the preplacodal lens ectoderm, indicating Pax6 in the developing lens is also activated by Six3 [26,42]. Bmp7 has also been implicated in the initiation of Pax6 in lens induction, as in Bmp7 null mice, Pax6 expression is lost in the lens placode, resulting in lens defects [3,239].
Moreover, PAX6 is required for the dorso-ventral patterning of the optic cup, where PAX6 is highly expressed in the dorsal region and PAX2 is expressed in the ventral region [3,235]. In Pax6 mutant mice, there is dorsal expansion of Pax2 expression, while in Pax2 mutants there is ventral expansion of Pax6, demonstrating the antagonistic function of Pax6 and Pax2 in the optic cup [3,72]. In addition, this patterning is regulated in mice by Shh which promotes ventral expression of Vax1 and Pax2, while inhibiting expression of Pax6 and Rax [24,73,235,240]. In Vax1/Vax2 double-mutant mice, the optic nerve is transformed into retinal tissue, and Pax6 expression is increased in the ventral region of the optic cup, demonstrating the importance of Vax1/Vax2 in regulating Pax6 expression in the optic cup [75]. Lhx2 mutant mouse optic vesicles show lack of downstream target Vsx2 and display expansion of Pax6 ventrally, indicating Pax6 is partially regulated in the optic cup through Lhx2-mediated Vsx2 expression [32,46].
3.2. Retinoic Acid Signalling Pathway Components
3.2.1. STRA6
Biallelic loss-of-function mutations in STRA6, including missense, nonsense, frameshift and whole gene deletions, can lead to AM, usually manifesting bilaterally [8,12,97,241]. Commonly, pathogenic recessive STRA6 variants result in Microphthalmia, Syndromic 9 (MCOPS9), also known as Matthew Wood Syndrome, where syndromic features include pulmonary, diaphragmatic and cardiac defects, resulting in death within the first 2 years of life [12,17,96,242]. However, milder phenotypes have been observed, including a patient with bilateral anophthalmia and mild syndromic features, in addition to isolated AM in patients with homozygous missense STRA6 variants [19,20]. However, no genotype–phenotype correlation has yet been established [8,96,98].
STRA6 is a transmembrane receptor for retinol-binding protein, responsible for mediating vitamin A uptake into cells [8,20,98,243,244]. Cellular analysis showed that a missense mutation in STRA6 resulted in loss of vitamin A uptake [20]. Furthermore, the clinical phenotype is recapitulated in zebrafish models, with inhibition of retinoic acid resulting in microphthalmia, and developmental defects including heart morphogenesis [20]. Patients with STRA6 variants display variation in phenotype, such as one family where phenotype ranged from bilateral anophthalmia to unilateral microphthalmia with contralateral ocular coloboma, indicating phenotypic variation may result from environmental factors such as variable vitamin A uptake, as well as genetic background [8,20,98].
The severity of phenotypes associated with STRA6 variants demonstrate the importance of retinoic acid signalling in development [98]. Retinoids can regulate development through enhancing or suppressing developmental gene expression [20,245,246]. In eye organogenesis, retinoic acid is required to signal the formation of the lens placode, in addition to the invagination of the optic vesicle [43,48,49]. Absence of Stra6 in zebrafish impairs retinoic acid receptor signalling and gene regulation, resulting in craniofacial and cardiac developmental defects, as well as microphthalmia [245]. The results demonstrated in animal and cellular modelling indicate a conserved function of STRA6 in mediating vitamin A uptake, and that disruption of STRA6 results in AM due to disruption of the retinoic acid signalling pathway, and consequently, disrupted expression of genes required for eye development, including lens placode development and optic cup invagination [245].
3.2.2. RARβ
AM is associated with recessive loss-of-function or dominant gain-of-function pathogenic variants of RARβ [8,21,87,88]. Variants include missense, nonsense and frameshift mutations [87]. Gain-of-function missense RARβ variants found in AM patients were shown to exhibit increased response to retinoic acid ligands compared to the wild-type, indicating an increase in retinoic acid signalling mediated by RARβ is sufficient to disrupt eye development [87,88]. The clinical manifestation of RARβ mutations resemble those of STRA6, including pulmonary and cardiac defects, often resulting in termination of pregnancy or neonatal death [8,12,87,88].
RARβ is one of three retinoic acid receptors, with RARα and RARγ, which transduce retinoic acid signalling to allow retinoic acid to regulate specific target genes in development [51,88]. The retinoic acid receptors form heterodimers which are key to oculogenesis [48,51]. A mouse model with all three retinoic acid receptors inactivated resulted in clinical anophthalmia, including lack of optic nerve [51]. In mouse models, Rarβ-mediated retinoic acid signalling in the periocular mesenchyme has been shown to regulate apoptosis and the expression of Foxc1 and Pitx2, which coordinate the development of the anterior eye segment [51,247,248].
3.2.3. ALDH1A3
Mutations in the retinaldehyde dehydrogenase gene ALDH1A3 are associated with an estimated 10% of AM [21,99]. Disease-associated variants include missense, nonsense and exon skipping [99,100,101,249]. Biallelic ALDH1A3 mutations tend to be associated with bilateral AM, and can be isolated or complex (Table 1) [12]. Biallelic pathogenic AM ALDH1A3 alleles are commonly associated with a neurocognitive phenotype, but additional extraocular findings are rare [8,12,99]. Non-penetrance has been reported with ALDH1A3 disease-associated variants, such as a case of autosomal recessive ALDH1A3 variants where an asymptomatic sister carried the same homozygous mutation as two siblings affected with bilateral AM [101,249].
ALDH1A3 is part of the retinoic acid synthesis pathway, through oxidation of retinaldehyde to retinoic acid [8,48,99]. ALDH1A3 is one of three human retinaldehyde dehydrogenases, which have specific spatial and temporal expression patterns [48,50,250]. Pax6 mutant rat embryos are deficient in retinaldehyde dehydrogenase Raldh3 (Human ALDH1A3), indicating Raldh3 is genetically regulated by key oculogenesis transcription factor Pax6 [48,251].
The mouse retinaldehyde dehydrogenase gene Raldh2 (human ALDH1A2) is expressed in the periocular mesenchyme [50,250]. Loss-of-function mutations in Raldh2 inhibit optic cup invagination [49]. Consequently, ALDH1A2 in humans is thought to regulate retinoic acid synthesis signalling to the invaginating optic vesicle through signalling from the periocular mesenchyme [48]. However, in Raldh1-/-(Human ALDH1A1)/Raldh2-/- mouse embryos, retinoic acid synthesised by Raldh3 in the RPE acts on the neural retina to aid ventral invagination [48,50,250]. Raldh3-/- mouse mutant embryos initiate optic cup formation, but lack retinoic acid in the ventral optic cup, resulting in ventral retina shortening [250,252]. Consequently, there appears to be a conserved role for ALDH1A3 in synthesis of retinoic acid in the developing ventral optic cup, with mutations resulting in defects in optic cup formation, including invagination [48,250].
4. Current Research and Future Directions
4.1. Modelling Eye Development Using iPSCs
Current understanding of eye development is heavily based on evidence from animal modelling, focusing on zebrafish, Xenopus and mouse [76,253,254]. While conserved molecular pathways indicate a fundamental similarity across vertebrate eye development, conflicting evidence from experiments performed on different model organisms indicates a degree of divergence in the morphogenesis of the eye [255,256]. For example, the development of the cornea is known to vary by species; stratification of the human corneal epithelium occurs early in development, resembling chick development, while the development of the mouse corneal epithelium does not fully stratify until 10 weeks after birth [255]. Furthermore, the migration of neural crest cells which form the corneal endothelium and stromal keratocytes occurs in two waves in reptiles, birds and primates, while in rodents, cats, rabbits and cattle, this occurs in a single wave of cell migration [256]. To comprehensively understand the molecular basis of human developmental eye defects and ultimately, develop functionally viable therapies, it is vital to obtain an accurate understanding of human eye development, and appreciate how it differs from that of model organisms.
One method for understanding human biology is the use of human induced pluripotent stem cells (hiPSCs) [257,258]. Long-term modelling of hiPSCs can enhance our understanding of normal human cellular behaviour, as well as better comprehending diseases by deriving patient iPSCs which can be used for investigating the basis of phenotypic heterogeneity, in addition to therapy screening and personalised medicine [257,259]. Current work in developing methodologies for organoid modelling of eye development show potential for gaining a working understanding of human eye morphogenesis, and developing therapies [259,260]. RNA-seq transcriptome analysis indicates that stem-cell derived optic cups recapitulate human eye development in vivo, through temporal expression of cell differentiation markers and retinal disease genes, as well as mRNA alternative splicing [261,262,263]. Early-stage optic vesicle-like structures generated from hiPSCs derived from a microphthalmic patient with a VSX2 null variant showed upregulation of WNT pathway components and RPE marker, MITF, which were rescued by pharmacological inhibition of WNT signalling [232]. These results showed a key role for VSX2 in WNT signalling and maintenance of the neural retina through WNT pathway suppression [232]. This study shows the potential utility of hiPSC modelling in improving understanding of human eye development, and developing novel therapies. However, a constraint of in vitro modelling is the lack of vasculature and surrounding tissues, which may result in loss of vital external cues for recapitulating in vivo development [264]. Additionally, whole organism modelling is necessary for understanding the effect of gene activity on organogenesis of multiple organs, which is particularly important when studying syndromic disorders [265]. Utilising knowledge gleaned from in vivo animal modelling in conjunction with human cellular modelling is key to obtaining a complete picture of the molecular basis of human eye development and the role of perturbed genetic pathways in congenital disorders, such as AM.
4.2. Genomics and Epigenetics
The functionality and availability of genetic tools has progressed rapidly in recent years. Next-generation sequencing is a powerful tool for understanding development and disease, allowing relatively cheap, high-throughput methods for analysing the genome, transcriptome and epigenome [266]. Whole exome/genome sequencing can identify novel disease-associated coding and non-coding variants, as well as be a useful tool in diagnosing patients with heterogeneous ocular disorders with numerous underlying genetic causes, such as AM (Table 1) [81,267]. Increasing the proportion of known variants associated with AM will improve the frequency of genetic diagnosis, which is important for directing disease management, including screening for potential syndromic features, as well as providing reliable genetic counselling and family planning [11,85]. RNA-seq has aided in characterising gene expression in the developing retina, as well as elucidating genetic pathways affected in ocular disorders through differential transcriptomic analysis, such as the role of VSX2 in regulation of the WNT pathway in retinal organoid differentiation [232,268,269]. Improved understanding of temporal gene expression throughout eye development will aid in identifying potential disease-associated variants, as well as putative therapy targets.
A potential source of disruption to the molecular regulation of the developmental pathway is epigenetic disturbance. Epigenetic changes, such as chromatin remodelling and histone modification, result in altered gene regulation, and therefore, have a great potential for disrupting the ocular developmental pathway [270,271]. Heterogeneity in epigenetic patterning in stem cell cultures has been shown to impact the ability of cells to differentiate into retinae, providing reasonable evidence that epigenetic regulation is important to normal eye development, and consequentially, may play a role in ocular malformations [272]. Epigenetic changes altering the secondary structure of DNA have been shown to induce neurodevelopmental disorders including autism [273]. Utilising epigenetic profiling techniques such as CHIP-seq to identify epigenetic variation associated with developmental ocular disorders will provide a better understanding of how these disorders arise, in addition to providing a new avenue of putative treatments to explore [266,274,275].
4.3. Proteomics and Metabolomics
Post-translational modifications can have a significant impact on protein activity, and consequently, cellular function, for example, post-translational modifications play a key role in the regulation of zebrafish retina regeneration [276,277,278]. Consequently, investigating the proteome alongside the transcriptome is key to understanding cellular behaviour in eye development. Additionally, the impact of downstream metabolites in temporal mediation of cellular behaviour has been revealed in recent years [279]. Advances in metabolomic investigation, such as metabolome profiling, will provide greater insight into different aspects of cellular processes and how metabolites impact development, in addition to providing new biomarkers for understanding the pathways disrupted in disease and targets for neonatal screening therapies [280,281].
4.4. Therapeutics
One of the fundamental reasons for investigating the molecular basis of human eye development and disease is to manipulate the knowledge gained into targeted therapies that will benefit patients. As AM is a structural eye defect arising from three weeks gestation, developing prenatal treatments has been difficult as the mother may not know they are pregnant and compounds may have an adverse effect on both the mother or the rest of the developing foetus. However, with the first in utero exploratory multicentre phase I/II clinical trial (NCT03706482) evaluating safety and efficacy of postnatal or prenatal intravenous administration of allogeneic expanded foetal mesenchymal stem cells for the treatment of severe osteogenesis imperfecta underway, there may be potential applicability for congenital ocular disorders in the future.
Small-molecule drugs, such as Ataluren, target in-frame nonsense mutations by interfering with ribosomal fidelity to enable near-cognate amino acids to be substituted rather than a release factor, thus enabling the production of full-length functional proteins. This drug had positive results as a postnatal treatment of aniridia in mouse models, demonstrating developmental plasticity of the eye. A phase 2 multicentre randomized, double-masked, placebo-controlled clinical trial (NCT02647359) is currently underway to assess the safety and efficacy of Ataluren for nonsense-mediated aniridia. If successful, this presents another avenue of post-natal treatment for certain genetic congenital diseases such as microphthalmia, where growth and visual maturation may be encouraged [282].
5. Conclusions
This review has highlighted the key molecular coordinators of early-eye development, and summarized current understanding of how disruption of these precise spatiotemporal regulators can lead to disruption of eye organogenesis, resulting in anophthalmia or microphthalmia (AM). Recent advances in the field of genetics, including high-throughput next-generation sequencing technology has progressed knowledge of the genetic basis of AM, as well as improving diagnosis and genetic counselling [85]. Increasing genetic screening of AM patients using whole exome/genome sequencing will continue to improve the rate of diagnosis, through identifying novel disease-associated genes and isoforms [85]. Furthermore, increased genetic diagnosis will provide larger cohorts to analyse the AM genotype/phenotype relationship, including identifying environmental factors or genetic modifiers which may result in heterogeneity and variable expressivity observed in AM [85]. Additionally, utilising animal and cellular models of AM, in conjunction with genome-editing technology such as CRISPR, will further advance understanding of function and interactions of molecular regulators of eye development, and how pathogenic alleles result in AM, with the ultimate aim of creating therapies that may aid patients with AM [85,232].
Funding
This research was funded by The Wellcome Trust, grant number 205174/Z/16/Z; and Moorfields Eye Charity.
