
A Syringe-Based and Centrifugation-Free DNA Extraction Procedure for the Rapid Detection of Bacteria


Department of Biological Engineering, College of Engineering, Konkuk University, Seoul 05029, Korea
*
Author to whom correspondence should be addressed.
Chemosensors 2021, 9(7), 167; https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070167
Submission received: 1 June 2021
/
Revised: 25 June 2021
/
Accepted: 30 June 2021
/
Published: 3 July 2021
(This article belongs to the Collection pH Sensors, Biosensors and Systems)
Abstract
:Several bacteria are known to cause food poisoning; therefore, diagnostic systems that detect bacteria are essential. Nucleic acid-based testing methods that involve polymerase chain reaction (PCR) amplification are of great interest due to their high sensitivity and specificity. Herein, we developed a syringe-based one-step DNA extraction device that streamlines the extraction of genomic DNA (gDNA) from bacteria within 2 min, enabling versatile application of nucleic acid-based testing in the field. Notably, the bolt-nut structured case coupled with the syringe enables control of the volume of solution dispensed for enabling DNA extraction without the need for bulky centrifuge equipment. Using the proposed system, the gDNA of a model bacterium, Escherichia coli, was extracted at a good quantity and quality and amplified via PCR. The DNA extracted was comparable to that extracted via a centrifugation-based procedure. In addition, bacteria that were artificially spiked in common samples, including a work cloth, a work bench, and meat, were successfully detected with high accuracy.
1. Introduction
Food poisoning caused by bacteria or viruses is a major public health problem that additionally incurs various social problems [1]. To prevent the risk of food poisoning, the development of diagnostic systems that detect specific target bacteria is essential. In particular, coliforms that produce the enzyme β-galactosidase are widely used as an indicator of the microbial safety of food, processed products, and drinking water [2,3]. Escherichia coli (E. coli) is one of the most studied coliforms and several studies have attempted to develop diagnostic systems for E. coli. The standard method used to discriminate E. coli involves microbial culturing using selective media, such as MacConkey agar, that contains different chromogenic or fluorogenic substrates [4,5,6]. However, culture-based methods require 1–2 days or more to generate results [7], and may produce false-positive results. For example, lactose-fermenting bacteria, such as Lactobacillus spp., which are non-coliform bacteria, can generate the same results as those obtained using E. coli, resulting in false-positive results because MacConkey agar can only differentiate bacteria based on their ability to ferment lactose. Therefore, polymerase chain reaction (PCR)-based nucleic acid testing, especially quantitative PCR (qPCR), owing to its high accuracy, is widely used as an alternative method to detect bacteria, such as E. coli and other coliforms, that cause food poisoning [8,9,10,11]. The first step in qPCR involves the extraction of genomic DNA (gDNA) from target bacteria [12,13]. Two representative methods are widely used for gDNA extraction: organic extraction and solid-phase extraction [14]. The organic extraction method is inexpensive and can extract a large amount of gDNA. However, it requires toxic chemicals and is time-consuming, owing to several steps, such as phenol–chloroform extraction, ethanol precipitation, and washing. In contrast, the solid-phase extraction method, which uses a silica membrane for interacting with DNA in the presence of chaotropic salts, presents a better option for the rapid extraction of gDNA because it is performed using a single tube and is a relatively simple procedure [15,16]. Several commercial kits manufactured and marketed by different companies are available. In principle, gDNA that is bound to silica membranes in the presence of chaotropic salts via the formation of salt bridges is eluted by changing the salt concentration after multiple washing steps [17]. While it is a simple and quick operation, centrifugation is required in each step, which is not desirable in field applications, especially in a resource-limited setting [18]. In addition, there is an attempt to skip DNA extraction, which is termed Direct-PCR. As the name indicates, this method can directly amplify target nucleic acids without the need for DNA extraction from the sample [19,20]. Because it simplifies the whole assay procedure and shortens the total analysis time, it has attracted special attention as an excellent alternative to the traditional DNA detection methods. However, Direct-PCR buffer that effectively lyses the sample and neutralizes PCR inhibitors is not compatible with other DNA amplification methods, such as recombinase polymerase amplification and helicase dependent amplification. Furthermore, Direct-PCR reagents are expensive and cannot completely minimize some PCR inhibitors, leading to inefficient PCR amplification and reduced detection sensitivity.
