
Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cancer Stem Cells in Tumor Growth, Metastasis, and Treatment Resistance
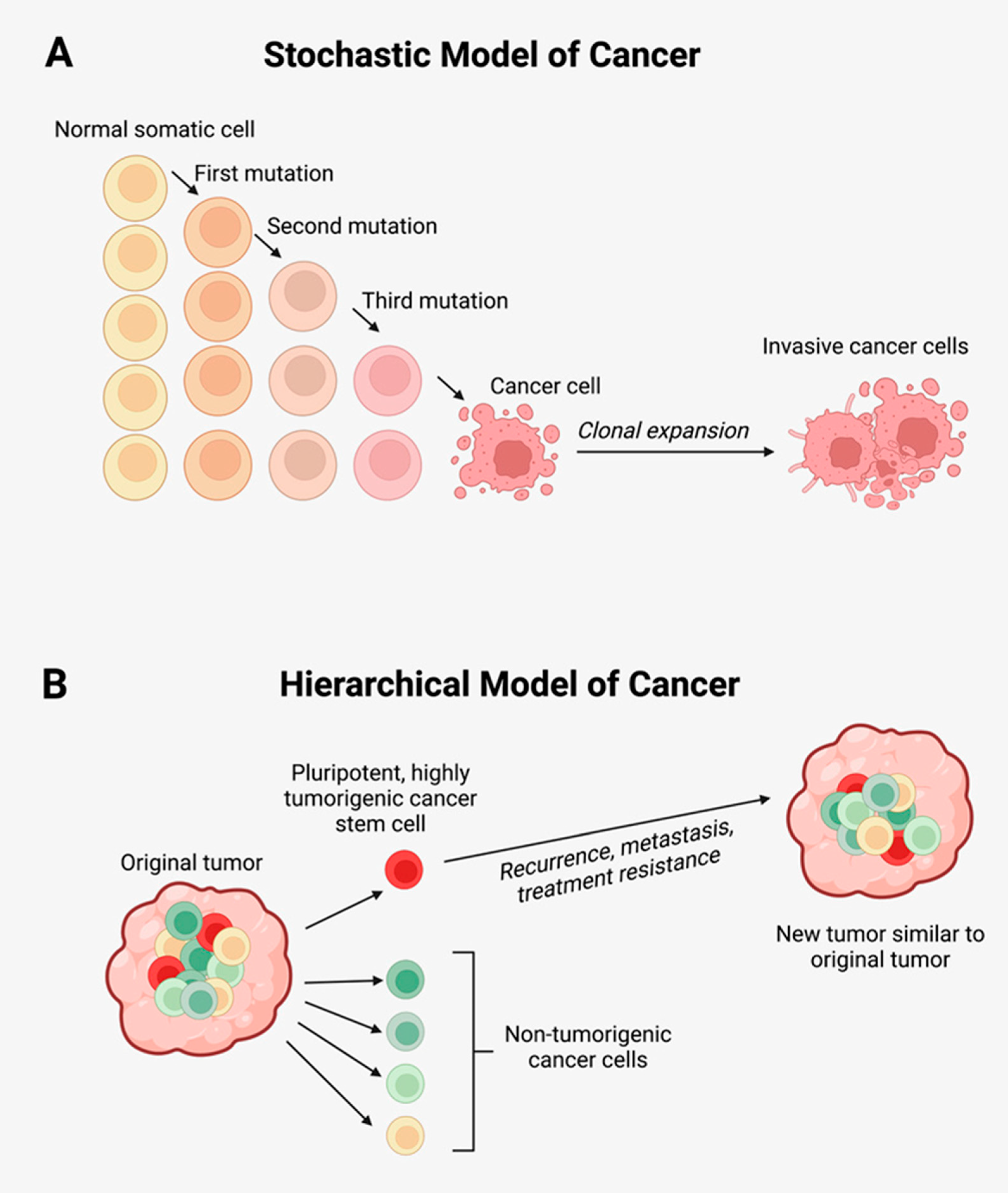
2.1. Cancer Stem Cells
2.2. Cancer Stem Cells and Metastasis and Treatment Resistance
2.3. Cancer Stem Cells and Multi-Drug Resistance
3. Mechanisms of Metastasis
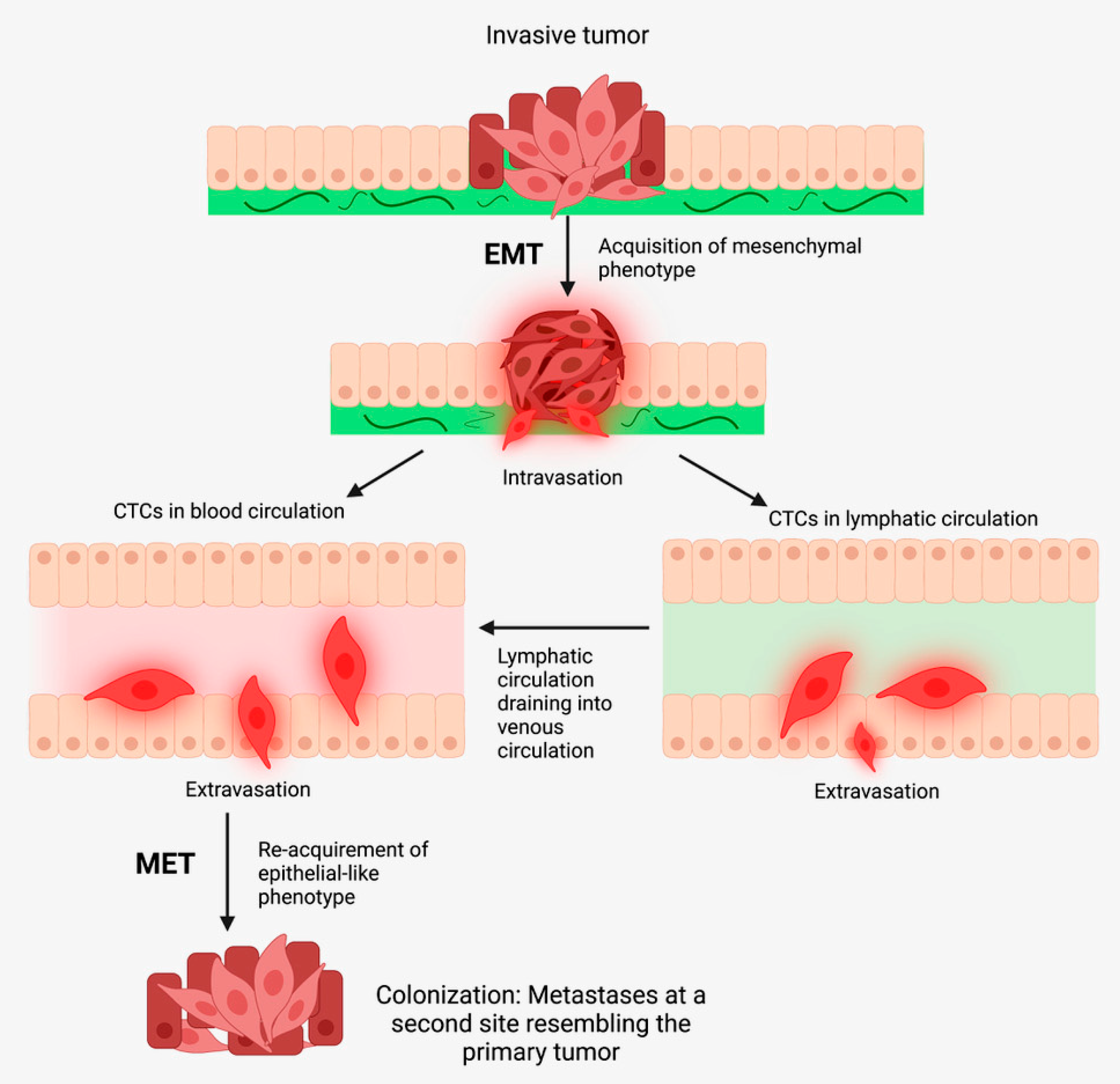
3.1. The Metastatic Cascade
3.2. Epithelial-to-Mesenchymal Transition
3.3. JAK/STAT3 Signaling Pathway
4. The Tumor Microenvironment and Its Role in Tumor Recurrence and Metastasis
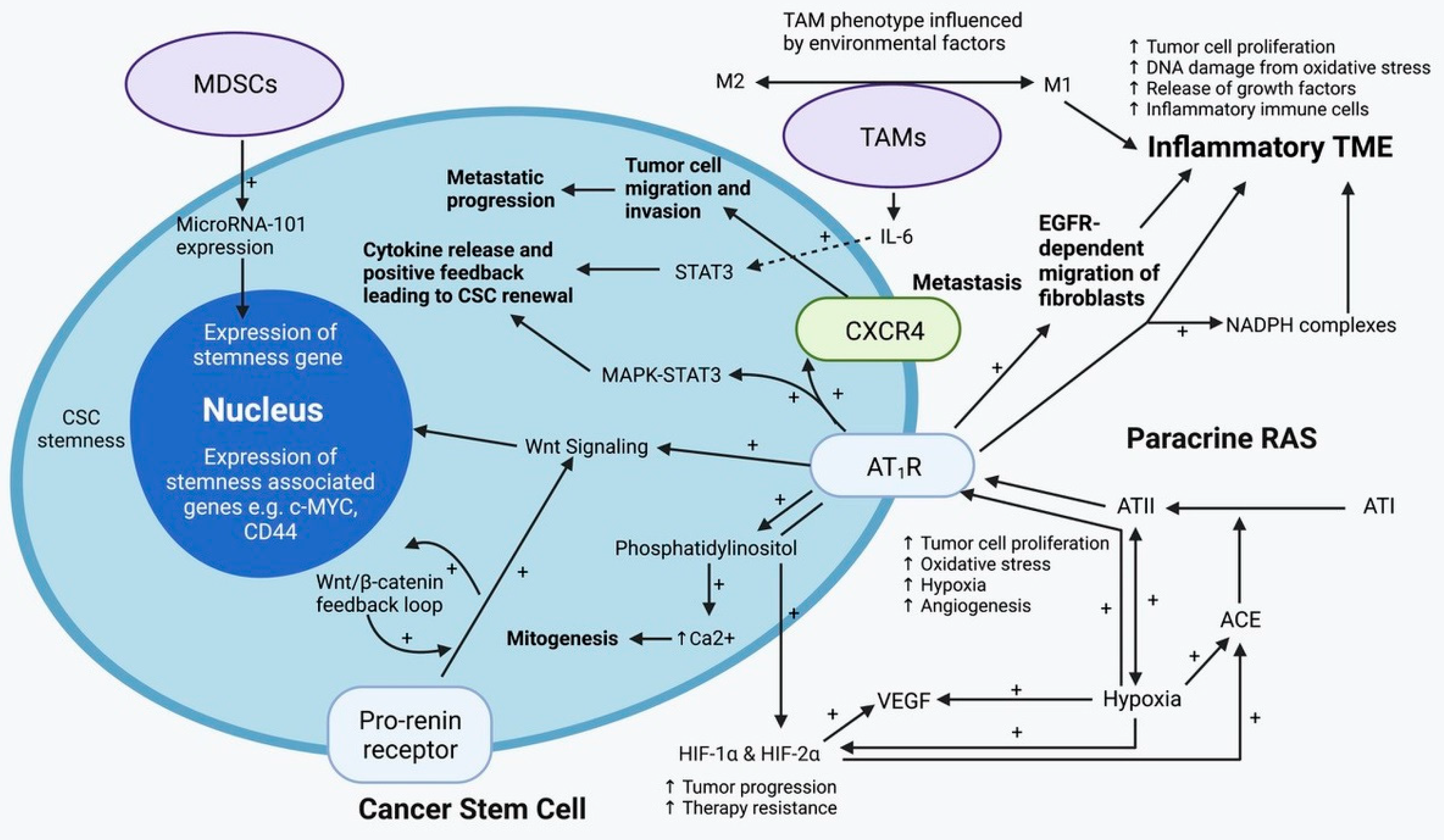
4.1. Tumor-Associated Macrophages
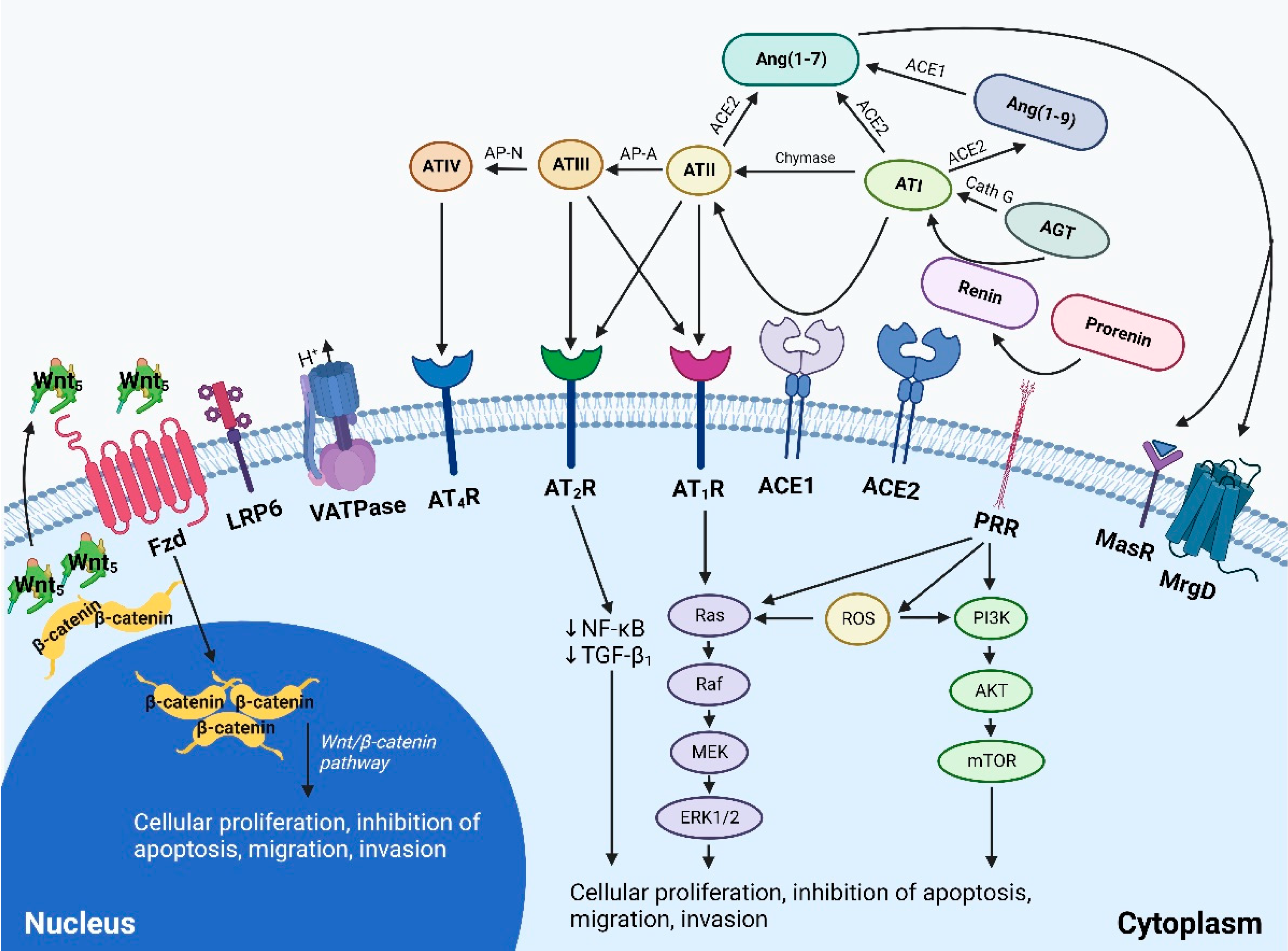
4.2. The Paracrine Renin-Angiotensin System
4.3. Signaling Pathways Converging on the Renin-Angiotensin System
4.4. Notch Signaling Pathway
4.5. Sonic Hedgehog Signaling Pathway
5. Therapeutic Interventions for Cancer Metastasis and Treatment Resistance
5.1. Single-Point Inhibition of Cancer Stem Cells and Signaling Pathways
5.1.1. Targeting the Notch Signaling Pathway
5.1.2. Targeting the Wnt/β-catenin Signaling Pathway
5.1.3. Targeting the Sonic Hedgehog Signaling Pathway
5.1.4. Strategies to Overcome Targeting Multi-Drug Resistance
5.2. Single-Point Inhibition of the Tumor Microenvironment
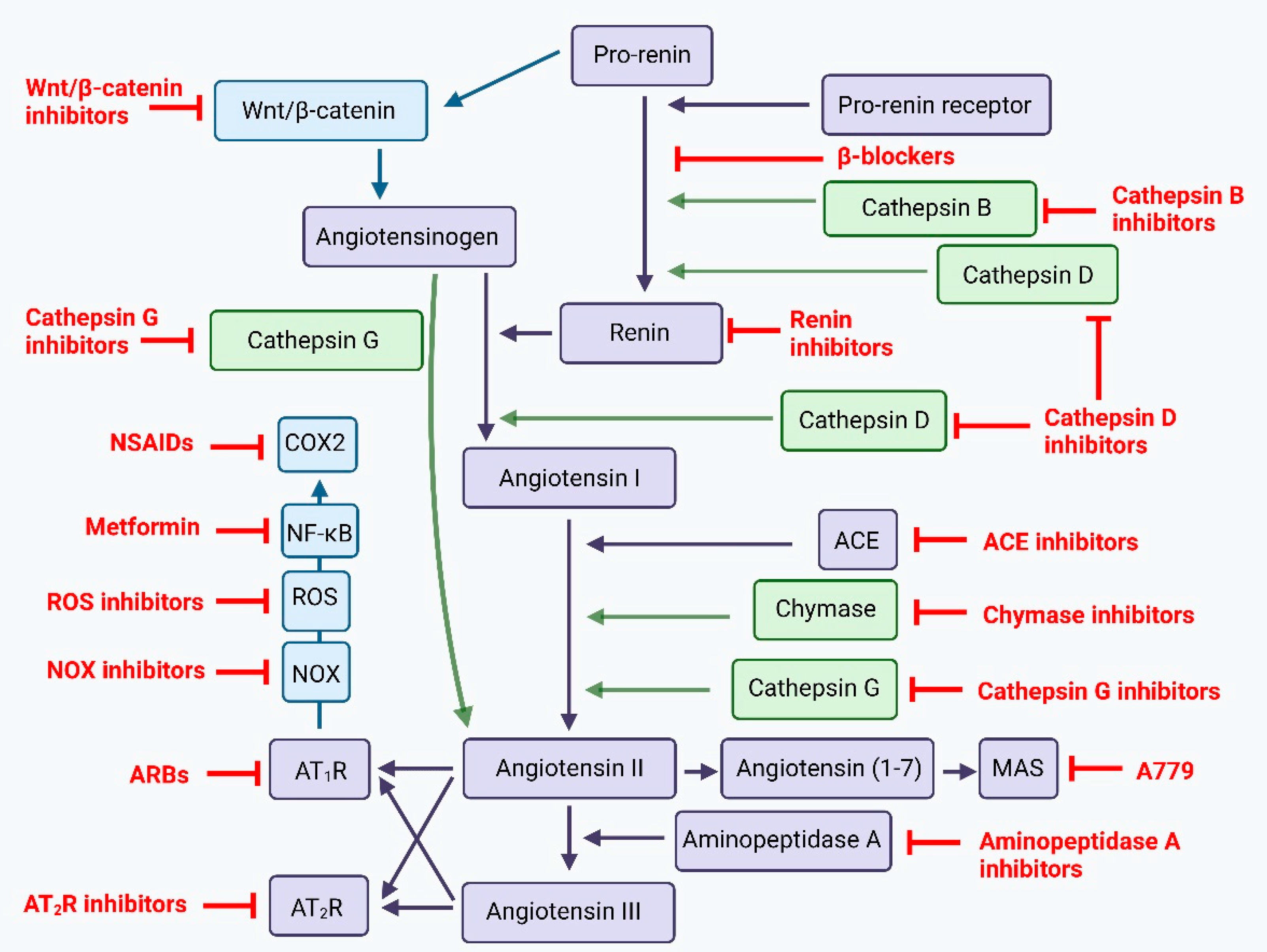
5.3. Targeting the Renin-Angiotensin System
5.4. Photosensitizer-Based Therapies
5.5. A Multi-Target Approach in the Treatment of Cancer
6. Conclusions
7. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem. Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations: Acquired genetic lability permits stepwise selection of variant sublines and underlies tumor progression. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Kilmister, E.J.; Tan, S.T. Cancer stem cells in head and neck cancers. In Atlas of Extreme Facial Cancer; Burton, L., Klaassen, M.F., Eds.; Springer Nature AG: Cham, Switzerland, 2022. [Google Scholar]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. (Eds.) A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. (Eds.) Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; Lleonart, M.E. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwerdtfeger, M.; Desiderio, V.; Kobold, S.; Regad, T.; Zappavigna, S.; Caraglia, M. Long non-coding RNAs in cancer stem cells. Transl. Oncol 2021, 14, 101134. [Google Scholar] [CrossRef] [PubMed]
- Eptaminitaki, G.C.; Wolff, N.; Stellas, D.; Sifakis, K.; Baritaki, S. Long Non-Coding RNAs (lncRNAs) in Response and Resistance to Cancer Immunosurveillance and Immunotherapy. Cells 2021, 10, 3313. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Tan, S.T. Insights Into Vascular Anomalies, Cancer, and Fibroproliferative Conditions: The Role of Stem Cells and the Renin-Angiotensin System. Front Surg 2022, 9, 868187. [Google Scholar] [CrossRef]
- Riganti, C.; Contino, M. New strategies to overcome resistance to chemotherapy and immune system in cancer. Int. J. Mol. Sci. 2019, 20, 4783. [Google Scholar] [CrossRef] [Green Version]