Acknowledgments
Mariya Moosajee gratefully acknowledges the support of the Wellcome Trust, Moorfields Eye Charity and National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Graw, J. Eye Development. Curr. Top. Dev. Biol. 2010, 90, 343–386. [Google Scholar] [CrossRef]
- Sinn, R. An eye on eye development. Mech. Dev. 2013, 130, 347–358. [Google Scholar] [CrossRef]
- Chow, R.L.; Lang, R.A. Early Eye Development in Vertebrates. Annu. Rev. Cell Dev. Biol. 2001, 17, 255–296. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, S. Eye Morphogenesis and Patterning of the Optic Vesicle. Curr. Top. Dev. Biol. 2010, 93, 61–84. [Google Scholar] [CrossRef] [Green Version]
- FitzPatrick, D.R.; van Heyningen, V. Developmental eye disorders. Curr. Opin. Genet. Dev. 2005, 15, 348–353. [Google Scholar] [CrossRef]
- Plaisancié, J.; Ceroni, F.; Holt, R.; Zazo Seco, C.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia. Hum. Genet. 2019, 1–32. [Google Scholar] [CrossRef]
- Chassaing, N.; Causse, A.; Vigouroux, A.; Delahaye, A.; Alessandri, J.-L.; Boespflug-Tanguy, O.; Boute-Benejean, O.; Dollfus, H.; Duban-Bedu, B.; Gilbert-Dussardier, B.; et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin. Genet. 2014, 86, 326–334. [Google Scholar] [CrossRef]
- Slavotinek, A. Genetics of anophthalmia and microphthalmia. Part 2: Syndromes associated with anophthalmia–microphthalmia. Hum. Genet. 2018, 1–16. [Google Scholar] [CrossRef]
- Verma, A.S.; FitzPatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef]
- Schneider, A.; Bardakjian, T.; Reis, L.M.; Tyler, R.C.; Semina, E.V. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am. J. Med. Genet. Part A 2009, 149A, 2706–2715. [Google Scholar] [CrossRef]
- Richardson, R.; Sowden, J.; Gerth-Kahlert, C.; Moore, A.T.; Moosajee, M. Clinical utility gene card for: Non-Syndromic Microphthalmia Including Next-Generation Sequencing-Based Approaches. Eur. J. Hum. Genet. 2017, 25. [Google Scholar] [CrossRef]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Shah, S.P.; Taylor, A.E.; Sowden, J.C.; Ragge, N.K.; Russell-Eggitt, I.; Rahi, J.S.; Gilbert, C.E. Anophthalmos, Microphthalmos, and Typical Coloboma in the United Kingdom: A Prospective Study of Incidence and Risk. Investig. Opthalmol. Vis. Sci. 2011, 52, 558–564. [Google Scholar] [CrossRef]
- Bernstein, C.S.; Anderson, M.T.; Gohel, C.; Slater, K.; Gross, J.M.; Agarwala, S. The cellular bases of choroid fissure formation and closure. Dev. Biol. 2018, 440, 137–151. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Williams, M.J.; Halford, S.; Gregory-Evans, K. Ocular coloboma: A reassessment in the age of molecular neuroscience. J. Med. Genet. 2004, 41, 881–891. [Google Scholar] [CrossRef]
- Ragge, N.K.; Subak-Sharpe, I.D.; Collin, J.R.O. A practical guide to the management of anophthalmia and microphthalmia. Eye 2007, 21, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Slavotinek, A.M. Eye development genes and known syndromes. Mol. Genet. Metab. 2011, 104, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.; Bakrania, P.; Bunyan, D.J.; Osborne, R.J.; Crolla, J.A.; Salt, A.; Ayuso, C.; Newbury-Ecob, R.; Abou-Rayyah, Y.; Collin, J.R.O.; et al. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia, microphthalmia and coloboma. Hum. Mutat. 2008, 29, E278–E283. [Google Scholar] [CrossRef]
- Gerth-Kahlert, C.; Williamson, K.; Ansari, M.; Rainger, J.K.; Hingst, V.; Zimmermann, T.; Tech, S.; Guthoff, R.F.; van Heyningen, V.; FitzPatrick, D.R. Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Mol. Genet. Genomic Med. 2013, 1, 15–31. [Google Scholar] [CrossRef]
- Casey, J.; Kawaguchi, R.; Morrissey, M.; Sun, H.; McGettigan, P.; Nielsen, J.E.; Conroy, J.; Regan, R.; Kenny, E.; Cormican, P.; et al. First implication of STRA6 mutations in isolated anophthalmia, microphthalmia, and coloboma: A new dimension to the STRA6 phenotype. Hum. Mutat. 2011, 32, 1417–1426. [Google Scholar] [CrossRef]
- Slavotinek, A.M.; Garcia, S.T.; Chandratillake, G.; Bardakjian, T.; Ullah, E.; Wu, D.; Umeda, K.; Lao, R.; Ling, P.; Tang, P.L.-F.; et al. Exome Sequencing in 32 Patients with Anophthalmia/Microphthalmia and Developmental Eye Defects. Clin. Genet. 2015, 88, 468–473. [Google Scholar] [CrossRef]
- Givens, K.T.; Lee, D.A.; Jones, T.; Ilstrup, D.M. Congenital rubella syndrome: Ophthalmic manifestations and associated systemic disorders. Br. J. Ophthalmol. 1993, 77, 358–363. [Google Scholar] [CrossRef]
- Busby, A.; Dolk, H.; Armstrong, B. Eye anomalies: Seasonal variation and maternal viral infections. Epidemiology 2005, 16, 317–322. [Google Scholar] [CrossRef]
- Zagozewski, J.; Zhang, Q.; Eisenstat, D. Genetic regulation of vertebrate eye development. Clin. Genet. 2014, 86, 453–460. [Google Scholar] [CrossRef]
- Zuber, M.E.; Gestri, G.; Viczian, A.S.; Barsacchi, G.; Harris, W.A. Specification of the vertebrate eye by a network of eye field transcription factors. Development 2003, 130, 5155–5167. [Google Scholar] [CrossRef] [Green Version]
- Loosli, F.; Winkler, S.; Wittbrodt, J. Six3 overexpression initiates the formation of ectopic retina. Genes Dev. 1999, 13, 649–654. [Google Scholar] [CrossRef]
- Chuang, J.C.; Raymond, P.A. Embryonic origin of the eyes in teleost fish. BioEssays 2002, 24, 519–529. [Google Scholar] [CrossRef]
- Danno, H.; Michiue, T.; Hitachi, K.; Yukita, A.; Ishiura, S.; Asashima, M. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate Rax expression. Proc. Natl. Acad. Sci. USA 2008, 105, 5408–5413. [Google Scholar] [CrossRef]
- Medina-Martinez, O.; Amaya-Manzanares, F.; Liu, C.; Mendoza, M.; Shah, R.; Zhang, L.; Behringer, R.R.; Mahon, K.A.; Jamrich, M. Cell-Autonomous Requirement for Rx Function in the Mammalian Retina and Posterior Pituitary. PLoS ONE 2009, 4, e4513. [Google Scholar] [CrossRef]
- Mathers, P.H.; Grinberg, A.; Mahon, K.A.; Jamrich, M. The Rx homeobox gene is essential for vertebrate eye development. Nature 1997, 387, 603–607. [Google Scholar] [CrossRef]
- Lagutin, O.V.; Zhu, C.C.; Kobayashi, D.; Topczewski, J.; Shimamura, K.; Puelles, L.; Russell, H.R.C.; McKinnon, P.J.; Solnica-Krezel, L.; Oliver, G. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 2003, 17, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagozewski, J.; Zhang, Q.; Pinto, V.; Wigle, J.; Eisenstat, D. The role of homeobox genes in retinal development and disease. Dev. Biol. 2014, 393, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazin-Lopez, N.; Valdivia, L.E.; Wilson, S.W.; Gestri, G. Watching eyes take shape. Curr. Opin. Genet. Dev. 2015, 32, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Speirs, C.; Lagutin, O.; Inbal, A.; Liu, W.; Solnica-Krezel, L.; Jeong, Y.; Epstein, D.J.; Oliver, G. Haploinsufficiency of Six3 Fails to Activate Sonic hedgehog Expression in the Ventral Forebrain and Causes Holoprosencephaly. Dev. Cell 2008, 15, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carl, M.; Wittbrodt, J. Graded interference with FGF signalling reveals its dorsoventral asymmetry at the mid-hindbrain boundary. Development 1999, 126, 5659–5667. [Google Scholar] [PubMed]
- Rebagliati, M.R.; Toyama, R.; Haffter, P.; Dawid, I.B. Cyclops encodes a nodal-related factor involved in midline signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 9932–9937. [Google Scholar] [CrossRef]
- Varga, Z.M.; Wegner, J.; Westerfield, M. Anterior movement of ventral diencephalic precursors separates the primordial eye field in the neural plate and requires cyclops. Development 1999, 126, 5533–5546. [Google Scholar]
- Brown, K.E.; Keller, P.J.; Ramialison, M.; Rembold, M.; Stelzer, E.H.K.; Loosli, F.; Wittbrodt, J. Nlcam modulates midline convergence during anterior neural plate morphogenesis. Dev. Biol. 2010, 339, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Rembold, M.; Loosli, F.; Adams, R.J.; Wittbrodt, J. Individual Cell Migration Serves as the Driving Force for Optic Vesicle Evagination. Science 2006, 313, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Cvekl, A.; Ashery-Padan, R. The cellular and molecular mechanisms of vertebrate lens development. Development 2014, 141, 4432–4447. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Lagutin, O.V.; Mende, M.; Streit, A.; Oliver, G. Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J. 2006, 25, 5383–5395. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; McGreal, R.; Liu, W. Lens Development and Crystallin Gene Expression. Prog. Mol. Biol. Transl. Sci. 2015, 134, 129–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-T.; Kim, J.W. Compartmentalization of vertebrate optic neuroephithelium: External cues and transcription factors. Mol. Cells 2012, 33, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Furuta, Y.; Hogan, B.L.M. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998, 12, 3764–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.; Saijoh, Y.; Hirokawa, K.E.; Kopinke, D.; Murtaugh, L.C.; Monuki, E.S.; Levine, E.M. Lhx2 links the intrinsic and extrinsic factors that control optic cup formation. Development 2009, 136, 3895–3906. [Google Scholar] [CrossRef] [PubMed]
- Hyer, J.; Kuhlman, J.; Afif, E.; Mikawa, T. Optic cup morphogenesis requires pre-lens ectoderm but not lens differentiation. Dev. Biol. 2003, 259, 351–363. [Google Scholar] [CrossRef]
- Cvekl, A.; Wang, W.-L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Mic, F.A.; Molotkov, A.; Molotkova, N.; Duester, G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev. Dyn. 2004, 231, 270–277. [Google Scholar] [CrossRef]
- Molotkov, A.; Molotkova, N.; Duester, G.; Lemke, G.; Bertuzzi, S. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Matt, N.; Ghyselinck, N.B.; Pellerin, I.; Dupé, V. Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev. Biol. 2008, 320, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, A.T.; Robertson, E.J. Overlapping expression domains of bone morphogenetic protein family members potentially account for limited tissue defects in BMP7 deficient embryos. Dev. Dyn. 1997, 208, 349–362. [Google Scholar] [CrossRef]
- Chauhan, B.K.; Disanza, A.; Choi, S.-Y.; Faber, S.C.; Lou, M.; Beggs, H.E.; Scita, G.; Zheng, Y.; Lang, R.A. Cdc42-and IRSp53-dependent contractile filopodia tether presumptive lens and retina to coordinate epithelial invagination. Development 2009, 136, 3657–3667. [Google Scholar] [CrossRef] [PubMed]
- Oltean, A.; Huang, J.; Beebe, D.C.; Taber, L.A. Tissue growth constrained by extracellular matrix drives invagination during optic cup morphogenesis. Biomech. Model. Mechanobiol. 2016, 15, 1405–1421. [Google Scholar] [CrossRef] [PubMed]
- Plageman, T.F.; Chauhan, B.K.; Yang, C.; Jaudon, F.; Shang, X.; Zheng, Y.; Lou, M.; Debant, A.; Hildebrand, J.D.; Lang, R.A. A Trio-RhoA-Shroom3 pathway is required for apical constriction and epithelial invagination. Development 2011, 138, 5177–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rajagopal, R.; Liu, Y.; Dattilo, L.K.; Shaham, O.; Ashery-Padan, R.; Beebe, D.C. The mechanism of lens placode formation: A case of matrix-mediated morphogenesis. Dev. Biol. 2011, 355, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plageman, T.F.; Chung, M.-I.; Lou, M.; Smith, A.N.; Hildebrand, J.D.; Wallingford, J.B.; Lang, R.A. Pax6-dependent Shroom3 expression regulates apical constriction during lens placode invagination. Development 2010, 137, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, S.; Levine, E.M.; Reh, T.A. Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development 2000, 127, 4599–4609. [Google Scholar]
- Hägglund, A.-C.; Berghard, A.; Carlsson, L. Canonical Wnt/β-Catenin Signalling Is Essential for Optic Cup Formation. PLoS ONE 2013, 8, e81158. [Google Scholar] [CrossRef]
- Nguyen, M.; Arnheiter, H.; Fuhrmann, S. Signaling and transcriptional regulation in early mammalian eye development: A link between FGF and MITF. Development 2000, 127, 3581–3591. [Google Scholar]
- Capowski, E.E.; Simonett, J.M.; Clark, E.M.; Wright, L.S.; Howden, S.E.; Wallace, K.A.; Petelinsek, A.M.; Pinilla, I.; Phillips, M.J.; Meyer, J.S.; et al. Loss of MITF expression during human embryonic stem cell differentiation disrupts retinal pigment epithelium development and optic vesicle cell proliferation. Hum. Mol. Genet. 2014, 23, 6332–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Morales, J.R.; Dolez, V.; Rodrigo, I.; Zaccarini, R.; Leconte, L.; Bovolenta, P.; Saule, S. OTX2 activates the molecular network underlying retina pigment epithelium differentiation. J. Biol. Chem. 2003, 278, 21721–21731. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morales, J.R.; Signore, M.; Acampora, D.; Simeone, A.; Bovolenta, P. Otx genes are required for tissue specification in the developing eye. Development 2001, 128, 2019–2030. [Google Scholar] [PubMed]