In this study, we developed a centrifugation-free and syringe-based one-step DNA extraction device for in-field detection of bacteria that cause food poisoning, which maximizes user convenience and reduces the duration of the overall assay. A bolt and nut-structured case was designed to fit a syringe, allowing the user to control the volume of the solution dispensed while avoiding the need for bulky centrifugation equipment. As a model target bacterium, E. coli was selected and its gDNA was extracted using both the proposed and standard, centrifugation-based procedures. We confirmed that gDNA extracted using the syringe-based system was of a suitable quantity and quality required for subsequent qPCR analysis, and was comparable to gDNA extracted via centrifugation-based assays. In addition, bacteria spiked in various samples were successfully detected with high accuracy.
2. Materials and Methods
2.1. Primer Design
DNA primers (forward primer: 5′-GCCATTGCACCGACAAAACT-3′; reverse primer: 5′-ACCAAGCATTCCGCCGATAA-3′) were designed using the Basic Logical Alignment Search Tool of the National Center for Biotechnology Information (National Institutes of Health, Bethesda, MD, USA) and were synthesized by Bionics (Seoul, Korea). The DNA primers were specific to the ybbW gene of E. coli that encodes a putative allantoin transporter and is recently found to be present in most strains of E. coli [21,22].
2.2. Bacterial Cultivation and Centrifugation-Based DNA Extraction
All bacterial strains used in this study (E. coli (KCTC 2441), Enterobacter cloacae (KCTC 2519), Klebsiella pneumoniae (ATCC 70063), Pseudomonas aeruginosa (ATCC 27853), and Staphylococcus aureus (ATCC 29213)) were purchased from the Korean Collection for Type Cultures (Daejeon, Korea). After the bacteria were grown in Luria-Bertani (LB) medium (BD, Franklin Lakes, NJ, USA) at 37 °C for 24 h, the culture solution was centrifuged at 5000× g for 5 min to obtain the cell pellet. The gDNA was then isolated using the Total DNA Extraction S&V kit (Bionics) according to the manufacturer’s instructions. Briefly, a bacterial cell lysate was prepared by adding 200 μL of lysis buffer and 20 μL of Proteinase K to the bacteria cells, which were incubated at 56 °C for 30 min and subsequently mixed with 500 μL of GDX buffer. Subsequently, binding, washing, and elution were performed to obtain the purified gDNA. Between each step, centrifugation was performed at 13,000× g for 2 min. The amount of extracted DNA was evaluated using a Nanodrop instrument (Spectramax iD5 multi-mode microplate reader; Molecular Devices, San José, CA, USA).
2.3. Fabrication of Syringe-Based DNA Extraction Device
The syringe-based DNA extraction device contained three major components. The first component is a silica membrane filter that binds to gDNA at a high salt concentration. The second component is a syringe (Korea Vaccine, Seoul, Korea) whose inner space is compartmentalized to sequentially flush the bacterial cell lysate, wash buffer, and elution buffer. A punctuated rubber packing at the center with an inverted bowl shape was placed inside the syringe (Figure 1A). The upper compartment was filled with 200 μL of elution buffer, and the lower compartment was filled with 750 μL of wash buffer. The final component involved a bolt and nut-structured case constructed using a Creality Ender 5 3D printer (Shenzhen, China), and was coupled with the syringe for enabling consistent control of the solution dispensed. The silica membrane filter, wash buffer, and elution buffer were all obtained from the Total DNA extraction S&V Kit.
2.4. Operation of Syringe-Based DNA Extraction Device
Bacterial cell lysates were prepared as described in Section 2.2. The prepared bacterial cell lysates were then drawn using the syringe, which was then assembled together with the bolt and nut-structured case and silica membrane filter. By turning the bolt and nut-structured case, the bacterial cell lysate and 750 μL of wash buffer present in the syringe were sequentially ejected through the silica membrane filter. Finally, 200 μL of elution buffer present in the syringe was ejected through the silica membrane filter to collect the purified gDNA, which was then analyzed via qPCR.
2.5. qPCR Assay
qPCR assays were performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA); the reaction mix (total volume, 10 μL) comprised TOPreal qPCR 1X PreMIX (SYBR Green with low ROX) (Enzynomics, Korea), forward/reverse DNA primers (100 nM), and the extracted gDNA at different concentrations. Amplification conditions were as follows: 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 15 s at 58 °C, and 30 s at 72 °C.