- Mina, L.A.; Sledge, G.W. Rethinking the metastatic cascade as a therapeutic target. Nat. Rev. Clin. Oncol. 2011, 8, 325–332. [Google Scholar] [CrossRef]
- Klein, C.A. The metastasis cascade. Science 2008, 321, 1785–1787. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.-I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea--a paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, A.; Vereb, G.; Szollosi, J. The hitchhikers guide to cancer stem cell theory: Markers, pathways and therapy. Cytom. A 2013, 83, 62–71. [Google Scholar] [CrossRef]
- Islam, F.; Qiao, B.; Smith, R.A.; Gopalan, V.; Lam, A.K. Cancer stem cell: Fundamental experimental pathological concepts and updates. Exp. Mol. Pathol. 2015, 98, 184–191. [Google Scholar] [CrossRef]
- Vermeulen, L.; Sprick, M.; Kemper, K.; Stassi, G.; Medema, J. Cancer stem cells–old concepts, new insights. Cell Death Differ. 2008, 15, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, L.; e Melo, F.d.S.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet. Oncol. 2012, 13, e83–e89. [Google Scholar] [CrossRef]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The cancer stem cell hypothesis: A work in progress. Lab. Investig. 2006, 86, 1203–1207. [Google Scholar] [CrossRef] [Green Version]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.; Kaplan, M.; Dalerba, P.; Weissman, I.; Clarke, M.; Ailles, L. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilmister, E.J.; Patel, J.; van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Cancer stem cell subpopulations are present within metastatic head and neck cutaneous squamous cell carcinoma. Front. Oncol. 2020, 10, 1091. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.P.; Brasch, H.D.; de Jongh, J.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in moderately differentiated head and neck cutaneous squamous cell carcinoma. Heliyon 2019, 5, e02257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.H.; Featherston, T.; Tan, S.T.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Itinteang, T. Characterization of Cancer Stem Cells in Moderately Differentiated Buccal Mucosal Squamous Cell Carcinoma. Front. Surg. 2016, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Brasch, H.D.; Dunne, J.C.; Davis, P.F.; Tan, S.T.; Itinteang, T. The Identification of Three Cancer Stem Cell Subpopulations within Moderately Differentiated Lip Squamous Cell Carcinoma. Front. Surg. 2017, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Baillie, R.; Itinteang, T.; Yu, H.H.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma. J. Clin. Pathol. 2016, 69, 742–744. [Google Scholar] [CrossRef] [Green Version]
- Yoganandarajah, V.; Patel, J.; van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T. Identification of cancer stem cell subpopulations in head and neck metastatic malignant melanoma. Cells 2020, 9, 324. [Google Scholar] [CrossRef] [Green Version]
- Wickremesekera, A.C.; Brasch, H.D.; Lee, V.M.; Davis, P.F.; Parker, A.; Koeck, H.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in metastatic melanoma to the brain express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 62. [Google Scholar] [CrossRef]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Glioblastoma Multiforme. Front. Surg. 2016, 3, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Cane, R.; Kennedy-Smith, A.; Brasch, H.D.; Savage, S.; Marsh, R.W.; Itinteang, T.; Tan, S.T.; Itinteang, T. Characterization of cancer stem cells in renal clear cell carcinoma. Stem. Cell Regen. Biol. 2019, 5, 6–17. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Marsh, R.W.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in primary colon adenocarcinoma. PLoS ONE 2019, 14, e0221963. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem. Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.; Leite, S.; Sauvageot, N.; Sarkisjan, D. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Walter, R.J.; Sonnentag, S.J.; Orian-Rousseau, V.; Munoz-Sagredo, L. Plasticity in colorectal cancer: Why cancer cells differentiate. Cancers 2021, 13, 918. [Google Scholar] [CrossRef]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, L.-J.; Gong, X.; Zhang, W.; Zhang, H.; Zhao, L. Co-expression of ATP binding cassette transporters is associated with poor prognosis in acute myeloid leukemia. Oncol. Lett. 2018, 15, 6671–6677. [Google Scholar] [CrossRef] [Green Version]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem. Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2—Cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Squartro, M.; Holland, E.C. Radiation resistance and stem-like cells in brain tumors. Cancer Cell 2006, 10, 454–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008, 68, 4674–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Lee, T.; Zheng, B.; Chan, K.; Guan, X. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammareri, P.; Scopelliti, A.; Todaro, M.; Eterno, V.; Francescangeli, F.; Moyer, M.P.; Agrusa, A.; Dieli, F.; Zeuner, A.; Stassi, G. Aurora-A is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res. 2010, 70, 4655–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020, 5, 1–35. [Google Scholar]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating notch signaling cisplatin enriches CD133+ cells via notch signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Jiang, Y.; Zou, F.; Liu, Y.; Wang, S.; Xu, N.; Xu, W.; Cui, C.; Xing, Y.; Liu, Y. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.-H. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, A.H.; Assadian, S.; Couture, F.; Lefebvre, K.J.; El-Assaad, W.; Barrès, V.; Ouellet, V.; Boulay, P.-L.; Yang, J.; Latour, M. V-ATPase-associated prorenin receptor is upregulated in prostate cancer after PTEN loss. Oncotarget 2019, 10, 4923. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dong, S.; Zhao, S.-C.; Liu, K.; Tan, Y.; Jiang, X.; Assaraf, Y.G.; Qin, B.; Chen, Z.-S.; Zou, C. Novel nanomedicines to overcome cancer multidrug resistance. Drug Resist. Updat. 2021, 58, 100777. [Google Scholar] [CrossRef] [PubMed]