- Vogel-Höpker, A.; Momose, T.; Rohrer, H.; Yasuda, K.; Ishihara, L.; Rapaport, D.H. Multiple functions of fibroblast growth factor-8 (FGF-8) in chick eye development. Mech. Dev. 2000, 94, 25–36. [Google Scholar] [CrossRef]
- Liang, L.; Sandell, J.H. Focus on molecules: Homeobox protein Chx10. Exp. Eye Res. 2008, 86, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Burkitt Wright, E.M.M.; Perveen, R.; Bowers, N.; Ramsden, S.; McCann, E.; O’Driscoll, M.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C.M. VSX2 in microphthalmia: A novel splice site mutation producing a severe microphthalmia phenotype. Br. J. Ophthalmol. 2010, 94, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Conger, S.B.; Burmeister, M. Mapping of genetic modifiers affecting the eye phenotype of ocular retardation (Chx10or-J) mice. Mamm. Genome 2006, 17, 518–525. [Google Scholar] [CrossRef]
- Horsford, D.J.; Nguyen, M.-T.T.; Sellar, G.C.; Kothary, R.; Arnheiter, H.; McInnes, R.R. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 2005, 132, 177–187. [Google Scholar] [CrossRef]
- Burmeister, M.; Novak, J.; Liang, M.-Y.; Basu, S.; Ploder, L.; Hawes, N.L.; Vidgen, D.; Hoover, F.; Goldman, D.; Kalnins, V.I.; et al. Ocular retardation mouse caused by Chx10 homeobox null allele: Impaired retinal progenitor proliferation and bipolar cell differentiation. Nat. Genet. 1996, 12, 376–384. [Google Scholar] [CrossRef]
- Marquardt, T.; Ashery-Padan, R.; Andrejewski, N.; Scardigli, R.; Guillemot, F.; Gruss, P. Pax6 Is Required for the Multipotent State of Retinal Progenitor Cells. Cell 2001, 105, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.; Gómez-Pardo, E.; Gruss, P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development 1996, 122, 3381–3391. [Google Scholar] [PubMed]
- Schwarz, M.; Cecconi, F.; Bernier, G.; Andrejewski, N.; Kammandel, B.; Wagner, M.; Gruss, P. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 2000, 127, 4325–4334. [Google Scholar]
- Hallonet, M.; Hollemann, T.; Pieler, T.; Gruss, P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999, 13, 3106–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Take-uchi, M.; Clarke, J.D.W.; Wilson, S.W. Hedgehog signalling maintains the optic stalk-retinal interface through the regulation of Vax gene activity. Development 2003, 130, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mui, S.H.; Kim, J.W.; Lemke, G.; Bertuzzi, S. Vax genes ventralize the embryonic eye. Genes Dev. 2005, 19, 1249–1259. [Google Scholar] [CrossRef]
- Richardson, R.; Tracey-White, D.; Webster, A.; Moosajee, M. The zebrafish eye-a paradigm for investigating human ocular genetics. Eye 2017, 31, 68–86. [Google Scholar] [CrossRef]
- Liu, C.; Nathans, J.; Bovolenta, P.; Alfano, G.; Marchitiello, A.; Mocchetti, C.; Crippa, L.; Bulfone, A.; Marigo, V.; Ballabio, A.; et al. An essential role for frizzled 5 in mammalian ocular development. Development 2008, 135, 3567–3576. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Bakeri, H.; Li, T.; Swaroop, A. Regulation of retinal progenitor expansion by Frizzled receptors: Implications for microphthalmia and retinal coloboma. Hum. Mol. Genet. 2012, 21, 1848–1860. [Google Scholar] [CrossRef]
- Knickmeyer, M.D.; Mateo, J.L.; Eckert, P.; Roussa, E.; Rahhal, B.; Zuniga, A.; Krieglstein, K.; Wittbrodt, J.; Heermann, S. TGFβ-facilitated optic fissure fusion and the role of bone morphogenetic protein antagonism. Open Biol. 2018, 8, 170134. [Google Scholar] [CrossRef]
- Ragge, N.K.; Brown, A.G.; Poloschek, C.M.; Lorenz, B.; Henderson, R.A.; Clarke, M.P.; Russell-Eggitt, I.; Fielder, A.; Gerrelli, D.; Martinez-Barbera, J.P.; et al. Heterozygous Mutations of OTX2 Cause Severe Ocular Malformations. Am. J. Hum. Genet. 2005, 76, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Riera, M.; Wert, A.; Nieto, I.; Pomares, E. Panel-based whole exome sequencing identifies novel mutations in microphthalmia and anophthalmia patients showing complex Mendelian inheritance patterns. Mol. Genet. Genomic Med. 2017, 5, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Gilbert-Dussardier, B.; Nicot, F.; Fermeaux, V.; Encha-Razavi, F.; Fiorenza, M.; Toutain, A.; Calvas, P. Germinal mosaicism and familial recurrence of aSOX2 mutation with highly variable phenotypic expression extending from AEG syndrome to absence of ocular involvement. Am. J. Med. Genet. Part A 2007, 143A, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Williamson, K.A.; Faber, V.; Laurent, N.; Grimaldi, M.; Thauvin-Robinet, C.; Durand, C.; Mugneret, F.; Gouyon, J.-B.; Bron, A.; et al. Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. Am. J. Med. Genet. Part A 2006, 140A, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Plaisancie, J.; Calvas, P.; Chassaing, N. Genetic Advances in Microphthalmia. J. Pediatr. Genet. 2016, 5, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Taylor, A.E.; Sowden, J.C.; Ragge, N.; Russell-Eggitt, I.; Rahi, J.S.; Gilbert, C.E. Anophthalmos, Microphthalmos, and Coloboma in the United Kingdom: Clinical Features, Results of Investigations, and Early Management. Ophthalmology 2012, 119, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Chitayat, D.; Caron, V.; Chassaing, N.; Bitoun, P.; Patry, L.; Cordier, M.-P.; Capo-Chichi, J.-M.; Francannet, C.; Calvas, P.; et al. Recessive and Dominant Mutations in Retinoic Acid Receptor Beta in Cases with Microphthalmia and Diaphragmatic Hernia. Am. J. Hum. Genet. 2013, 93, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Srour, M.; Caron, V.; Pearson, T.; Nielsen, S.B.; Lévesque, S.; Delrue, M.-A.; Becker, T.A.; Hamdan, F.F.; Kibar, Z.; Sattler, S.G.; et al. Gain-of-Function Mutations in RARB Cause Intellectual Disability with Progressive Motor Impairment. Hum. Mutat. 2016, 37, 786–793. [Google Scholar] [CrossRef]
- Ramirez-Botero, A.F.; Pachajoa, H. Syndromic microphthalmia-3 caused by a mutation on gene SOX2 in a Colombian male patient. Congenit. Anom. 2016, 56, 250–252. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, J.; Pelcastre, E.L.; Tovilla-Canales, J.L.; Garcia-Ortiz, J.E.; Amato-Almanza, M.; Villanueva-Mendoza, C.; Espinosa-Mattar, Z.; Zenteno, J.C. Mutational screening of CHX10, GDF6, OTX2, RAX and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br. J. Ophthalmol. 2010, 94, 1100–1104. [Google Scholar] [CrossRef]
- Schilter, K.F.; Schneider, A.; Bardakjian, T.; Soucy, J.-F.; Tyler, R.C.; Reis, L.M.; Semina, E.V. OTX2 microphthalmia syndrome: Four novel mutations and delineation of a phenotype. Clin. Genet. 2011, 79, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Ohtake, A.; Hoshino, M.; Amemiya, S.; Sasaki, N.; Ishizu, K.; Fujieda, K. OTX2 Loss of Function Mutation Causes Anophthalmia and Combined Pituitary Hormone Deficiency with a Small Anterior and Ectopic Posterior Pituitary. J. Clin. Endocrinol. Metab. 2009, 94, 314–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronina, V.A.; Kozhemyakina, E.A.; O’Kernick, C.M.; Kahn, N.D.; Wenger, S.L.; Linberg, J.V.; Schneider, A.S.; Mathers, P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2003, 13, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Iseri, S.U.; Wyatt, A.W.; Nürnberg, G.; Kluck, C.; Nürnberg, P.; Holder, G.E.; Blair, E.; Salt, A.; Ragge, N.K. Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum. Genet. 2010, 128, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Ragge, N.; Kariminejad, A.; Buffet, A.; Ghaderi-Sohi, S.; Martinovic, J.; Calvas, P. Mutation analysis of the STRA6 gene in isolated and non-isolated anophthalmia/microphthalmia. Clin. Genet. 2013, 83, 244–250. [Google Scholar] [CrossRef]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; FitzPatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 Cause a Broad Spectrum of Malformations Including Anophthalmia, Congenital Heart Defects, Diaphragmatic Hernia, Alveolar Capillary Dysplasia, Lung Hypoplasia, and Mental Retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681. [Google Scholar] [CrossRef]
- Fares-Taie, L.; Gerber, S.; Chassaing, N.; Clayton-Smith, J.; Hanein, S.; Silva, E.; Serey, M.; Serre, V.; Gérard, X.; Baumann, C.; et al. ALDH1A3 Mutations Cause Recessive Anophthalmia and Microphthalmia. Am. J. Hum. Genet. 2013, 92, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Abouzeid, H.; Favez, T.; Schmid, A.; Agosti, C.; Youssef, M.; Marzouk, I.; El Shakankiry, N.; Bayoumi, N.; Munier, F.L.; Schorderet, D.F. Mutations in ALDH1A3 Represent a Frequent Cause of Microphthalmia/Anophthalmia in Consanguineous Families. Hum. Mutat. 2014, 35, 949–953. [Google Scholar] [CrossRef]
- Plaisancié, J.; Brémond-Gignac, D.; Demeer, B.; Gaston, V.; Verloes, A.; Fares-Taie, L.; Gerber, S.; Rozet, J.-M.; Calvas, P.; Chassaing, N. Incomplete penetrance of biallelic ALDH1A3 mutations. Eur. J. Med. Genet. 2016, 59, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Bakrania, P.; Efthymiou, M.; Klein, J.C.; Salt, A.; Bunyan, D.J.; Wyatt, A.; Ponting, C.P.; Martin, A.; Williams, S.; Lindley, V.; et al. Mutations in BMP4 Cause Eye, Brain, and Digit Developmental Anomalies: Overlap between the BMP4 and Hedgehog Signaling Pathways. Am. J. Hum. Genet. 2008, 82, 304–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, L.M.; Tyler, R.C.; Schilter, K.F.; Abdul-Rahman, O.; Innis, J.W.; Kozel, B.A.; Schneider, A.S.; Bardakjian, T.M.; Lose, E.J.; Martin, D.M.; et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011, 130, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.W.; Osborne, R.J.; Stewart, H.; Ragge, N.K. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum. Mutat. 2010, 31, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Berry-Wynne, K.M.; Asai-Coakwell, M.; Sundaresan, P.; Footz, T.; French, C.R.; Abitbol, M.; Fleisch, V.C.; Corbett, N.; Allison, W.T.; et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010, 19, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, F.; Bu, J.; Liu, X.; Du, W.; Dong, J.; Cooney, J.D.; Dubey, S.K.; Shi, Y.; Gong, B.; et al. ABCB6 Mutations Cause Ocular Coloboma. Am. J. Hum. Genet. 2012, 90, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.; Logan, C.V.; McKibbin, M.; Sheridan, E.; Elçioglu, N.H.; Yenice, O.; Parry, D.A.; Fernandez-Fuentes, N.; Abdelhamed, Z.I.A.; Al-Maskari, A.; et al. Next generation sequencing identifies mutations in Atonal homolog 7 (ATOH7) in families with global eye developmental defects. Hum. Mol. Genet. 2012, 21, 776–783. [Google Scholar] [CrossRef]
- Zahrani, F.; Aldahmesh, M.A.; Alshammari, M.J.; Al-Hazzaa, S.A.F.; Alkuraya, F.S. Mutations in C12orf57 Cause a Syndromic Form of Colobomatous Microphthalmia. Am. J. Hum. Genet. 2013, 92, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, N.; Ragge, N.; Plaisancié, J.; Patat, O.; Geneviève, D.; Rivier, F.; Malrieu-Eliaou, C.; Hamel, C.; Kaplan, J.; Calvas, P. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Am. J. Med. Genet. Part A 2016, 170, 1895–1898. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Mohammed, J.Y.; Al-Hazzaa, S.; Alkuraya, F.S. Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genet. Med. 2012, 14, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Slavotinek, A.M.; Chao, R.; Vacik, T.; Yahyavi, M.; Abouzeid, H.; Bardakjian, T.; Schneider, A.; Shaw, G.; Sherr, E.H.; Lemke, G.; et al. VAX1 mutation associated with microphthalmia, corpus callosum agenesis, and orofacial clefting: The first description of a VAX1 phenotype in humans. Hum. Mutat. 2012, 33, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Okada, I.; Hamanoue, H.; Terada, K.; Tohma, T.; Megarbane, A.; Chouery, E.; Abou-Ghoch, J.; Jalkh, N.; Cogulu, O.; Ozkinay, F.; et al. SMOC1 Is Essential for Ocular and Limb Development in Humans and Mice. Am. J. Hum. Genet. 2011, 88, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Koshimizu, E.; Megarbane, A.; Hamanoue, H.; Okada, I.; Nishiyama, K.; Kodera, H.; Miyatake, S.; Tsurusaki, Y.; Nakashima, M.; et al. Whole-exome sequencing identified a homozygous FNBP4 mutation in a family with a condition similar to microphthalmia with limb anomalies. Am. J. Med. Genet. Part A 2013, 161, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A.; de la Cruz, J.; Lewis, R.A.; Karkera, J.D.; Manligas, G.S.; Roessler, E.; Muenke, M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. 2003, 116, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Bakrania, P.; Ugur Iseri, S.A.; Wyatt, A.W.; Bunyan, D.J.; Lam, W.W.K.; Salt, A.; Ramsay, J.; Robinson, D.O.; Ragge, N.K. Sonic hedgehog mutations are an uncommon cause of developmental eye anomalies. Am. J. Med. Genet. Part A 2010, 152A, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Esmailpour, T.; Riazifar, H.; Liu, L.; Donkervoort, S.; Huang, V.H.; Madaan, S.; Shoucri, B.M.; Busch, A.; Wu, J.; Towbin, A.; et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 2014, 51, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Ragge, N.; Isidor, B.; Bitoun, P.; Odent, S.; Giurgea, I.; Cogné, B.; Deb, W.; Vincent, M.; Le Gall, J.; Morton, J.; et al. Expanding the phenotype of the X-linked BCOR microphthalmia syndromes. Hum. Genet. 2018, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B.; et al. Mutations of the Mitochondrial Holocytochrome c–Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef]