2.6. Spike-and-Recovery Analysis
Three model samples (size, 10 cm × 10 cm) found in the food industry, including work clothes (sample 1), work bench (sample 2), and meat (sample 3), were spiked with cultures of E. coli at different concentrations. Specifically, the solution containing E. coli was poured onto our model samples, which were then distributed with a pipette and dried. Subsequently, the spiked E. coli were collected using pipette swabs, which were immediately submerged in 10 mL of saline containing 0.85% sodium chloride (3M Pipette Swab Plus; 3M, Saint Paul, MN, USA). Finally, 1 mL of the solution was spread onto LB agar plates, and the number of cells was determined as colony-forming units (CFU)/mL by plating 10-fold serial dilutions. Simultaneously, 1 mL of solution was used to extract gDNA using the syringe-based DNA extraction device, which was analyzed via qPCR as described in Section 2.5.
3. Results and Discussion
3.1. Design of the Syringe-Based DNA Extraction Device
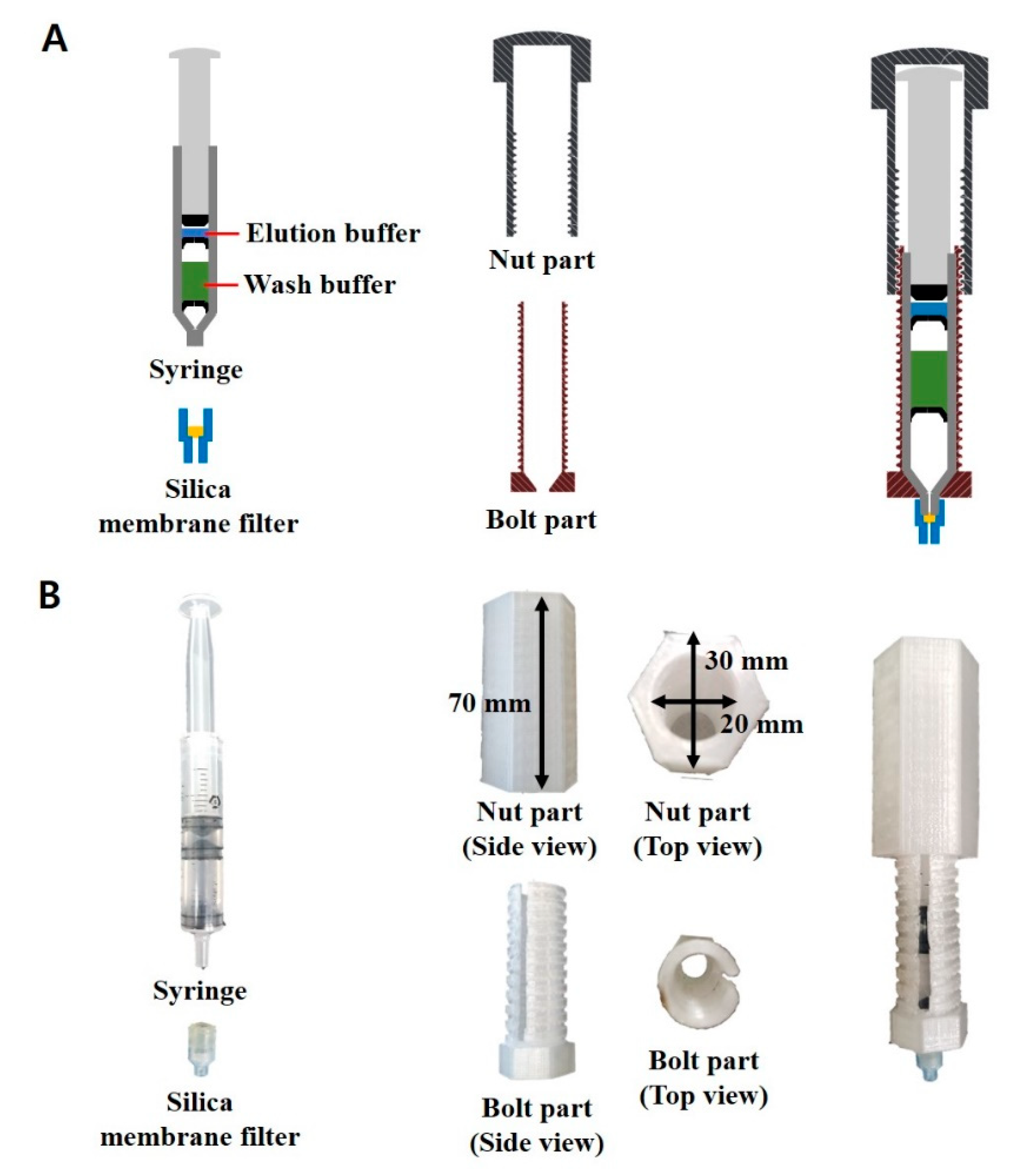
Figure 1A shows the schematic illustration of the syringe-based one-step DNA extraction device comprising three major components: (i) a silica membrane filter that is capable of binding to gDNA at a high salt concentration; (ii) a syringe where the inner space is filled with wash and elution buffers in each compartment by the inverted bowl-shaped rubber packing with a small hole at the center; (iii) a bolt and nut-structured case that enables the users to eject the solution through a silica membrane filter in a controlled and consistent manner. Figure 1B shows photographs of the syringe-based DNA extraction device. It is a portable, one-step device for the in-field detection of target bacteria.
The operational procedure for extracting gDNA from bacterial cell lysates is illustrated in Figure 2A. In the first step (Step 1), the bacterial cell lysate is sucked into a syringe. Next, a silica membrane filter is assembled to the syringe inlet, and the cell lysate and wash buffer present in the syringe are sequentially ejected through a silica membrane filter by turning the bolt and nut-structured case (Steps 2–4). The rubber packing that contains a small hole at the center was designed to contain an inverted bowl (arcuate) shape to prevent the solution from leaking in the absence of pressure. The arch-shaped rubber packing moves to the bottom of the syringe and changes into the original bowl shape only when pressure is applied, thereby discharging the solution through the silica membrane filter (Figure S1). In addition, we created an empty space for air in the compartment containing the wash buffer because it is advisable to dry the silica membrane filter after the washing step. As shown in Figure 2B, the proposed syringe-based DNA extraction system assembled with the bolt and nut-structured case allows for simple and consistent control of solution dispensed during the DNA extraction procedure. Overall, all the steps are performed using a single tube within 2 min, and the assay does not require bulky centrifuge devices. Therefore, the syringe-based one-step DNA extraction device is suitable for in-field diagnosis of target bacteria.
3.2. Performance of Syringe-Based DNA Extraction Device
We used the proposed system to extract gDNA from a model bacterium, E. coli, and estimated the gDNA amount (45.23 ng/µL) and the ratio of absorbance at 260 and 280 nm (A260/280; 1.64) (Table 1). For comparison of efficiency, we extracted gDNA from E. coli at the same number (2.2 × 108 CFU) using a commercial centrifugation-based DNA extraction kit (see the details in Section 2). As shown in Table 1, there was no significant difference in gDNA extracted using the syringe-based and centrifugation-based methods. Although the accuracy of the syringe-based method was relatively lower than the centrifugation-based one, there were no issues with the subsequent PCR amplification.