- Rainger, J.; Pehlivan, D.; Johansson, S.; Bengani, H.; Sanchez-Pulido, L.; Williamson, K.A.; Ture, M.; Barker, H.; Rosendahl, K.; Spranger, J.; et al. Monoallelic and Biallelic Mutations in MAB21L2 Cause a Spectrum of Major Eye Malformations. Am. J. Hum. Genet. 2014, 94, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.M.; Nelson, C.; Tarlé, S.A.; Pribila, J.T.; Bardakjian, T.; Woods, S.; Schneider, A.; Glaser, T. Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell 2015, 161, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Bertolacini, C.D.P.; Ribeiro-Bicudo, L.A.; Petrin, A.; Richieri-Costa, A.; Murray, J.C. Clinical findings in patients with GLI2 mutations-phenotypic variability. Clin. Genet. 2012, 81, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Guion-Almeida, M.L.; Richieri-Costa, A.; Zechi-Ceide, R.M. Holoprosencephaly spectrum, ano/microphthalmia, and first branchial arch defects: Evidence for a new disorder. Clin. Dysmorphol. 2008, 17, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L.A.; Guerini Rde, C.; Richieri-Costa, A. Holoprosencephaly with microphthalmia, hypoplastic ears, vertebral segmentation defects, and congenital heart defects. Am. J. Med. Genet. Part A 2005, 136A, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Govaerts, K.; Van Esch, H.; Verbeeck, J.; Tuomi, M.-L.; HeikkilÃ, H.; Torniainen, S.; Devriendt, K.; Fryns, J.-P.; Marynen, P.; et al. Novel PORCN mutations in focal dermal hypoplasia. Clin. Genet. 2009, 76, 535–543. [Google Scholar] [CrossRef]
- Leoyklang, P.; Suphapeetiporn, K.; Wananukul, S.; Shotelersuk, V. Three novel mutations in the PORCN gene underlying focal dermal hypoplasia. Clin. Genet. 2008, 73, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.; Li, C.; Sherr, E.H.; Chudley, A.E. Mutation analysis of the FRAS1 gene demonstrates new mutations in a propositus with Fraser syndrome. Am. J. Med. Genet. Part A 2006, 140A, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Baranzini, S.E.; Schanze, D.; Labelle-Dumais, C.; Short, K.M.; Chao, R.; Yahyavi, M.; Bijlsma, E.K.; Chu, C.; Musone, S.; et al. Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J. Med. Genet. 2011, 48, 375–382. [Google Scholar] [CrossRef]
- Shaw, N.D.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Erdin, S.; Williamson, K.A.; Rainger, J.; Stortchevoi, A.; et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, M.E.; Rodríguez de Córdoba, S.; Schneider, A.S.; Dwyer, M.A.; Ayuso, C.; Bovolenta, P. Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am. J. Med. Genet. Part A 2004, 129, 92–94. [Google Scholar] [CrossRef]
- Gestri, G.; Osborne, R.J.; Wyatt, A.W.; Gerrelli, D.; Gribble, S.; Stewart, H.; Fryer, A.; Bunyan, D.J.; Prescott, K.; Collin, J.R.O.; et al. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Hum. Genet. 2009, 126, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Faqeih, E.; Seidahmed, M.Z.; Sunker, A.; Alali, F.E.; Khadijah, A.; Alkuraya, F.S. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum. Mutat. 2011, 32, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Shamseldin, H.E.; Loucks, C.M.; Seidahmed, M.Z.; Ansari, S.; Ibrahim Khalil, M.; Al-Yacoub, N.; Davis, E.E.; Mola, N.A.; Szymanska, K.; et al. Mutations in CSPP1, Encoding a Core Centrosomal Protein, Cause a Range of Ciliopathy Phenotypes in Humans. Am. J. Hum. Genet. 2014, 94, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Deml, B.; Reis, L.M.; Maheshwari, M.; Griffis, C.; Bick, D.; Semina, E.V. Whole exome analysis identifies dominant COL4A1 mutations in patients with complex ocular phenotypes involving microphthalmia. Clin. Genet. 2014, 86, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Ellard, S.; Kneen, R.; Lim, M.; Osborne, N.; Rankin, J.; Stoodley, N.; Van Der Knaap, M.; Whitney, A.; Jardine, P. Childhood presentation of COL4A1 mutations. Dev. Med. Child Neurol. 2012, 54, 569–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, N.; Davis, E.E.; McKnight, K.L.; Niederriter, A.R.; Causse, A.; David, V.; Desmaison, A.; Lamarre, S.; Vincent-Delorme, C.; Pasquier, L.; et al. Targeted resequencing identifies PTCH1 as a major contributor to ocular developmental anomalies and extends the SOX2 regulatory network. Genome Res. 2016, 26, 474–485. [Google Scholar] [CrossRef]
- Adly, N.; Alhashem, A.; Ammari, A.; Alkuraya, F.S. Ciliary Genes TBC1D32/C6orf170 and SCLT1 are Mutated in Patients with OFD Type IX. Hum. Mutat. 2014, 35, 36–40. [Google Scholar] [CrossRef]
- Matías-Pérez, D.; García-Montaño, L.A.; Cruz-Aguilar, M.; García-Montalvo, I.A.; Nava-Valdéz, J.; Barragán-Arevalo, T.; Villanueva-Mendoza, C.; Villarroel, C.E.; Guadarrama-Vallejo, C.; la Cruz, R.V.; et al. Identification of novel pathogenic variants and novel gene-phenotype correlations in Mexican subjects with microphthalmia and/or anophthalmia by next-generation sequencing. J. Hum. Genet. 2018, 63, 1169–1180. [Google Scholar] [CrossRef]
- Aramaki, M.; Udaka, T.; Kosaki, R.; Makita, Y.; Okamoto, N.; Yoshihashi, H.; Oki, H.; Nanao, K.; Moriyama, N.; Oku, S.; et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J. Pediatr. 2006, 148, 410–414. [Google Scholar] [CrossRef]
- Nishina, S.; Kosaki, R.; Yagihashi, T.; Azuma, N.; Okamoto, N.; Hatsukawa, Y.; Kurosawa, K.; Yamane, T.; Mizuno, S.; Tsuzuki, K.; et al. Ophthalmic features of CHARGE syndrome with CHD7 mutations. Am. J. Med. Genet. Part A 2012, 158A, 514–518. [Google Scholar] [CrossRef]
- Crespí, J.; Buil, J.A.; Bassaganyas, F.; Vela-Segarra, J.I.; Díaz-Cascajosa, J.; Ayala-Ramírez, R.; Zenteno, J.C. A Novel Mutation Confirms MFRP as the Gene Causing the Syndrome of Nanophthalmos–Renititis Pigmentosa–Foveoschisis–Optic Disk Drusen. Am. J. Ophthalmol. 2008, 146, 323–328. [Google Scholar] [CrossRef]
- Nair, K.S.; Hmani-Aifa, M.; Ali, Z.; Kearney, A.L.; Ben Salem, S.; Macalinao, D.G.; Cosma, I.M.; Bouassida, W.; Hakim, B.; Benzina, Z.; et al. Alteration of the serine protease PRSS56 causes angle-closure glaucoma in mice and posterior microphthalmia in humans and mice. Nat. Genet. 2011, 43, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Rau, I.; El Matri, L.; Kreienkamp, H.-J.; Fehr, S.; Baklouti, K.; Chouchane, I.; Li, Y.; Rehbein, M.; Fuchs, J.; et al. Autosomal-Recessive Posterior Microphthalmos Is Caused by Mutations in PRSS56, a Gene Encoding a Trypsin-Like Serine Protease. Am. J. Hum. Genet. 2011, 88, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorram, D.; Choi, M.; Roos, B.R.; Stone, E.M.; Kopel, T.; Allen, R.; Alward, W.L.M.; Scheetz, T.E.; Fingert, J.H. Novel TMEM98 mutations in pedigrees with autosomal dominant nanophthalmos. Mol. Vis. 2015, 21, 1017–1023. [Google Scholar] [PubMed]
- Awadalla, M.S.; Burdon, K.P.; Souzeau, E.; Landers, J.; Hewitt, A.W.; Sharma, S.; Craig, J.E. Mutation in TMEM98 in a Large White Kindred With Autosomal Dominant Nanophthalmos Linked to 17p12-q12. JAMA Ophthalmol. 2014, 132, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.F.; Mohr, D.W.; Kasch, L.M.; Barton, J.A.; Pittiglio, R.; Ingersoll, R.; Craig, B.; Marosy, B.A.; Doheny, K.F.; Bromley, W.C.; et al. Identification of an HMGB3 Frameshift Mutation in a Family With an X-linked Colobomatous Microphthalmia Syndrome Using Whole-Genome and X-Exome Sequencing. JAMA Ophthalmol. 2014, 132, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.; Schneider, A.; Slavotinek, A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur. J. Hum. Genet. 2015, 23, 337–341. [Google Scholar] [CrossRef]
- Chao, R.; Nevin, L.; Agarwal, P.; Riemer, J.; Bai, X.; Delaney, A.; Akana, M.; JimenezLopez, N.; Bardakjian, T.; Schneider, A.; et al. A Male with Unilateral Microphthalmia Reveals a Role for TMX3 in Eye Development. PLoS ONE 2010, 5, e10565. [Google Scholar] [CrossRef]
- Williamson, K.A.; Rainger, J.; Floyd, J.A.B.; Ansari, M.; Meynert, A.; Aldridge, K.V.; Rainger, J.K.; Anderson, C.A.; Moore, A.T.; Hurles, M.E.; et al. Heterozygous Loss-of-Function Mutations in YAP1 Cause Both Isolated and Syndromic Optic Fissure Closure Defects. Am. J. Hum. Genet. 2014, 94, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-F.; Xiang, L.; Cheng, W.; Cheng, F.-F.; He, K.-W.; Zhang, B.-W.; Zheng, S.-S.; Han, R.-Y.; Zheng, Y.-H.; Xu, X.-T.; et al. Mutation of IPO13 causes recessive ocular coloboma, microphthalmia, and cataract. Exp. Mol. Med. 2018, 50, 53. [Google Scholar] [CrossRef]
- Bidinost, C.; Matsumoto, M.; Chung, D.; Salem, N.; Zhang, K.; Stockton, D.W.; Khoury, A.; Megarbane, A.; Bejjani, B.A.; Traboulsi, E.I. Heterozygous and Homozygous Mutations in PITX3 in a Large Lebanese Family with Posterior Polar Cataracts and Neurodevelopmental Abnormalities. Investig. Opthalmol. Vis. Sci. 2006, 47, 1274–1280. [Google Scholar] [CrossRef]
- George, A.; Zand, D.J.; Hufnagel, R.B.; Sharma, R.; Sergeev, Y.V.; Legare, J.M.; Rice, G.M.; Scott Schwoerer, J.A.; Rius, M.; Tetri, L.; et al. Biallelic Mutations in MITF Cause Coloboma, Osteopetrosis, Microphthalmia, Macrocephaly, Albinism, and Deafness. Am. J. Hum. Genet. 2016, 99, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Ragge, N.K.; Ragoussis, J. Molecular analysis of FOXC1 in subjects presenting with severe developmental eye anomalies. Mol. Vis. 2009, 15, 1366–1373. [Google Scholar] [PubMed]
- Willer, T.; Lee, H.; Lommel, M.; Yoshida-Moriguchi, T.; de Bernabe, D.B.V.; Venzke, D.; Cirak, S.; Schachter, H.; Vajsar, J.; Voit, T.; et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012, 44, 575–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetro, A.; Iascone, M.; Limongelli, I.; Ameziane, N.; Gana, S.; Della Mina, E.; Giussani, U.; Ciccone, R.; Forlino, A.; Pezzoli, L.; et al. Loss-of-Function FANCL Mutations Associate with Severe Fanconi Anemia Overlapping the VACTERL Association. Hum. Mutat. 2015, 36, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.F.; Hufnagel, R.B.; Miller, K.A.; Zhou, Y.; McGowan, S.J.; Taylor, J.; Craft, J.; Taylor, J.C.; Santoro, S.L.; Huang, T.; et al. A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 2016, 98, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Sukalo, M.; Tilsen, F.; Kayserili, H.; Müller, D.; Tüysüz, B.; Ruddy, D.M.; Wakeling, E.; Ørstavik, K.H.; Snape, K.M.; Trembath, R.; et al. DOCK6 Mutations Are Responsible for a Distinct Autosomal-Recessive Variant of Adams-Oliver Syndrome Associated with Brain and Eye Anomalies. Hum. Mutat. 2015, 36, 593–598. [Google Scholar] [CrossRef]
- Song, Z.; Si, N.; Xiao, W. A novel mutation in the CRYAA gene associated with congenital cataract and microphthalmia in a Chinese family. BMC Med. Genet. 2018, 19, 190. [Google Scholar] [CrossRef]
- Asgari, N.; Akbari, M.T.; Deilamani, F.K.; Babamohammadi, G. Molecular Genetic Analysis of FOXL2 Gene in Two Iranian Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. J. Hum. Genet. Genomics 2017, 1. [Google Scholar] [CrossRef]
- Billingsley, G.; Santhiya, S.T.; Paterson, A.D.; Ogata, K.; Wodak, S.; Hosseini, S.M.; Manisastry, S.M.; Vijayalakshmi, P.; Gopinath, P.M.; Graw, J.; et al. CRYBA4, a Novel Human Cataract Gene, Is Also Involved in Microphthalmia. Am. J. Hum. Genet. 2006, 79, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Kou, Y.; Shboul, M.; Wang, Z.; Shersheer, Q.; Lyu, Z.; Liu, P.; Zhao, X.; Tian, J. Novel frame shift mutation in ERCC6 leads to a severe form of Cockayne syndrome with postnatal growth failure and early death: A case report and brief literature review. Medicine 2018, 97, e11636. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Tobias, E.S.; Tolmie, J.L.; Martin-Coignard, D.; Drouin-Garraud, V.; Valayannopoulos, V.; Sarasin, A.; Dollfus, H. Cerebro-oculo-facio-skeletal syndrome: Three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J. Med. Genet. 2008, 45, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Drury, S.; Boustred, C.; Tekman, M.; Stanescu, H.; Kleta, R.; Lench, N.; Chitty, L.S.; Scott, R.H. A Novel Homozygous ERCC5 Truncating Mutation in a Family With Prenatal Arthrogryposis-Further Evidence of Genotype-Phenotype Correlation. Am. J. Med. Genet. Part A 2014, 164, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, N.G.J.; Raams, A.; Silengo, M.C.; Wijgers, N.; Niedernhofer, L.J.; Robinson, A.R.; Giglia-Mari, G.; Hoogstraten, D.; Kleijer, W.J.; Hoeijmakers, J.H.J.; et al. First Reported Patient with Human ERCC1 Deficiency Has Cerebro-Oculo-Facio-Skeletal Syndrome with a Mild Defect in Nucleotide Excision Repair and Severe Developmental Failure. Am. J. Hum. Genet. 2007, 80, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantagrel, V.; Lefeber, D.J.; Ng, B.G.; Guan, Z.; Silhavy, J.L.; Bielas, S.L.; Lehle, L.; Hombauer, H.; Adamowicz, M.; Swiezewska, E.; et al. SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell 2010, 142, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, E.; Wu, D.; Madireddy, L.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Bardakjian, T.; Kopinsky, S.; Kwok, P.-Y.; Schneider, A.; et al. Two missense mutations in SALL4 in a patient with microphthalmia, coloboma, and optic nerve hypoplasia. Ophthalmic Genet. 2017, 38, 371–375. [Google Scholar] [CrossRef]
- De Bernardo, G.; Giordano, M.; Di Toro, A.; Sordino, D.; De Brasi, D. Prenatal diagnosis of Fraser syndrome: A matter of life or death? Ital. J. Pediatr. 2015, 41, 86. [Google Scholar] [CrossRef]
- Devuyst, O.; Arnould, V.J. Mutations in RPGRIP1L: Extending the clinical spectrum of ciliopathies. Nephrol. Dial. Transplant. 2008, 23, 1500–1503. [Google Scholar] [CrossRef]
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Unger, S.; Górna, M.W.; Le Béchec, A.; Do Vale-Pereira, S.; Bedeschi, M.F.; Geiberger, S.; Grigelioniene, G.; Horemuzova, E.; Lalatta, F.; Lausch, E.; et al. FAM111A Mutations Result in Hypoparathyroidism and Impaired Skeletal Development. Am. J. Hum. Genet. 2013, 92, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Anazi, S.; Ben-Omran, T.; Seidahmed, M.Z.; Caddle, L.B.; Palmer, K.; Ali, R.; Alshidi, T.; Hagos, S.; Goodwin, L.; et al. Mutations in SMG9, Encoding an Essential Component of Nonsense-Mediated Decay Machinery, Cause a Multiple Congenital Anomaly Syndrome in Humans and Mice. Am. J. Hum. Genet. 2016, 98, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Wallis, D.E.; Roessler, E.; Hehr, U.; Nanni, L.; Wiltshire, T.; Richieri-Costa, A.; Gillessen-Kaesbach, G.; Zackai, E.H.; Rommens, J.; Muenke, M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat. Genet. 1999, 22, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Wright, K.J.; Le Corre, S.; Micalizzi, A.; Romani, M.; Abhyankar, A.; Saada, J.; Perrault, I.; Amiel, J.; Litzler, J.; et al. A Homozygous PDE6D Mutation in Joubert Syndrome Impairs Targeting of Farnesylated INPP5E Protein to the Primary Cilium. Hum. Mutat. 2014, 35, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Altunoglu, U.; Beleggia, F.; Yigit, G.; Kayserili, H.; Nürnberg, P.; Li, Y.; Altmüller, J.; Wollnik, B. An unusual presentation of Kabuki syndrome with orbital cysts, microphthalmia, and cholestasis with bile duct paucity. Am. J. Med. Genet. Part A 2016, 170, 3282–3288. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Audollent, S.; Joly, D.; Dureau, P.; Salomon, R.; Tellier, A.-L.; Augé, J.; Bouissou, F.; Antignac, C.; Gubler, M.-C.; et al. PAX2 mutations in renal-coloboma syndrome: Mutational hotspot and germline mosaicism. Eur. J. Hum. Genet. 2000, 8, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Audollent, S.; Martinovic, J.; Ozilou, C.; Babron, M.-C.; Sivanandamoorthy, S.; Saunier, S.; Salomon, R.; Gonzales, M.; Rattenberry, E.; et al. Pleiotropic Effects of CEP290 (NPHP6) Mutations Extend to Meckel Syndrome. Am. J. Hum. Genet. 2007, 81, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.E.; Ostergaard, P.; Moore, A.T.; Connell, F.C.; Williams, D.; Quarrell, O.; Brady, A.F.; Spier, I.; Hazan, F.; Moldovan, O.; et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): Review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 2014, 22, 881–887. [Google Scholar] [CrossRef]
- Li, J.-K.; Fei, P.; Li, Y.; Huang, Q.-J.; Zhang, Q.; Zhang, X.; Rao, Y.-Q.; Li, J.; Zhao, P. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep. 2016, 6, 26564. [Google Scholar] [CrossRef] [Green Version]
- Vervoort, V.S.; Viljoen, D.; Smart, R.; Suthers, G.; DuPont, B.R.; Abbott, A.; Schwartz, C.E. Sorting nexin 3 (SNX3) is disrupted in a patient with a translocation t(6;13) (q21;q12) and microcephaly, microphthalmia, ectrodactyly, prognathism (MMEP) phenotype. J. Med. Genet. 2002, 39, 893–899. [Google Scholar] [CrossRef]
- Ariss, M.; Natan, K.; Friedman, N.; Traboulsi, E.I. Ophthalmologic Abnormalities in Mowat-Wilson Syndrome and a Mutation in ZEB2. Ophthalmic Genet. 2012, 33, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Valero De Bernabé, D.; Currier, S.; Steinbrecher, A.; Celli, J.; Van Beusekom, E.; Van Der Zwaag, B.; Lya Kayserili, H.; Merlini, L.; Chitayat, D.; Dobyns, W.B.; et al. Mutations in the O-Mannosyltransferase Gene POMT1 Give Rise to the Severe Neuronal Migration Disorder Walker-Warburg Syndrome. Am. J. Hum. Genet 2002, 71, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Hayashi, Y.K.; Matsumoto, H.; Ogawa, M.; Noguchi, S.; Murakami, N.; Sakuta, R.; Mochizuki, M.; Michele, D.E.; Campbell, K.P.; et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in alpha-DG. Neurology 2004, 62, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Van Reeuwijk, J.; Janssen, M.; Van Den Elzen, C.; Beltran-Valero De Bernabé, D.; Sabatelli, P.; Merlini, L.; Boon, M.; Scheffer, H.; Brockington, M.; Muntoni, F.; et al. POMT2 mutations cause a-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005, 42, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Teber, S.; Sezer, T.; Kafalı, M.; Chiara Manzini, M.; Konuk Yüksel, B.; Tekin, M.; Fitöz, S.; Walsh, C.A.; Deda, G. Severe muscle–eye–brain disease is associated with a homozygous mutation in the POMGnT1 gene. Eur. J. Paediatr. Neurol. 2008, 12, 133–136. [Google Scholar] [CrossRef]
- Kondo-Iida, E.; Kobayashi, K.; Watanabe, M.; Sasaki, J.; Kumagai, T.; Koide, H.; Saito, K.; Osawa, M.; Nakamura, Y.; Toda, T. Novel Mutations and Genotype-Phenotype Relationships in 107 Families With Fukuyama-Type Congenital Muscular Dystrophy (FCMD). Hum. Mol. Genet. 1999, 8, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero De Bernabé, D.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004, 41, 61. [Google Scholar] [CrossRef]
- Van Reeuwijk, J.; Olderode-Berends, M.J.W.; Van Den Elzen, C.; Brouwer, O.F.; Roscioli, T.; Van Pampus, M.G.; Scheffer, H.; Brunner, H.G.; Van Bokhoven, H.; Hol, F.A. A homozygous FKRP start codon mutation is associated with Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin. Genet. 2010, 78, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Leibovitz, Z.; Mandel, H.; Falik-Zaccai, T.C.; Ben Harouch, S.; Savitzki, D.; Krajden-Haratz, K.; Gindes, L.; Tamarkin, M.; Lev, D.; Dobyns, W.B.; et al. Walker-Warburg syndrome and tectocerebellar dysraphia: A novel association caused by a homozygous DAG1 mutation. Eur. J. Paediatr. Neurol. 2018, 22, 525–531. [Google Scholar] [CrossRef]
- Riemersma, M.; Mandel, H.; van Beusekom, E.; Gazzoli, I.; Roscioli, T.; Eran, A.; Gershoni-Baruch, R.; Gershoni, M.; Pietrokovski, S.; Vissers, L.E.; et al. Absence of α- and β-dystroglycan is associated with Walker-Warburg syndrome. Neurology 2015, 84, 2177–2182. [Google Scholar] [CrossRef]
- Stevens, E.; Carss, K.J.; Cirak, S.; Foley, A.R.; Torelli, S.; Willer, T.; Tambunan, D.E.; Yau, S.; Brodd, L.; Sewry, C.A.; et al. Mutations in B3GALNT2 Cause Congenital Muscular Dystrophy and Hypoglycosylation of a-Dystroglycan. Am. J. Hum. Genet. 2013, 92, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Handley, M.T.; Morris-Rosendahl, D.J.; Brown, S.; Macdonald, F.; Hardy, C.; Bem, D.; Carpanini, S.M.; Borck, G.; Martorell, L.; Izzi, C.; et al. Mutation Spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and Genotype-Phenotype Correlations in Warburg Micro Syndrome and Martsolf Syndrome. Hum. Mutat. 2013, 34, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Borck, G.; Wunram, H.; Steiert, A.; Volk, A.E.; Körber, F.; Roters, S.; Herkenrath, P.; Wollnik, B.; Morris-Rosendahl, D.J.; Kubisch, C. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum. Genet. 2011, 129, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Aligianis, I.A.; Morgan, N.V.; Mione, M.; Johnson, C.A.; Rosser, E.; Hennekam, R.C.; Adams, G.; Trembath, R.C.; Pilz, D.T.; Stoodley, N.; et al. Mutation in Rab3 GTPase-Activating Protein (RAB3GAP) Noncatalytic Subunit in a Kindred with Martsolf Syndrome. Am. J. Hum. Genet. 2006, 78, 702–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esch, H.; Jansen, A.; Bauters, M.; Froyen, G.; Fryns, J.-P. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am. J. Med. Genet. Part A 2007, 143A, 364–369. [Google Scholar] [CrossRef]
- Gillespie, R.L.; Urquhart, J.; Lovell, S.C.; Biswas, S.; Parry, N.R.A.; Schorderet, D.F.; Lloyd, I.C.; Clayton-Smith, J.; Black, G.C. Abrogation of HMX1 Function Causes Rare Oculoauricular Syndrome Associated With Congenital Cataract, Anterior Segment Dysgenesis, and Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorderet, D.F.; Nichini, O.; Boisset, G.; Polok, B.; Tiab, L.; Mayeur, H.; Raji, B.; de la Houssaye, G.; Abitbol, M.M.; Munier, F.L. Mutation in the Human Homeobox Gene NKX5-3 Causes an Oculo-Auricular Syndrome. Am. J. Hum. Genet. 2008, 82, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, C.; D’Adamo, P.; Gentile, F.; Vingolo, E.M.; Gasparini, P.; Banfi, S. A novel GJA1 mutation causes oculodentodigital dysplasia without syndactyly. Am. J. Med. Genet. Part A 2005, 133A, 58–60. [Google Scholar] [CrossRef]
- Richardson, R.J.; Joss, S.; Tomkin, S.; Ahmed, M.; Sheridan, E.; Dixon, M.J. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J. Med. Genet. 2006, 43, e37. [Google Scholar] [CrossRef]
- Brice, G.; Ostergaard, P.; Jeffery, S.; Gordon, K.; Mortimer, P.S.; Mansour, S. A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clin. Genet. 2013, 84, 378–381. [Google Scholar] [CrossRef]
- Narumi, S.; Numakura, C.; Shiihara, T.; Seiwa, C.; Nozaki, Y.; Yamagata, T.; Momoi, M.Y.; Watanabe, Y.; Yoshino, M.; Matsuishi, T.; et al. Various types of LRP5 mutations in four patients with osteoporosis-pseudoglioma syndrome: Identification of a 7.2-kb microdeletion using oligonucleotide tiling microarray. Am. J. Med. Genet. Part A 2010, 152A, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Garay, I.; Tomás, M.; Oltra, S.; Ramser, J.; Moltó, M.D.; Prieto, F.; Meindl, A.; Kutsche, K.; Martínez, F. A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly, and mental retardation. Eur. J. Hum. Genet. 2007, 15, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Isrie, M.; Breuss, M.; Tian, G.; Hansen, A.H.; Cristofoli, F.; Morandell, J.; Kupchinsky, Z.A.; Sifrim, A.; Rodriguez-Rodriguez, C.M.; Dapena, E.P.; et al. Mutations in Either TUBB or MAPRE2 Cause Circumferential Skin Creases Kunze Type. Am. J. Hum. Genet. 2015, 97, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuss, M.; Heng, J.I.-T.; Poirier, K.; Tian, G.; Jaglin, X.H.; Qu, Z.; Braun, A.; Gstrein, T.; Ngo, L.; Haas, M.; et al. Mutations in the β-Tubulin Gene TUBB5 Cause Microcephaly with Structural Brain Abnormalities. Cell Rep. 2012, 2, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Bardakjian, T.M.; Schneider, A.S.; Ng, D.; Johnston, J.J.; Biesecker, L.G. Association of a de novo16q copy number variant with a phenotype that overlaps with Lenz microphthalmia and Townes-Brocks syndromes. BMC Med. Genet. 2009, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Siani, V.; Carles, D.; Delezoide, A.L.; Alberti, E.M.; Battin, J.; Chateil, J.F.; Gilbert-Dussardier, B.; Coupry, I.; Arveiler, B.; et al. X-linked dominant chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia. Am. J. Med. Genet. Part A 2005, 136A, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Laloo, B.; Barillot, M.; Barnetche, T.; Blanchard, C.; Rooryck, C.; Marche, M.; Burgelin, I.; Coupry, I.; Chassaing, N.; et al. A mutation in the 3′-UTR of the HDAC6 gene abolishing the post-transcriptional regulation mediated by hsa-miR-433 is linked to a new form of dominant X-linked chondrodysplasia. Hum. Mol. Genet. 2010, 19, 2015–2027. [Google Scholar] [CrossRef]