3.3. qPCR Analysis of Extracted gDNA
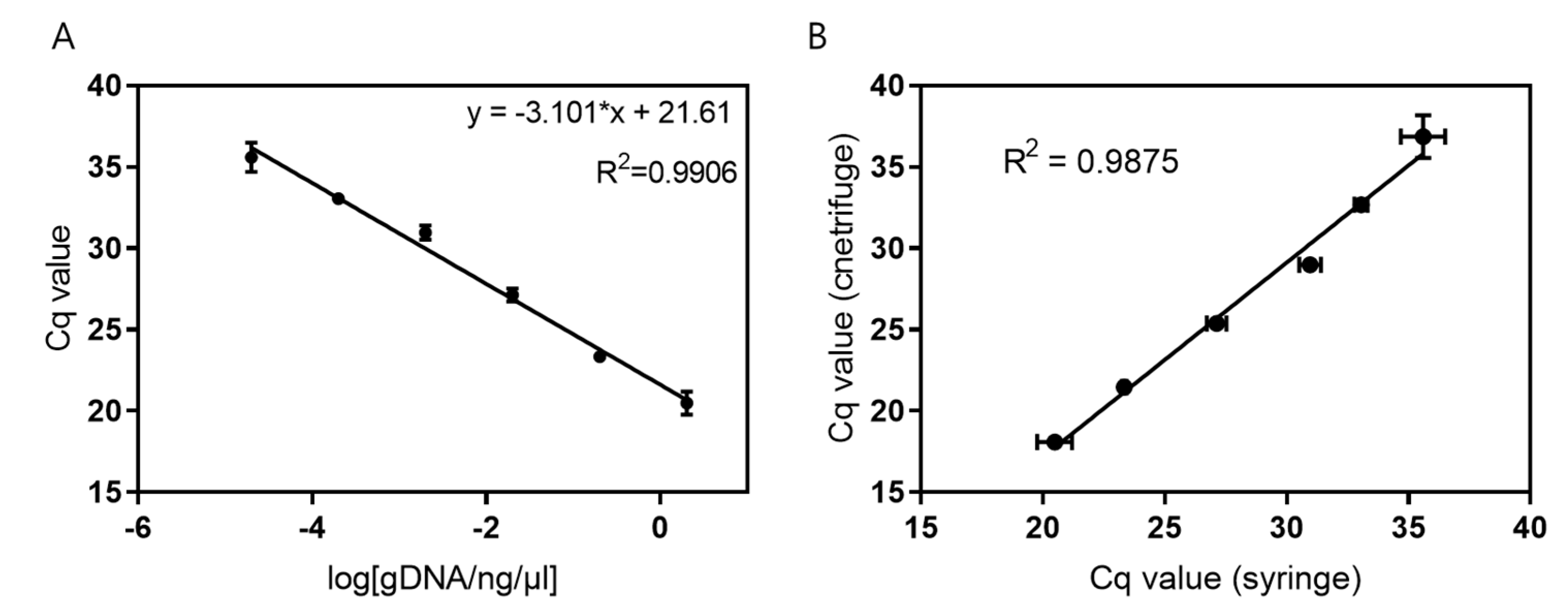
The extracted E. coli gDNA at different amounts was analyzed via qPCR. As shown in Figure 3A, a linear relationship was observed for the logarithmic values of E. coli gDNA concentrations ranging from 20 fg/μL to 2 ng/μL over 6 orders of magnitude (R2 = 0.9906). Similarly, qPCR was performed using different amounts of the extracted E. coli gDNA, which were compared using the syringe-based DNA extraction device. Both systems were found to have a high correlation (R2 = 0.9875) and could detect a very low concentration of DNA (20 fg/μL = 4 copies/μL) based on qPCR analysis (Figure 3B). Therefore, gDNA extracted using the syringe-based device was of excellent quality, and there was a minor loss of gDNA during the extraction process.
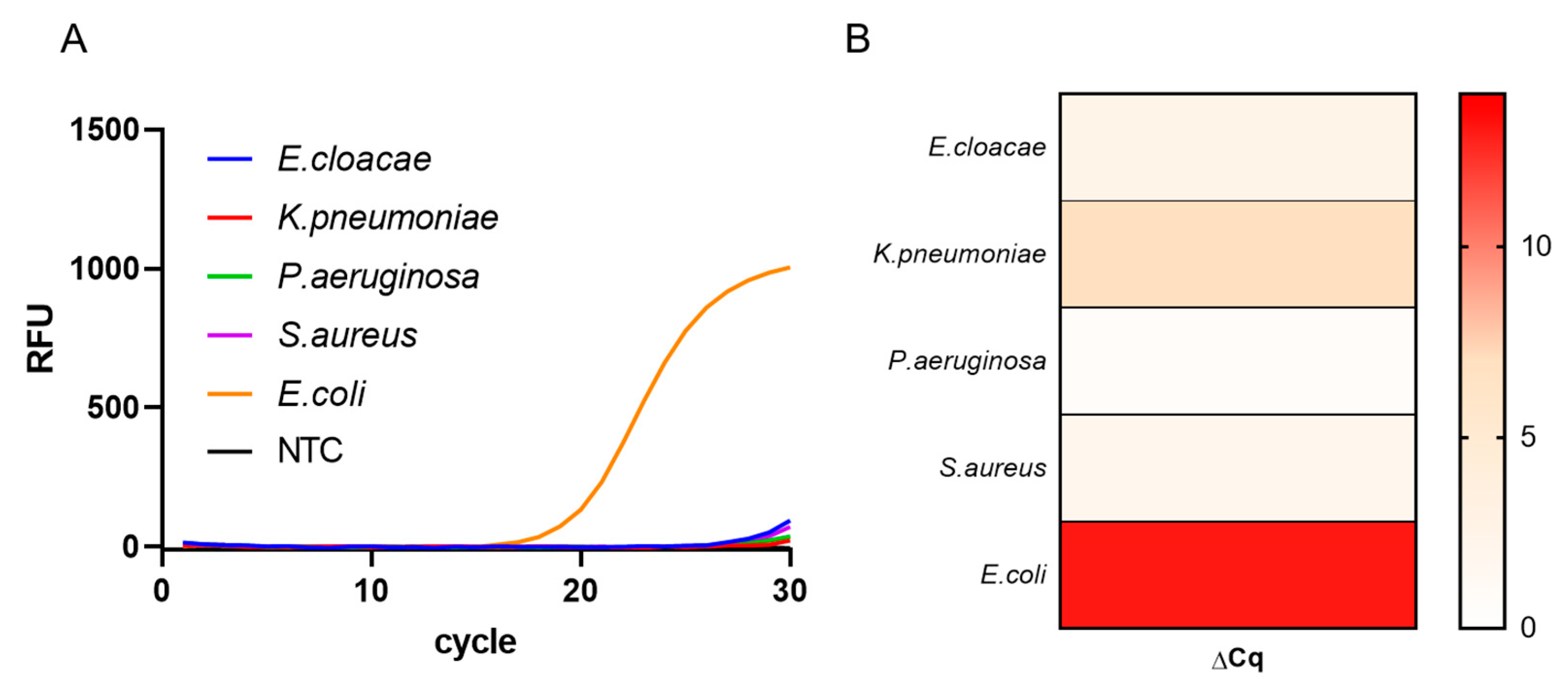
To evaluate the selectivity of the procedure, we performed qPCR using gDNA extracted from different bacteria (E. cloacae, K. pneumoniae, P. aeruginosa, S. aureus, and E. coli). Since the DNA primer was specifically designed for the ybbW gene of E. coli, it was expected that PCR amplification would occur only in the presence of E. coli gDNA. As shown in Figure 4, PCR amplification products were only obtained for E. coli, and were clearly distinguished from the PCR products of other bacteria (E. cloacae, K. pneumoniae, P. aeruginosa, and S. aureus). In addition, the change in the quantification cycle (ΔCq) value of E. coli, calculated by subtracting the Cq value in the presence of E. coli gDNA from 40 in a non-template control, was found to be 13.07, which is significantly higher than the ΔCq values of other bacteria. These results indicate that gDNA extracted using the syringe-based device is suitable for performing subsequent amplification using specific DNA primers, demonstrating that the extracted gDNA can be used in downstream applications.
3.4. Spike-and-Recovery Analysis