- Uz, E.; Alanay, Y.; Aktas, D.; Vargel, I.; Gucer, S.; Tuncbilek, G.; von Eggeling, F.; Yilmaz, E.; Deren, O.; Posorski, N.; et al. Disruption of ALX1 Causes Extreme Microphthalmia and Severe Facial Clefting: Expanding the Spectrum of Autosomal-Recessive ALX-Related Frontonasal Dysplasia. Am. J. Hum. Genet. 2010, 86, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Fregeau, B.; Kim, B.J.; Hernández-García, A.; Jordan, V.K.; Cho, M.T.; Schnur, R.E.; Monaghan, K.G.; Juusola, J.; Rosenfeld, J.A.; Bhoj, E.; et al. De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions. Am. J. Hum. Genet. 2016, 98, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Bem, D.; Yoshimura, S.-I.; Nunes-Bastos, R.; Bond, F.F.; Kurian, M.A.; Rahman, F.; Handley, M.T.W.; Hadzhiev, Y.; Masood, I.; Straatman-Iwanowska, A.A.; et al. Loss-of-Function Mutations in RAB18 Cause Warburg Micro Syndrome. Am. J. Hum. Genet. 2011, 88, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, M.; Hanson, I.; van Heyningen, V. Aniridia. Eur. J. Hum. Genet. 2012, 20, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Thomas, M.G.; Andrews, C.; Chan, W.-M.; Proudlock, F.A.; McLean, R.J.; Pradeep, A.; Engle, E.C.; Gottlob, I. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur. J. Hum. Genet. 2014, 22, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.N.; Williamson, K.A.; Hanson, I.M.; Owen, L.J.; Bengani, H.; Van, V.; Marsh, J.A.; Fitzpatrick, D.R. Recurrent heterozygous missense variants in the PAX6 paired domain cause severe microphthalmia. In Proceedings of the Association for Research in Vision and Ophthalmology (ARVO)-Poster Presented at Gene Variants and Regulation of Ocular Genes Expression in Health and Disease Session, Vancouver, BC, Canada, 28 April–2 May 2019; p. A0373. [Google Scholar]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, T.; Ishizu, K.; Nakamura, A. Molecular and Clinical Findings in Patients with LHX4 and OTX2 Mutations. Clin. Pediatr. Endocrinol. Case Reports Clin. Investig. Off. J. Jpn. Soc. Pediatr. Endocrinol. 2013, 22, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Acampora, D.; Mazan, S.; Lallemand, Y.; Avantaggiato, V.; Maury, M.; Simeone, A.; Brulet, P. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 1995, 121, 3279–3290. [Google Scholar] [PubMed]
- Ang, S.L.; Jin, O.; Rhinn, M.; Daigle, N.; Stevenson, L.; Rossant, J. A targeted mouse Otx2 mutation leads to severe defects in gastrulation and formation of axial mesoderm and to deletion of rostral brain. Development 1996, 122, 243–252. [Google Scholar] [PubMed]
- Matsuo, I.; Kuratani, S.; Kimura, C.; Takeda, N.; Aizawa, S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev. 1995, 9, 2646–2658. [Google Scholar] [CrossRef]
- Hide, T.; Hatakeyama, J.; Kimura-Yoshida, C.; Tian, E.; Takeda, N.; Ushio, Y.; Shiroishi, T.; Aizawa, S.; Matsuo, I. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development 2002, 129, 4347–4357. [Google Scholar]
- Muranishi, Y.; Terada, K.; Inoue, T.; Katoh, K.; Tsujii, T.; Sanuki, R.; Kurokawa, D.; Aizawa, S.; Tamaki, Y.; Furukawa, T. An essential role for RAX homeoprotein and NOTCH-HES signaling in Otx2 expression in embryonic retinal photoreceptor cell fate determination. J. Neurosci. 2011, 31, 16792–16807. [Google Scholar] [CrossRef]
- Lupo, G.; Andreazzoli, M.; Gestri, G.; Liu, Y.; He, R.Q.; Barsacchi, G. Homeobox genes in the genetic control of eye development. Int. J. Dev. Biol. 2000, 44, 627–636. [Google Scholar]
- Andreazzoli, M.; Gestri, G.; Angeloni, D.; Menna, E.; Barsacchi, G. Role of Xrx1 in Xenopus eye and anterior brain development. Development 1999, 126, 2451–2460. [Google Scholar] [PubMed]
- Chow, R.L.; Altmann, C.R.; Lang, R.A.; Hemmati-Brivanlou, A. Pax6 induces ectopic eyes in a vertebrate. Development 1999, 126, 4213–4222. [Google Scholar] [PubMed]
- Nishihara, D.; Yajima, I.; Tabata, H.; Nakai, M.; Tsukiji, N.; Katahira, T.; Takeda, K.; Shibahara, S.; Nakamura, H.; Yamamoto, H. Otx2 Is Involved in the Regional Specification of the Developing Retinal Pigment Epithelium by Preventing the Expression of Sox2 and Fgf8, Factors That Induce Neural Retina Differentiation. PLoS ONE 2012, 7, e48879. [Google Scholar] [CrossRef] [PubMed]
- Tucker, P.; Laemle, L.; Munson, A.; Kanekar, S.; Oliver, E.R.; Brown, N.; Schlecht, H.; Vetter, M.; Glaser, T. The eyeless mouse mutation (ey1) removes an alternative start codon from theRx/rax homeobox gene. Genesis 2001, 31, 43–53. [Google Scholar] [CrossRef]
- Loosli, F.; Staub, W.; Finger-Baier, K.C.; Ober, E.A.; Verkade, H.; Wittbrodt, J.; Baier, H. Loss of eyes in zebrafish caused by mutation of chokh/rx3. EMBO Rep. 2003, 4, 894–899. [Google Scholar] [CrossRef]
- Muranishi, Y.; Terada, K.; Furukawa, T. An essential role for Rax in retina and neuroendocrine system development. Dev. Growth Differ. 2012, 54, 341–348. [Google Scholar] [CrossRef]
- Zhang, L.; Mathers, P.H.; Jamrich, M. Function of Rx, but not Pax6, is essential for the formation of retinal progenitor cells in mice. Genesis 2000, 28, 135–142. [Google Scholar] [CrossRef]
- Barabino, S.M.L.; Spada, F.; Cotelli, F.; Boncinelli, E. Inactivation of the zebrafish homologue of Chx10 by antisense oligonucleotides causes eye malformations similar to the ocular retardation phenotype. Mech. Dev. 1997, 63, 133–143. [Google Scholar] [CrossRef]
- Rowan, S.; Chen, C.-M.A.; Young, T.L.; Fisher, D.E.; Cepko, C.L.; McInnes, R.R. Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 2004, 131, 5139–5152. [Google Scholar] [CrossRef]
- Green, E.S.; Stubbs, J.L.; Levine, E.M. Genetic rescue of cell number in a mouse model of microphthalmia: Interactions between Chx10 and G1-phase cell cycle regulators. Development 2003, 130, 539–552. [Google Scholar] [CrossRef]
- Capowski, E.E.; Wright, L.S.; Liang, K.; Phillips, M.J.; Wallace, K.; Petelinsek, A.; Hagstrom, A.; Pinilla, I.; Borys, K.; Lien, J.; et al. Regulation of WNT Signaling by VSX2 During Optic Vesicle Patterning in Human Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Moosajee, M.; Hingorani, M.; Moore, A.T. PAX6-Related Aniridia. In GeneReviews®; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.; Kondoh, H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Grindley, J.C.; Davidson, D.R.; Hill, R.E. The role of Pax-6 in eye and nasal development. Development 1995, 121, 1433–1442. [Google Scholar] [PubMed]
- Hill, R.E.; Favor, J.; Hogan, B.L.M.; Ton, C.C.T.; Saunders, G.F.; Hanson, I.M.; Prosser, J.; Jordan, T.; Hastie, N.D.; Heyningen, V. Van Mouse Small eye results from mutations in a paired-like homeobox-containing gene. Nature 1991, 354, 522–525. [Google Scholar] [CrossRef]
- Tétreault, N.; Champagne, M.-P.; Bernier, G. The LIM homeobox transcription factor Lhx2 is required to specify the retina field and synergistically cooperates with Pax6 for Six6 trans-activation. Dev. Biol. 2009, 327, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Wawersik, S.; Purcell, P.; Rauchman, M.; Dudley, A.T.; Robertson, E.J.; Maas, R. BMP7 Acts in Murine Lens Placode Development. Dev. Biol. 1999, 207, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Tao, C.; Li, H.; Ladher, R.; Gotoh, N.; Feng, G.-S.; Wang, F.; Zhang, X.; Marigo, V.; Ballabio, A.; et al. Deficient FGF signaling causes optic nerve dysgenesis and ocular coloboma. Development 2013, 140, 2711–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segel, R.; Levy-Lahad, E.; Pasutto, F.; Picard, E.; Rauch, A.; Alterescu, G.; Schimmel, M.S. Pulmonary hypoplasia-diaphragmatic hernia-anophthalmia-cardiac defect (PDAC) syndrome due to STRA6 mutations-What are the minimal criteria? Am. J. Med. Genet. Part A 2009, 149A, 2457–2463. [Google Scholar] [CrossRef]
- Chitayat, D.; Sroka, H.; Keating, S.; Colby, R.S.; Ryan, G.; Toi, A.; Blaser, S.; Viero, S.; Devisme, L.; Boute-Bénéjean, O.; et al. The PDAC syndrome (pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect) (Spear syndrome, Matthew-Wood syndrome): Report of eight cases including a living child and further evidence for autosomal. Am. J. Med. Genet. Part A 2007, 143A, 1268–1281. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kawaguchi, R. The Membrane Receptor for Plasma Retinol-Binding Protein, A New Type of Cell-Surface Receptor. Int. Rev. Cell Mol. Biol. 2011, 288, 1–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 Disrupts Vitamin A Uptake Homeostasis in a STRA6-Deficient Animal Model for Matthew-Wood Syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.D.; Mao, G.E. Teratology of Retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef] [PubMed]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvekl, A.; Tamm, E.R. Anterior eye development and ocular mesenchyme: New insights from mouse models and human diseases. BioEssays 2004, 26, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Semerci, C.N.; Kalay, E.; Yıldırım, C.; Dinçer, T.; Ölmez, A.; Toraman, B.; Koçyiğit, A.; Bulgu, Y.; Okur, V.; Şatıroğlu-Tufan, L.; et al. Novel splice-site and missense mutations in the ALDH1A3 gene underlying autosomal recessive anophthalmia/microphthalmia. Br. J. Ophthalmol. 2014, 98, 832–840. [Google Scholar] [CrossRef]
- Duester, G. Keeping an eye on retinoic acid signaling during eye development. Chem. Biol. Interact. 2009, 178, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Shintani, T.; Sakuta, H.; Kato, A.; Ohkawara, T.; Osumi, N.; Noda, M. Identification of RALDH-3, a novel retinaldehyde dehydrogenase, expressed in the ventral region of the retina. Mech. Dev. 2000, 98, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Dupé, V.; Matt, N.; Garnier, J.-M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [Green Version]
- Zuber, M.E. Eye Field Specification in Xenopus laevis. Curr. Top. Dev. Biol. 2010, 93, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Slijkerman, R.W.N.; Song, F.; Astuti, G.D.N.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W.J. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Lwigale, P.Y. Corneal Development: Different Cells from a Common Progenitor. Prog. Mol. Biol. Transl. Sci. 2015, 134, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Zieske, J.D. Corneal development associated with eyelid opening. Int. J. Dev. Biol. 2004, 48, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschi, K.K.; Li, S.; Roy, K. Induced pluripotent stem cells for regenerative medicine. Annu. Rev. Biomed. Eng. 2014, 16, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.; Hyun, J.; McArdle, A.; Senarath-Yapa, K.; Hu, M.; Chung, M.; Wong, V.; Longaker, M.; Wan, D. Induced Pluripotent Stem Cells in Regenerative Medicine and Disease Modeling. Curr. Stem Cell Res. Ther. 2014, 9, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.C.; Khan, S.; Lo, C.Y.; Hewitt, A.W.; Wong, R.C.B. Drug discovery using induced pluripotent stem cell models of neurodegenerative and ocular diseases. Pharmacol. Ther. 2017, 177, 32–43. [Google Scholar] [CrossRef]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 201901572. [Google Scholar] [CrossRef]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.L.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef] [Green Version]
- Kaewkhaw, R.; Kaya, K.D.; Brooks, M.; Homma, K.; Zou, J.; Chaitankar, V.; Rao, M.; Swaroop, A. Transcriptome Dynamics of Developing Photoreceptors in Three-Dimensional Retina Cultures Recapitulates Temporal Sequence of Human Cone and Rod Differentiation Revealing Cell Surface Markers and Gene Networks. Stem Cells 2015, 33, 3504–3518. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.P.; Collin, G.B.; Hicks, W.L.; Yu, M.; Charette, J.R.; Shi, L.Y.; Wang, J.; Naggert, J.K.; Peachey, N.S.; Nishina, P.M. Mouse models of human ocular disease for translational research. PLoS ONE 2017, 12, e0183837. [Google Scholar] [CrossRef] [PubMed]
- Chaitankar, V.; Karakülah, G.; Ratnapriya, R.; Giuste, F.O.; Brooks, M.J.; Swaroop, A. Next generation sequencing technology and genomewide data analysis: Perspectives for retinal research. Prog. Retin. Eye Res. 2016, 55, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Chatterjee, S.; Mukherjee, A.; Mutsuddi, M. Whole exome sequencing: Uncovering causal genetic variants for ocular diseases. Exp. Eye Res. 2017, 164, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Langouet-Astrie, C.J.; Meinsen, A.L.; Grunwald, E.R.; Turner, S.D.; Enke, R.A. RNA sequencing analysis of the developing chicken retina. Sci. Data 2016, 3, 160117. [Google Scholar] [CrossRef] [PubMed]
- Farkas, M.H.; Au, E.D.; Sousa, M.E.; Pierce, E.A. RNA-Seq: Improving Our Understanding of Retinal Biology and Disease. Cold Spring Harb. Perspect. Med. 2015, 5, a017152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today 2015, 20, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Corso-Díaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef]
- Hiler, D.; Chen, X.; Hazen, J.; Kupriyanov, S.; Carroll, P.A.; Qu, C.; Xu, B.; Johnson, D.; Griffiths, L.; Frase, S.; et al. Quantification of Retinogenesis in 3D Cultures Reveals Epigenetic Memory and Higher Efficiency in iPSCs Derived from Rod Photoreceptors. Cell Stem Cell 2015, 17, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Miyake, K.; Hariya, N.; Mochizuki, K. Epigenetics as a basis for diagnosis of neurodevelopmental disorders: Challenges and opportunities. Expert Rev. Mol. Diagn. 2014, 14, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Mitton, K.P. Epigenetic regulatory mechanisms in vertebrate eye development and disease. Heredity (Edinb). 2010, 105, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdasco, M.; Gómez, A.; Rubio, M.J.; Català-Mora, J.; Zanón-Moreno, V.; Lopez, M.; Hernández, C.; Yoshida, S.; Nakama, T.; Ishikawa, K.; et al. DNA Methylomes Reveal Biological Networks Involved in Human Eye Development, Functions and Associated Disorders. Sci. Rep. 2017, 7, 11762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem, Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Eastlake, K.; Heywood, W.E.; Tracey-White, D.; Aquino, E.; Bliss, E.; Vasta, G.R.; Mills, K.; Khaw, P.T.; Moosajee, M.; Limb, G.A. Comparison of proteomic profiles in the zebrafish retina during experimental degeneration and regeneration. Sci. Rep. 2017, 7, 44601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh-Roy, A.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Kinesin-13 and Tubulin Posttranslational Modifications Regulate Microtubule Growth in Axon Regeneration. Dev. Cell 2012, 23, 716–728. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.Z.; Begley, P.; Mullard, G.; Hollywood, K.A.; Bishop, P.N. Introduction to metabolomics and its applications in ophthalmology. Eye 2016, 30, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, G.A.; Larrouy-Maumus, G.; de Carvalho, L.P.S. Metabolomic strategies for the identification of new enzyme functions and metabolic pathways. EMBO Rep. 2014, 15, 657–669. [Google Scholar] [CrossRef]
- Klupczyńska, A.; Dereziński, P.; Kokot, Z.J. Metabolomics in Medical Sciences--Trends, Challenges and Perspectives. Acta Pol. Pharm. 2015, 72, 629–641. [Google Scholar]
- Wang, X.; Gregory-Evans, K.; Wasan, K.M.; Sivak, O.; Shan, X.; Gregory-Evans, C.Y. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol. Ther. Nucleic Acids 2017, 7, 417–428. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Clinical images of anophthalmia and microphthalmia. (a) Bilateral anophthalmia. (b) Bilateral microphthalmia. (c) Unilateral anophthalmia with shell.
Figure 1.
Clinical images of anophthalmia and microphthalmia. (a) Bilateral anophthalmia. (b) Bilateral microphthalmia. (c) Unilateral anophthalmia with shell.

Figure 2.
The genetics of early optic vesicle development. (a) A single eye field is induced at the midline of the anterior neural plate, through OTX2 and SOX2 coregulation of RAX, along with SIX3-mediated repression of WNT, which allows upregulation of eye field transcription factors (EFTFs) including PAX6, RAX, LHX2, TBX3 and SIX6. (b) The eye field is formed expressing EFTFs, which form a self-regulating network sufficient to coordinate the development of the eye through suppression of genes that antagonize eye development and upregulation of genes required for eye development. (c) Secretion of factors from TGFβ, FGF and SHH families from the underlying axial mesoderm stimulate the anterior migration of prospective hypothalamic cells, causing the eye field to split in two to form bilateral optic primordia. (d) Cellular proliferation and bilateral migration of the optic primordia is regulated by RAX. (e) The optic primordia evaginate from the forebrain through the cephalic mesenchyme forming bilateral optic pits/grooves. (f) Extended evagination of the optic pits through the cephalic mesenchyme results in the formation of bilateral optic vesicles.
Figure 2.
The genetics of early optic vesicle development. (a) A single eye field is induced at the midline of the anterior neural plate, through OTX2 and SOX2 coregulation of RAX, along with SIX3-mediated repression of WNT, which allows upregulation of eye field transcription factors (EFTFs) including PAX6, RAX, LHX2, TBX3 and SIX6. (b) The eye field is formed expressing EFTFs, which form a self-regulating network sufficient to coordinate the development of the eye through suppression of genes that antagonize eye development and upregulation of genes required for eye development. (c) Secretion of factors from TGFβ, FGF and SHH families from the underlying axial mesoderm stimulate the anterior migration of prospective hypothalamic cells, causing the eye field to split in two to form bilateral optic primordia. (d) Cellular proliferation and bilateral migration of the optic primordia is regulated by RAX. (e) The optic primordia evaginate from the forebrain through the cephalic mesenchyme forming bilateral optic pits/grooves. (f) Extended evagination of the optic pits through the cephalic mesenchyme results in the formation of bilateral optic vesicles.

Figure 3.
The genetics of optic cup and lens formation. (a) A preplacodal region develops within the surface ectoderm overlying the optic pit/groove, stimulated by expression of SIX3, which activates PAX6 and SOX2. (b) The lens placode is signalled to thicken through LHX2-regulated BMP4 expression from the evaginating optic vesicle. (c) The developing lens placode releases BMP and retinoic acid (RA), which bind to the optic vesicle, stimulating coordinated invagination of the optic vesicle and lens placode, which forms the lens pit. (d) The invagination of the optic vesicle results in the formation of a bilayered optic cup. (e) TGFβ signalling from the extraocular mesenchyme induces and maintains MITF and OTX2 expression in the outer layer of the optic cup, which forms the presumptive retinal pigmented epithelium (RPE). FGF signals from the surface ectoderm stimulate the inner layer of the optic cup to form a VSX2-expressing presumptive neural retina (NR). The continued invagination of the lens pit results in the formation of the lens vesicle. (f) From 47 days gestation, retinal differentiation occurs, with the inner layer of the optic cup forming the neural retina (NR) and the outer layer forming the retinal pigmented epithelium (RPE). The lens vesicle detaches from the surface ectoderm and forms a definitive lens by 58 days gestation.
Figure 3.
The genetics of optic cup and lens formation. (a) A preplacodal region develops within the surface ectoderm overlying the optic pit/groove, stimulated by expression of SIX3, which activates PAX6 and SOX2. (b) The lens placode is signalled to thicken through LHX2-regulated BMP4 expression from the evaginating optic vesicle. (c) The developing lens placode releases BMP and retinoic acid (RA), which bind to the optic vesicle, stimulating coordinated invagination of the optic vesicle and lens placode, which forms the lens pit. (d) The invagination of the optic vesicle results in the formation of a bilayered optic cup. (e) TGFβ signalling from the extraocular mesenchyme induces and maintains MITF and OTX2 expression in the outer layer of the optic cup, which forms the presumptive retinal pigmented epithelium (RPE). FGF signals from the surface ectoderm stimulate the inner layer of the optic cup to form a VSX2-expressing presumptive neural retina (NR). The continued invagination of the lens pit results in the formation of the lens vesicle. (f) From 47 days gestation, retinal differentiation occurs, with the inner layer of the optic cup forming the neural retina (NR) and the outer layer forming the retinal pigmented epithelium (RPE). The lens vesicle detaches from the surface ectoderm and forms a definitive lens by 58 days gestation.
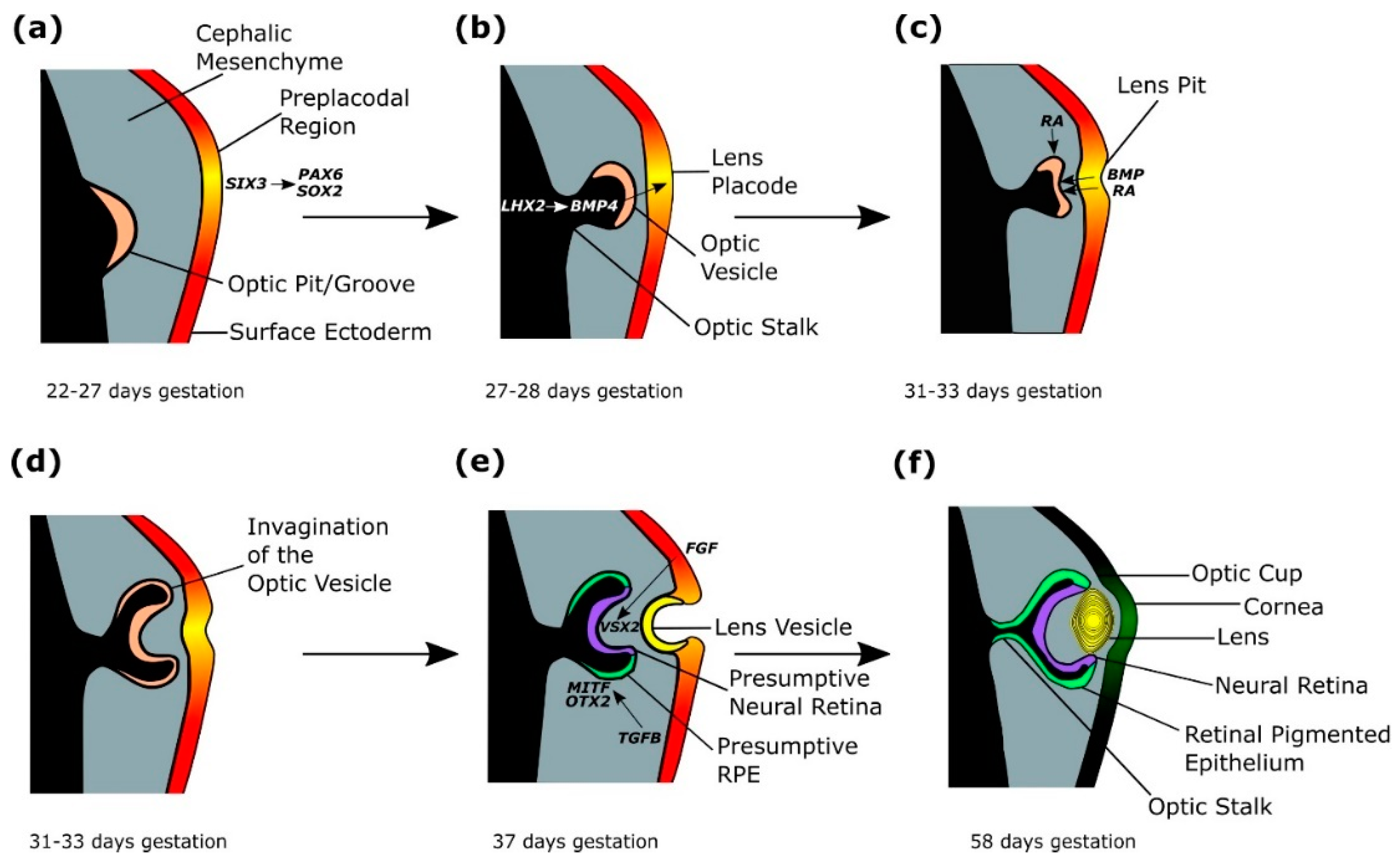

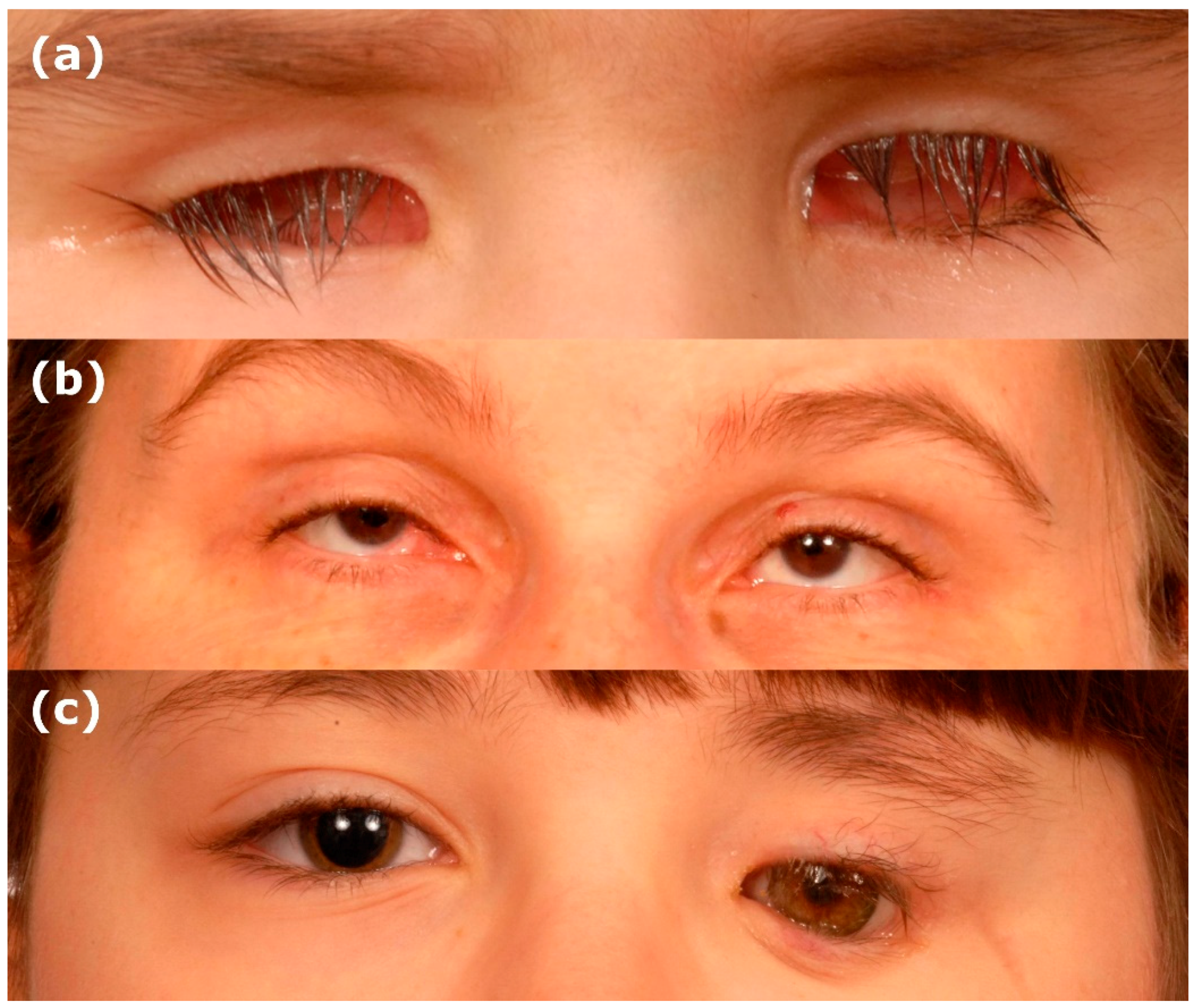{kind=link}
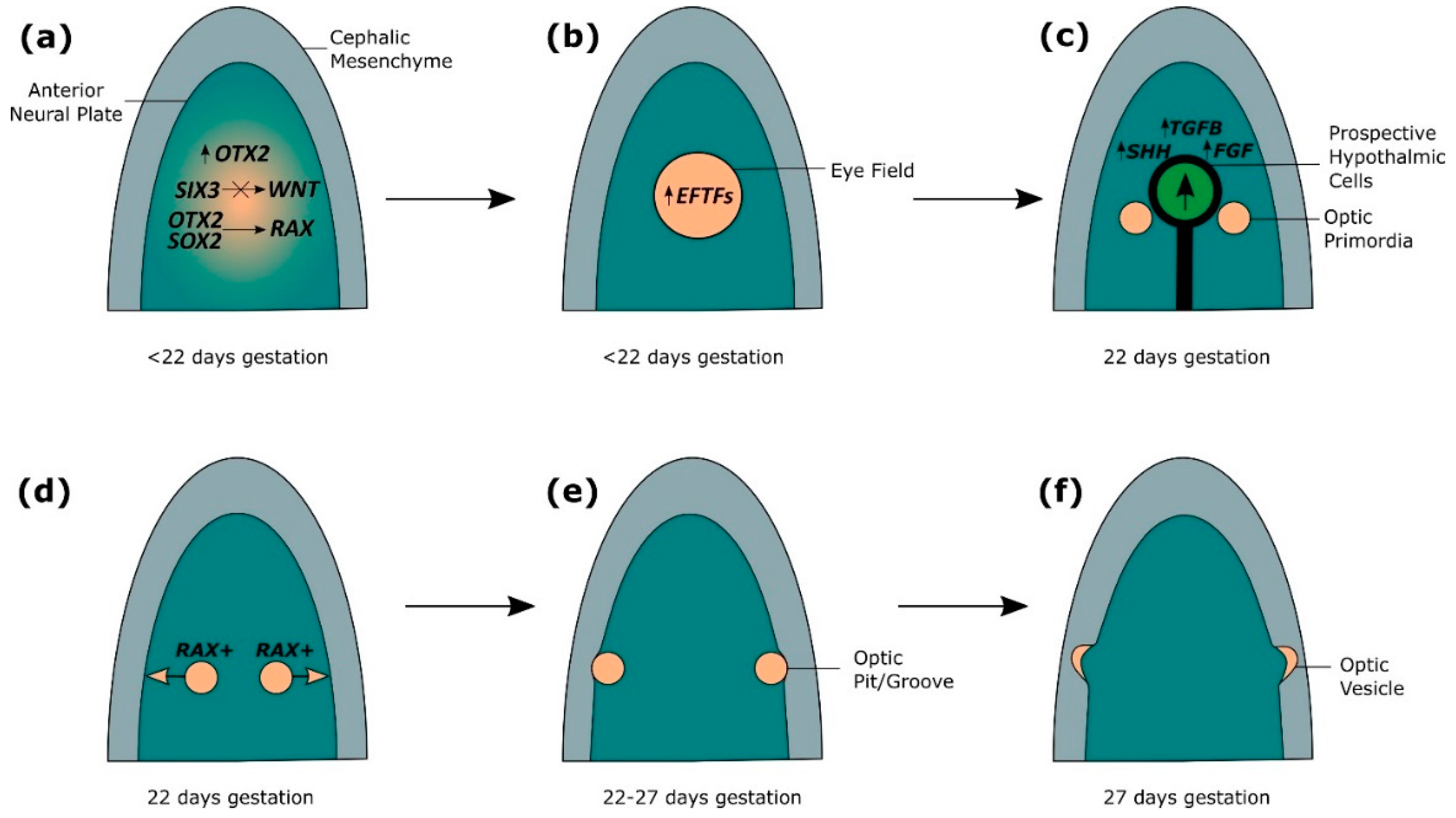{kind=link}
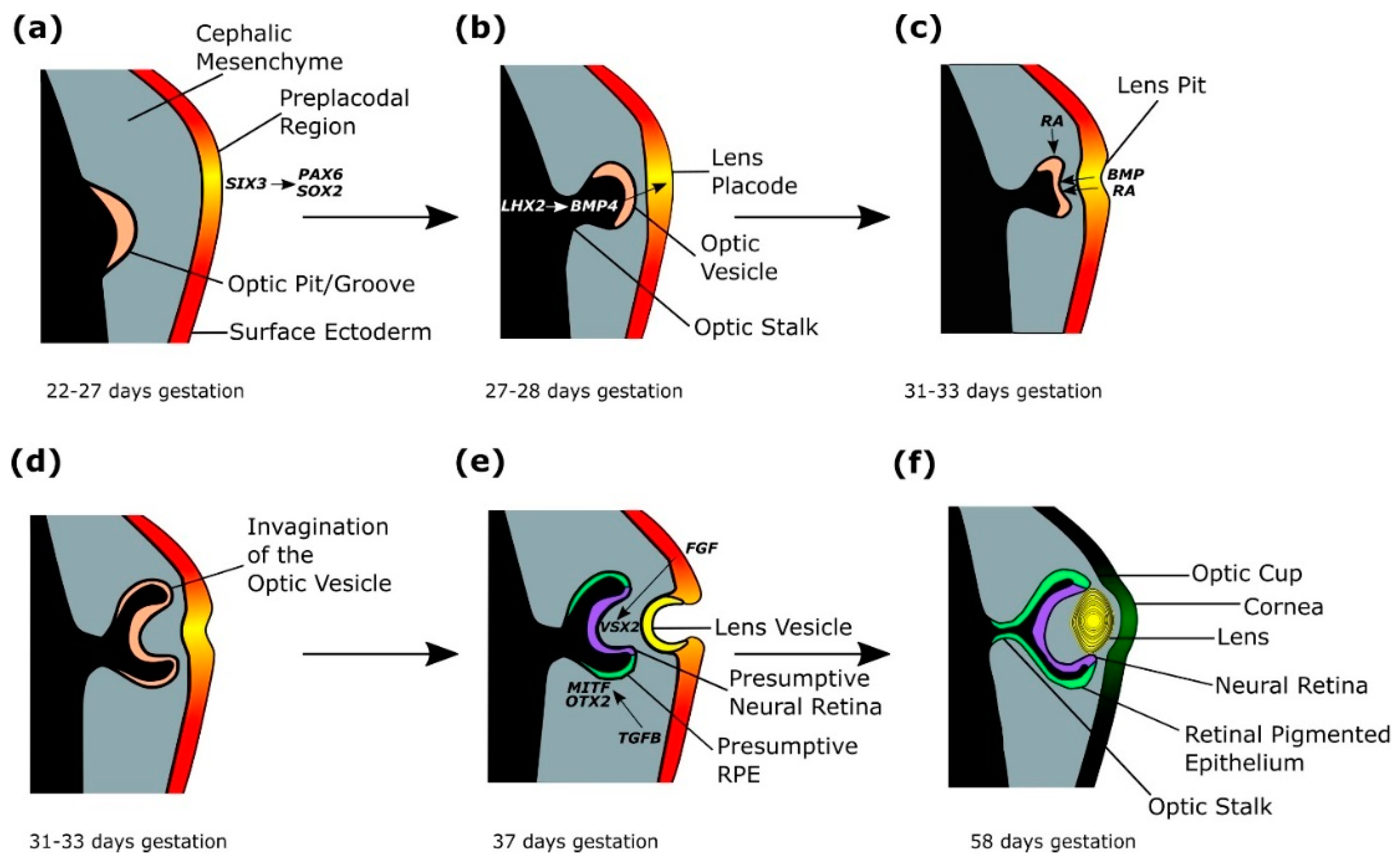{kind=link}
Table 1.
Genes associated with anophthalmia and microphthalmia. A—unilateral or bilateral anophthalmia. M—unilateral or bilateral microphthalmia. I—Isolated AM with no additional ocular features. C—complex AM (additional ocular features found in microphthalmic or contralateral eye of the patient listed). N—no syndromic features observed. S—systemic features observed.
Table 1.
Genes associated with anophthalmia and microphthalmia. A—unilateral or bilateral anophthalmia. M—unilateral or bilateral microphthalmia. I—Isolated AM with no additional ocular features. C—complex AM (additional ocular features found in microphthalmic or contralateral eye of the patient listed). N—no syndromic features observed. S—systemic features observed.
| Gene Name | OMIM# | Disease Name | Disease OMIM# | Anophthalmia/Microphthalmia (A/M) | Isolated/Complex (I/C) (Associated Ocular Features) | Non-Syndromic/Syndromic (N/S) | Reference(s) | |
|---|---|---|---|---|---|---|---|---|
| 1 | SOX2 | 184429 | Microphthalmia, syndromic 3 (MCOPS3) | 206900 | A, M | I, C (coloboma, microcornea, iris defect, retinal tuft, optic nerve hypoplasia, reduced palpebral fissure, congenital cataract, glaucoma, colobomatous cyst, synechiae, anterior segment dysgenesis, retinal/chorioretinal dystrophy, myopia) | N, S | [7,10,19,89,90] |
| 2 | OTX2 | 600037 | Microphthalmia, syndromic 5 (MCOPS5) | 610125 | A, M | I, C (coloboma, microcornea, retinal defect, optic nerve hypoplasia/aplasia, small/absent optic chiasm, LCA, early onset retinal dystrophy, hyperopia, amblyopia, cataract, focal retinal dysplasia, corectopia, synechiae, sclerocornea, persistent pupillary membrane, nystagmus, posterior vitreous opacity) | N, S | [7,18,19,21,80,90,91,92,93] |
| 3 | RAX | 601881 | Microphthalmia, isolated 3 (MCOP3) | 611038 | A, M | I, C (coloboma, sclerocornea, persistent fetal vasculature, retinal detachment, optic nerve atrophy/hypoplasia) | N, S | [7,90,94] |
| 4 | VSX2 | 142993 | Microphthalmia, isolated 2 (MCOP2) | 610093 | A, M | I, C (coloboma, congenital cataract/cloudy cornea, iris defect, microcornea, no pupillary aperture, retinal detachment, dislocated lens, small/underdeveloped optic nerve/chiasm, retinal dysfunction) | N, S | [7,66,95] |
| Microphthalmia, isolated with coloboma 3 (MCOPCB3) | 610092 | |||||||
| 5 | PAX6 | 607108 | Aniridia 1 (AN1) | 106210 | A, M | I, C (coloboma, aniridia/iris hypoplasia, anterior segment dysgenesis, agenesis of optic nerve/chiasm, primary aphakia, sclerocornea, congenital glaucoma) | N, S | [7,92] |
| 6 | STRA6 | 610745 | Microphthalmia, syndromic 9 (MCOPS9) | 601186 | A, M | I, C (coloboma, cyst, retinal detachment, abnormal cornea/iris) | N, S | [19,20,21,96,97,98] |
| 7 | RARβ | 180220 | Microphthalmia, syndromic 12 (MCOPS12) | 615524 | A, M | I, C (coloboma, sclerocornea, anterior segment dysgenesis) | S | [21,87,88] |
| 8 | ALDH1A3 | 600463 | Microphthalmia, isolated 8 (MCOP8) | 615113 | A, M | I, C (coloboma, microcornea corectopia, cyst, hypoplastic/small optic nerve/tract/chiasm, small/short palpebral fissure, conjunctival discoloration, symblepharon, nystagmus, iris attachment to the cornea) | N, S | [99,100,101] |
| 9 | FOXE3 | 601094 | Anterior segment dysgenesis 2 (ASD2) | 610256 | M | C (coloboma, anterior segment dysgenesis, sclerocornea, aphakia, aniridia) | N, S | [7] |
| 10 | BMP4 | 112262 | Microphthalmia, syndromic 6 (MCOPS6) | 607932 | A, M | I, C (coloboma, microcornea, retinal dystrophy, myopia, sclerocornea, anterior segment dysgenesis, corectopia, blepharophimosis, optic nerve hypoplasia, tilted/anomalous optic disc, cyst, nystagmus, cataract, glaucoma, aphakia, embryotoxon, persistent hypoplastic primary vitreous) | N, S | [102,103] |
| 11 | BMP7 | 112267 | - | - | A, M | I, C (coloboma) | S | [104] |
| 12 | GDF3 | 606522 | Microphthalmia, isolated 7 (MCOP7) | 613704 | M | I, C (coloboma, optic nerve hypoplasia, foveal hypoplasia, nystagmus) | N, S | [105] |
| Microphthalmia, isolated with coloboma 6 (MCOPCB6) | 613703 | |||||||
| 13 | GDF6 | 601147 | Microphthalmia, isolated 4 (MCOP4) | 613094 | A, M | I, C (coloboma, optic nerve hypoplasia, foveal hypoplasia, nystagmus) | N, S | [7,21,90] |
| Microphthalmia, isolated with coloboma 6, digenic (MCOPCB6) | 613703 | |||||||
| 14 | ABCB6 | 605452 | Microphthalmia, isolated with coloboma 7 (MCOPCB7) | 614497 | M | C (coloboma) | N | [106] |
| 15 | ATOH7 | 609875 | Persistent hyperplastic primary vitreous, autosomal recessive (PHPVAR) | 221900 | M | I, C (microcornea, congenital cataract/corneal opacity, optic nerve aplasia/hypoplasia, retinal detachment/nonattachment, persistent fetal vasculature, nystagmus, vitreous degeneration, glaucoma, shallow anterior chamber, anterior displacement of the iris, peripheral anterior synechiae, calcifications present on the hyaloid membranes/retina/vitreous, vitreoretinal dysplasia) | N | [107] |
| 16 | C12orf57 | 615140 | Temtamy syndrome (TEMTYS) | 218340 | M | C (coloboma) | S | [108] |
| 17 | TENM3 | 610083 | Microphthalmia, isolated with coloboma 9 (MCOPCB9) | 615145 | M | C (coloboma, microcornea, nystagmus, esotropia, myopia, retinal detachment) | N, S | [109,110] |
| 18 | VAX1 | 604294 | Microphthalmia, syndromic 11 (MCOPS11) | 614402 | A, M | C (optic nerve hypoplasia, small optic nerve, cyst) | S | [111] |
| 19 | SMOC1 | 608488 | Microphthalmia with limb anomalies (MLA) | 206920 | A | C (optic nerve/tract aplasia) | S | [112] |
| 20 | FNBP4 | 615265 | Microphthalmia with limb anomalies (MLA) | 206920 | A | I | S | [113] |
| 21 | SHH | 600725 | Microphthalmia, isolated with coloboma 5 (MCOPCB5) | 611638 | A, M | I, C (coloboma, funnel retinal detachment with sub-retinal opacity, microcornea, small optic nerve, retinal dystrophy, tilted optic disc, myopia, nystagmus, glaucoma, posterior embryotoxon) | N, S | [102,114,115] |