Finally, a spike-and-recovery test was performed to determine the utility of the syringe-based one-step DNA extraction device in practical applications. Three samples (work cloth, work bench, and meat) that are susceptible to contamination in food factories were selected and spiked with E. coli at different concentrations. After performing a lysis of the collected samples, the gDNA was extracted using our syringe-based device and analyzed via both qPCR and cell culturing (see the details in Section 2). The number of cells obtained using the qPCR and culturing methods was similar, with excellent recovery rates of 98–103% and a relative standard deviation (RSD) of <15% (Table 2). These results confirm that the proposed syringe-based device can be used to extract DNA and detect bacteria present in common samples.
4. Conclusions
In summary, we developed a simple, syringe-based one-step DNA extraction device comprising a silica membrane filter, a commercially available syringe, and a specially designed bolt and nut-structured case. The proposed system was used to extract gDNA of good quantity and quality, which was successfully used in a downstream application, qPCR. In addition, E. coli present in spiked samples were successfully detected with excellent recovery rates and RSD. The performance of our system is comparable to that of commercial, centrifugation-based procedures; however, the syringe-based method completes the extraction process within 2 min without the need for bulky centrifuge equipment. We believe that this portable, one-step device in combination with isothermal nucleic acid amplification methods has practical potential for use in the on-field detection of various bacteria, especially in resource-limited settings.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/chemosensors9070167/s1, Figure S1: The operational procedure showing how different solutions are sequentially dispensed. Green, red, and blue arrows indicate the pressure uniformly distributed over the rubber packing, the pressure concentrated on the center of the rugger packing, and the pressure that does not applied on the rubber packing, respectively.
Author Contributions
Conceptualization, T.Y. and S.K.; methodology, T.Y., S.K. and J.H.K.; validation, T.Y., S.K. and J.H.K.; formal analysis, T.Y. and S.K.; investigation, T.Y., S.K. and J.H.K.; writing-original draft preparation, T.Y.; writing-review and editing, K.S.P.; supervision, K.S.P.; funding acquisition, K.S.P. All authors have read and agreed to be published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2020R1C1C1012275), and Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry and Energy (MOTIE, 20194010201900).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. WHO Estimates of the Global Burden of Foodborne Diseases: Foodborne Disease Burden Epidemiology Reference Group; World Health Organization: Geneva, Switzerland, 2015; p. 225. [Google Scholar]
- Christen, L.; Davidson, J.; McAllister, S.; Roth, L. Coliform and Other Indicator Bacteria: In Standard Methods for the Examination of Dairy Products; RT Marshal; American Public Health Association: Washington, DC, USA, 1992. [Google Scholar]
- Leclerc, H.; Mossel, D.; Edberg, S.; Struijk, C. Advances in the bacteriology of the coliform group: Their suitability as markers of microbial water safety. Annu. Rev. Microbiol. 2001, 55, 201–234. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.; Siddons, C.A.; Zadik, P.; Jewes, L. An improved selective medium for the isolation of Escherichia coli O 157. J. Med. Microbiol. 1991, 35, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.; Fung, D.Y. Identification and quantification of fecal coliforms using violet red bile agar at elevated temperature. J. Milk Food Technol. 1976, 39, 768–770. [Google Scholar] [CrossRef]
- Edberg, S.C.; Allen, M.J.; Smith, D.B. National field evaluation of a defined substrate method for the simultaneous enumeration of total coliforms and Escherichia coli from drinking water: Comparison with the standard multiple tube fermentation method. Appl. Environ. Microbiol. 1988, 54, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, P.A. Further studies on the selectivity of violet red bile agar. J. Milk Food Technol. 1960, 23, 45–48. [Google Scholar] [CrossRef]
- Molina, F.; López-Acedo, E.; Tabla, R.; Roa, I.; Gómez, A.; Rebollo, J.E. Improved detection of Escherichia coli and coliform bacteria by multiplex PCR. BMC Biotechnol. 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reza, Z.M.; Mohammad, A.; Salomeh, K.; Reza, A.G.; Hossein, S.; Maryam, S.; Azam, A.; Mana, S.; Negin, N.; Reza, K.A. Rapid detection of coliforms in drinking water of Arak city using multiplex PCR method in comparison with the standard method of culture (Most Probably Number). Asian Pac. J. Trop. Biomed. 2014, 4, 404–409. [Google Scholar]
- Hwang, S.H.; Kwon, W.Y.; Eun, H.; Jeong, S.; Park, J.S.; Kim, K.J.; Kim, H.J.; Lee, S.H.; Park, K.; Yoon, J.-J. The use of a 2-aminopurine-containing split G-quadruplex for sequence-specific DNA detection. Artif. Cells Nanomed. Biotechnol. 2018, 46, S950–S955. [Google Scholar] [CrossRef] [PubMed]
- Holst-Jensen, A.; Rønning, S.B.; Løvseth, A.; Berdal, K.G. PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal. Bioanal. Chem. 2003, 375, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Cenis, J. Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res. 1992, 20, 2380. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Why the need for qPCR publication guidelines? —The case for MIQE. Methods 2010, 50, 217–226. [Google Scholar] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry 1987, 19, 11–15. [Google Scholar]
- Boom, R.; Sol, C.; Salimans, M.; Jansen, C.; Wertheim-van Dillen, P.; Van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill-Ambroz, K.L.; Brown-Guedira, G.L.; Fellers, J.P. Modified rapid DNA extraction protocol for high throughput microsatellite analysis in wheat. Crop Sci. 2002, 42, 2088–2091. [Google Scholar] [CrossRef] [Green Version]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Mogg, R.; Bond, J. A cheap, reliable and rapid method of extracting high-quality DNA from plants. Mol. Ecol. Notes 2003, 3, 666–668. [Google Scholar] [CrossRef]
- Ben-Amar, A.; Oueslati, S.; Mliki, A. Universal direct PCR amplification system: A time- and cost-effective tool for high-throughput applications. 3 Biotech. 2017, 7, 246. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, S.E.; Bathrick, A. Direct PCR amplification of forensic touch and other challenging DNA samples: A review. Forensic Sci. Int. Genet. 2018, 32, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.I.; McQuillan, J.; Taiwo, M.; Parks, R.; Stenton, C.A.; Morgan, H.; Mowlem, M.C.; Lees, D.N. A highly specific Escherichia coli qPCR and its comparison with existing methods for environmental waters. Water Res. 2017, 126, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Park, J.S.; Yoon, T.H.; Park, J.; Park, K.S. Nucleic acid lateral flow assay for simultaneous detection of hygiene indicator bacteria. Anal. Bioanal. Chem. 2021, 413, 1–9. [Google Scholar]
Figure 1.
Syringe-based one-step DNA extraction device. (A) Schematic illustration of each component of the syringe-based DNA extraction device. The elution buffer and wash buffer fill the syringe, compartmentalized with the inverted bowl-shaped rubber packing. (B) Photographs of each component of the syringe-based DNA extraction device.
Figure 1.
Syringe-based one-step DNA extraction device. (A) Schematic illustration of each component of the syringe-based DNA extraction device. The elution buffer and wash buffer fill the syringe, compartmentalized with the inverted bowl-shaped rubber packing. (B) Photographs of each component of the syringe-based DNA extraction device.

Figure 2.