| 22 | NAA10 | 300013 | Microphthalmia, syndromic 1 (MCOPS1) | 309800 | A | I | S | [116] |
| 23 | BCOR | 300485 | Microphthalmia, syndromic 2 (MCOPS2) | 300166 | A, M | I, C (congenital cataract, microcornea, posterior embryotoxon, secondary aphakia, secondary glaucoma, retinal detachment, persistent fetal vasculature, iris heterochromia, nystagmus, myopia, iris rubeosis, flat anterior chambers) | S | [117] |
| 24 | HCCS | 300056 | Linear skin defects with multiple congenital anomalies 1 (LSDMCA1) | 309801 | A, M | I, C (corneal opacity/cloudy and vascular cornea, cyst, sclerocornea, glaucoma,) | S | [118] |
| 25 | MAB21L2 | 604357 | Microphthalmia, syndromic 14 (MCSKS) | 615877 | A, M | I, C (coloboma, microcornea, exotropia, sclerocornea, strabismus) | S | [119] |
| 26 | RBP4 | 180250 | Microphthalmia, isolated with coloboma 10 (MCOPCB10) | 616428 | A, M | C (coloboma, small optic nerve/chiasm, cyst, underdeveloped extraocular muscles) | N, S | [81,120] |
| 27 | GLI2 | 165230 | Holoprosencephaly 9 (HPE9) | 610829 | A, M | I, C (coloboma, optic nerve agenesis) | S | [121,122,123] |
| 28 | PORCN | 300651 | Focal dermal hypoplasia (FDH) | 305600 | A, M | I, C (coloboma, aniridia, strabismus, ectopia lentis) | S | [124,125] |
| 29 | FRAS1 | 607830 | Fraser syndrome 1 (FRASRS1) | 219000 | A, M | C (fused/small palpebral fissure, cryptophthalmos) | S | [126] |
| 30 | FREM1 | 608944 | Manitoba oculotrichoanal syndrome (MOTA) | 248450 | A | I, C (coloboma, obstruction of the nasolacrimal duct) | S | [127] |
| 31 | SMCHD1 | 614982 | Bosma arhinia microphthalmia syndrome (BAMS) | 603457 | M | I, C (coloboma, hypertelorism, occluded or absent nasolacrimal duct, cataract) | S | [128] |
| 32 | SIX6 | 606326 | Microphthalmia, syndromic 6 (MCOPS6) | 607932 | A, M | C (coloboma, cataract, nystagmus, secondary glaucoma, optic nerve dysplasia/absence of optic nerve/chiasm/tract, retinal dystrophy, cyst) | S | [129] |
| 33 | TFAP2A | 107580 | Branchiooculofacial syndrome (BOFS) | 113620 | A, M | C (coloboma, cataract/corneal clouding, reduced corneal diameter, primary aphakia, sclerocornea, retinal detachment, lacrimal duct obstruction, cyst, subluxed cataractous lens, shallow anterior chamber, persistent pupillary membrane, iris hypoplasia, dysplastic optic disc) | S | [130] |
| 34 | TCTN2 | 613846 | Meckel syndrome, type 8 (MKS8) | 613885 | A, M | I | S | [131] |
| 35 | CSPP1 | 611654 | Joubert syndrome 21 (JBTS21) | 615636 | A | I | S | [132] |
| 36 | COL4A1 | 120130 | Brain small vessel disease with or without ocular anomalies (BSVD1) | 175780 | M | C (microcornea, Peter’s anomaly, retinal detachment, congenital cataract, glaucoma, anterior segment dysgenesis, hypermetropia, astigmatism) | S | [133,134] |
| 37 | PTCH1 | 601309 | Holoprosencephaly 7 (HPE7) | 610828 | M | C (coloboma, cataract, sclerocornea, anterior segment dysgenesis) | N, S | [135] |
| 38 | TBC1D32 | 615867 | Orofaciodigital syndrome IX (OFD9) | 258865 | A, M | C (coloboma) | S | [136] |
| 39 | CHD7 | 605806 | CHARGE syndrome | 214800 | A, M | I, C (coloboma, microcornea, cataract, persistent fetal vasculature) | N, S | [137,138,139] |
| 40 | MFRP | 606227 | Microphthalmia, isolated 5 (MCOP5) | 611040 | M | C (retinitis pigmentosa, foveoschisis, optic disc drusen, macular edema, glaucoma, hyperopia) | N | [140] |
| Nanophthalmos 2 (NN02) | 609549 | |||||||
| 41 | PRSS56 | 613858 | Microphthalmia, isolated 6 (MCOP6) | 613517 | M | C (hyperopia, elevated papillomacular retinal fold, shallow anterior chamber, thick lens, thickened scleral wall) | N | [141,142] |
| 42 | TMEM98 | 615949 | Nanophthalmos 4 (NN04) | 615972 | M | C (hyperopia, angle closure glaucoma, narrow iridocorneal angle, shallow anterior chamber depth, optic disc drusen) | N | [143,144] |
| 43 | HMGB3 | 300193 | Microphthalmia, syndromic 13 (MCOPS13) | 300915 | A, M | C (coloboma, congenital cataract) | S | [145] |
| 44 | PXDN | 605158 | Cornea opacification and other ocular anomalies (ASGD7) | 269400 | M | C (sclerocornea, anterior segment dysgenesis, iridocorneal dysgenesis, glaucoma, cataract) | N, S | [146] |
| 45 | TMX3 | 616102 | Microphthalmia with coloboma 1 (MCOPCB1) | 300345 | A, M | I, C (coloboma, cyst) | N, S | [147] |
| 46 | YAP1 | 606608 | Coloboma, ocular, with or without hearing impairment, cleft lip/palate, and/or mental retardation (COB1) | 120433 | A, M | I, C (coloboma, extraocular muscle defect, cataract, ectopic pupil) | N, S | [148] |
| 47 | IPO13 | 610411 | - | - | M | C (coloboma, cataract, narrowed palpebral fissure, nystagmus, microcornea) | N | [149] |
| 48 | PITX3 | 602669 | Cataract 11, multiple types (CTRCT11) | 610623 | M | C (cataract/corneal opacity) | S | [150] |
| 49 | NDP | 300658 | Norrie disease (ND) | 310600 | M | C (sclerocornea) | N | [92] |
| 50 | MITF | 156845 | COMMAD syndrome | 617306 | M | C (coloboma, microcornea with pannus, cataract, translucent irides, optic nerve/tract hypoplasia) | S | [151] |
| 51 | FOXC1 | 601090 | - | - | M | C (microcornea, sclerocornea, cyst, myopia, cataract, Rieger anomaly, retinal detachment) | N | [152] |
| 52 | CRPPA | 614631 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 7 (MDDGA7) | 614643 | M | C (cataract, optic nerve hypoplasia) | S | [153] |
| 53 | FANCL | 608111 | Fanconi anemia, complementation group L (FANCL) | 614083 | M | C (short upslant palpebral fissure, indiscernible pupil) | S | [154] |
| 54 | SMO | 601500 | Curry-Jones syndrome (CRJS) | 601707 | M | C (coloboma, unusually shaped pupil) | S | [155] |
| 55 | DOCK6 | 614194 | Adams-Oliver syndrome 2 (AOS2) | 614219 | M | I, C (retinal detachment) | S | [156] |
| 56 | CRYAA | 123580 | Cataract 9, multiple types (CTRCT9) | 604219 | M | C (congenital cataract) | N | [157] |
| 57 | FOXL2 | 605597 | Blepharophimosis, ptosis and epicanthus inversus (BPES) | 110100 | M | C (blepharophimosis, ptosis, epicanthus inversus, telecanthus, strabismus) | N, S | [158] |
| 58 | CRYBA4 | 123631 | Cataract 23, multiple types (CTRCT23) | 610425 | M | C (cataract, enophthalmia) | N | [159] |
| 59 | ERCC6 | 609413 | Cerebrooculofacioskeletal syndrome 1 (COFS1) | 214150 | M | C (congenital cataract, short palpebral fissure, blepharokeratoconjunctivitis) | S | [160,161] |
| 60 | ERCC5 | 133530 | Cerebrooculofacioskeletal syndrome 3 (COFS3) | 616570 | M | C (cataract) | S | [162] |
| 61 | ERCC1 | 126380 | Cerebrooculofacioskeletal syndrome 4 (COFS4) | 610758 | M | C (blepharophimosis) | S | [163] |
| 62 | SRD5A3 | 611715 | Congenital disorder of glycosylation, type 1q (CDG1q) | 612379 | M | C (coloboma, nystagmus, cataract, optic atrophy) | S | [164] |
| 63 | SALL4 | 607343 | Duane-radial ray syndrome (DRRS) | 607323 | M | C (coloboma, optic nerve hypoplasia) | S | [165] |
| 64 | FREM2 | 608945 | Fraser syndrome 2 (FRASRS2) | 617666 | M | C (coloboma, cyst) | S | [166] |
| 65 | RPGRIP1L | 610937 | Meckel syndrome 5 (MKS5) | 611561 | M | I | S | [167] |
| 66 | SLC25A24 | 608744 | Fontaine progeroid syndrome (FPS) | 612289 | M | I | S | [168] |
| 67 | FAM111A | 615292 | Gracile bone dysplasia (GCLEB) | 602361 | M | I | S | [169] |
| 68 | SMG9 | 613176 | Heart and brain malformation syndrome (HBMS) | 616920 | M | I | S | [170] |
| 69 | SIX3 | 603714 | Holoprosencephaly 2 (HPE2) | 157170 | M | C (coloboma, myopia, astigmatism, dysplastic optic nerve, nystagmus, exotropia, cataract, hypertropia) | S | [171] |
| 70 | PDE6D | 602676 | Joubert syndrome 22 (JBTS22) | 615665 | M | I, C (coloboma) | S | [172] |
| 71 | KMT2D | 602113 | Kabuki syndrome 1 (KABUK1) | 147920 | M | C (cyst) | S | [173] |
| 72 | PAX2 | 167409 | Papillorenal syndrome (PAPRS) | 120330 | M | C (coloboma, optic nerve dysplasia, retinal degeneration) | S | [174,175] |
| 73 | TMEM216 | 613277 | Meckel syndrome, type 2 (MKS2) | 603194 | M | I | S | [176] |
| 74 | CEP290 | 610142 | Meckel syndrome, type 4 (MKS4) | 611134 | M | I | S | [177] |
| 75 | KIF11 | 148760 | Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation (MCLMR) | 152950 | M | I, C (coloboma, cataract, chorioretinopathy, hypermetropia, persistent hyaloid artery, peripheral fibrovascular proliferation, retinal detachment) | S | [178,179] |
| 76 | SNX3 | 605930 | - | - | M | I | S | [180] |
| 77 | ZEB2 | 605802 | Mowat-Wilson syndrome (MOWS) | 235730 | M | C (cataract, retinal aplasia, corectopia, optic nerve hypoplasia/pallor, retinal atrophy) | S | [181] |
| 78 | POMT1 | 607423 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 1 (MDDGA1) | 236670 | M | C (anterior chamber dysgenesis, exophthalmia, buphthalmos, megalocornea, glaucoma, retinal dysplasia, congenital cataract/corneal clouding, retinal detachment) | S | [182,183] |
| 79 | POMT2 | 607439 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 2 (MDDGA2) | 613150 | M | C (Peter’s anomaly, cataract, buphthalmos) | S | [184] |
| 80 | POMGNT1 | 614828 | - | - | M | C (corneal clouding/cataract) | S | [185] |
| 81 | FKTN | 607440 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 4 (MDDGA4) | 253800 | M | C (retinal detachment) | S | [186] |
| 82 | FKRP | 606596 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 5 (MDDGA5) | 613153 | M | C (cataract, asymmetric pupils, persistent hyperplastic primary vitreous, anterior chamber abnormality) | S | [187,188] |
| 83 | DAG1 | 128239 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 9 (MDDGA9) | 616538 | M | C (buphthalmos, corneal opacity, glaucoma, retinal detachment) | S | [189,190] |
| 84 | B3GALNT2 | 610194 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies, type A, 11 (MDDGA11) | 615181 | M | C (cataract, optic nerve hypoplasia, myopia) | S | [191] |
| 85 | RAB3GAP1 | 602536 | Warburg micro syndrome 1 (WARBM1) | 600118 | M | C (microcornea, cataract) | S | [192] |
| 86 | RAB3GAP2 | 609275 | Warburg micro syndrome 2 (WARBM2) | 614225 | M | C (congenital cataract, small pupil, aphakia, hypermetropia, secondary glaucoma) | S | [193,194] |
| Martsolf syndrome | 212720 | |||||||
| 87 | NHS | 300457 | Nance-Horan syndrome (NHS) | 300672 | M | C (microcornea, congenital cataract) | S | [195] |
| Cataract 40, X-linked (CTRCT40) | 302200 | |||||||
| 88 | HMX1 | 142992 | Oculoauricular syndrome (OCACS) | 612109 | M | C (microcornea, coloboma, nystagmus, cataract, microphakia, synechiae, anterior segment dysgenesis, small dysplastic optic disc, strabismus, sclerocornea, posterior embryotoxon, stromal iris cyst, retinal dystrophy, dysplastic macropapillae, macular hypoplasia, irido-corneal adherences, enophthalmus, esotropia, calcified phthisis bulbi) | S | [196,197] |
| 89 | GJA1 | 121014 | Oculodentodigital dysplasia, autosomal recessive | 257850 | M | I, C (cataract, uveitis, glaucoma, persistent pupillary membrane) | S | [198,199,200] |
| Oculodentodigital dysplasia (ODDD) | 164200 | |||||||
| 90 | LRP5 | 603506 | Osteoporosis-pseudoglioma syndrome (OPPG) | 259770 | M | C (retinal detachment, persistent hyperplasia of the primary vitreous) | S | [201] |
| 91 | PQBP1 | 300463 | Renpenning syndrome (RENS1) | 309500 | M | C (coloboma) | S | [202] |
| 92 | TUBB | 191130 | Symmetric circumferential skin creases, congenital, 1 (CSCSC1) | 156610 | M | C (short palpebral fissure, retinal dysplasia, microcornea) | S | [203,204] |
| Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) | 615771 | M | ||||||
| 93 | MAPRE2 | 605789 | Symmetric circumferential skin creases, congenital, 2 (CSCSC2) | 616734 | M | C (short/slanting palpebral fissure, strabismus, ptosis) | S | [203] |
| 94 | SALL1 | 602218 | Townes-Brocks syndrome 1 (TBS1) | 107480 | A, M | C (abnormal lens, aplastic optic nerve, small optic chiasm) | S | [205] |
| 95 | HDAC6 | 300272 | Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia | 300863 | M | I | S | [206,207] |
| 96 | ALX1 | 601527 | Frontonasal dysplasia 3 (FND3) | 613456 | M | C (coloboma) | S | [208] |
| 97 | RERE | 605226 | Neurodevelopmental disorder with or without anomalies of the brain, eye, or heart (NEDBEH) | 616975 | M | C (coloboma, optic nerve hypoplasia, anisometropia) | S | [209] |
| 98 | RAB18 | 602207 | Warburg micro syndrome 3 | 614222 | M | C (microcornea, congenital cataract, small atonic pupil, progressive optic atrophy) | S | [210] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb7030016
AMA Style
Harding P, Moosajee M. The Molecular Basis of Human Anophthalmia and Microphthalmia. Journal of Developmental Biology. 2019; 7(3):16. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb7030016
Chicago/Turabian StyleHarding, Philippa, and Mariya Moosajee. 2019. "The Molecular Basis of Human Anophthalmia and Microphthalmia" Journal of Developmental Biology 7, no. 3: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb7030016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.