Operation procedure of the syringe-based one-step DNA extraction device. (A) Schematic illustration of the DNA extraction procedure using the syringe-based DNA extraction device. (B) Photographs of the syringe-based DNA extraction device at each step of the extraction procedure.
Figure 2.
Operation procedure of the syringe-based one-step DNA extraction device. (A) Schematic illustration of the DNA extraction procedure using the syringe-based DNA extraction device. (B) Photographs of the syringe-based DNA extraction device at each step of the extraction procedure.

Figure 3.
Detection sensitivity of Escherichia coli gDNA. (A) Linear correlation of the logarithmic value of Escherichia coli gDNA concentration with Cq values. (B) Comparison of Cq values obtained using the syringe-based (x-axis) and centrifugation-based (y-axis) extraction procedures. Data are expressed as mean ± standard deviation; n = 3 technical replicates. gDNA, genomic DNA; Cq, cycle quantification value.
Figure 3.
Detection sensitivity of Escherichia coli gDNA. (A) Linear correlation of the logarithmic value of Escherichia coli gDNA concentration with Cq values. (B) Comparison of Cq values obtained using the syringe-based (x-axis) and centrifugation-based (y-axis) extraction procedures. Data are expressed as mean ± standard deviation; n = 3 technical replicates. gDNA, genomic DNA; Cq, cycle quantification value.

Figure 4.
Detection specificity of Escherichia coli gDNA. (A) Real-time polymerase chain reaction amplification curves of gDNA (2 ng/μL) extracted from different bacteria. (B) Heatmap of delta Cq values of gDNA (2 ng/μL) extracted from different bacteria. The delta Cq was calculated by subtracting the Cq value of the control from the Cq value of each sample. Data are expressed as mean ± standard deviation; n = 3 technical replicates. gDNA, genomic DNA; Cq, cycle quantification; RFU, relative fluorescence unit; NTC, non-template control.
Figure 4.
Detection specificity of Escherichia coli gDNA. (A) Real-time polymerase chain reaction amplification curves of gDNA (2 ng/μL) extracted from different bacteria. (B) Heatmap of delta Cq values of gDNA (2 ng/μL) extracted from different bacteria. The delta Cq was calculated by subtracting the Cq value of the control from the Cq value of each sample. Data are expressed as mean ± standard deviation; n = 3 technical replicates. gDNA, genomic DNA; Cq, cycle quantification; RFU, relative fluorescence unit; NTC, non-template control.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
DNA extracted from Escherichia coli using syringe-based and centrifugation-based extraction methods.
Table 1.
DNA extracted from Escherichia coli using syringe-based and centrifugation-based extraction methods.
| DNA Extraction Method | E. coli (Cell Number, CFU) | A260/A280 | Concentration (ng/µL) | Time |
|---|---|---|---|---|
| Syringe-based method | 2.2 × 108 | 1.64 ± 0.23 | 45.23 ± 11.7 | ~2 min |
| Centrifuge-based method | 2.2 × 108 | 1.72 ± 0.09 | 30.83 ± 3.6 | ~30 min |
Data are expressed as mean ± standard deviation; n = 3 technical replicates. A260/A280, ratio of absorbance at 260 nm to absorbance at 280 nm; CFU, colony-forming unit.
Table 2.
Results of spike-and-recovery assay using syringe-based DNA extraction device.
| Samples | Cell Count in Spiked Sample (CFU/mL) | Cell Count Detected Via Syringe Method (CFU/mL) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Sample 1 (work cloth) | 2.6 × 105 | 2.68 × 105 | 103.00 | 0.37 |
| Sample 2 (work bench) | 3.3 × 107 | 3.34 × 107 | 101.23 | 14.93 |
| Sample 3 (meat) | 4.5 × 106 | 4.41 × 106 | 98.08 | 5.96 |
CFU, colony-forming unit; RSD, relative standard deviation.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yoon, T.; Kim, S.; Kim, J.H.; Park, K.S. A Syringe-Based and Centrifugation-Free DNA Extraction Procedure for the Rapid Detection of Bacteria. Chemosensors 2021, 9, 167. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070167
AMA Style
Yoon T, Kim S, Kim JH, Park KS. A Syringe-Based and Centrifugation-Free DNA Extraction Procedure for the Rapid Detection of Bacteria. Chemosensors. 2021; 9(7):167. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070167
Chicago/Turabian StyleYoon, Taehwi, Seokjoon Kim, Jung Ho Kim, and Ki Soo Park. 2021. "A Syringe-Based and Centrifugation-Free DNA Extraction Procedure for the Rapid Detection of Bacteria" Chemosensors 9, no. 7: 167. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9070167
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.