- Janni, W.; Vogl, F.D.; Wiedswang, G.; Synnestvedt, M.; Fehm, T.; Jückstock, J.; Borgen, E.; Rack, B.; Braun, S.; Sommer, H.; et al. Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse—A European pooled analysis. Clin. Cancer Res. 2011, 17, 2967–2976. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem. Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaei, G.; Aziz, S.G.-G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharm. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zheng, T.; Zhao, L.; Zhao, G. Resveratrol suppresses epithelial-mesenchymal transition in GBM by regulating smad-dependent signaling. Biomed. Res. Int. 2019, 2019, 1321973. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco. Targets Ther. 2015, 8, 2973–2980. [Google Scholar]
- Choi, J.E.; Bae, J.S.; Kang, M.J.; Chung, M.J.; Jang, K.Y.; Park, H.S.; Moon, W.S. Expression of epithelial-mesenchymal transition and cancer stem cell markers in colorectal adenocarcinoma: Clinicopathological significance. Oncol. Rep. 2017, 38, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug signaling axis simultaneously controls epithelial-mesenchymal transition and stemness maintenance in colorectal cancer. Stem. Cells 2016, 34, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Bryson, B.L.; Junk, D.J.; Cipriano, R.; Jackson, M.W. STAT3-mediated SMAD3 activation underlies Oncostatin M-induced Senescence. Cell Cycle 2017, 16, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javaid, A.; Zahra, D.; Rashid, F.; Mashraqi, M.; Alzamami, A.; Khurshid, M.; Ashfaq, U.A. Regulation of micro-RNA, epigenetic factor by natural products for the treatment of cancers: Mechanistic insight and translational association. Saudi J. Biol. Sci. 2022, 29, 103255. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Kilmister, E.J.; Tan, S.T. The Role of the Renin-Angiotensin System in the Cancer Stem Cell Niche. J. Histochem. Cytochem. 2021, 69, 835–847. [Google Scholar] [CrossRef]
- Mahmud, Z.; Rahman, A.; Mishu, I.D.; Kabir, Y. Mechanistic insights into the interplays between neutrophils and other immune cells in cancer development and progression. Cancer Metastasis Rev. 2022, 14, 1–28. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-conditioned macrophages secrete migration-stimulating factor: A new marker for M2-polarization, influencing tumor cell motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Raggi, C.; Mousa, H.S.; Correnti, M.; Sica, A.; Invernizzi, P. Cancer stem cells and tumor-associated macrophages: A roadmap for multitargeting strategies. Oncogene 2016, 35, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Ludin, A.; Itkin, T.; Gur-Cohen, S.; Mildner, A.; Shezen, E.; Golan, K.; Kollet, O.; Kalinkovich, A.; Porat, Z.; D’Uva, G.; et al. Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat. Immunol. 2012, 13, 1072–1082. [Google Scholar] [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afsar, B.; Afsar, R.E.; Ertuglu, L.A.; Kuwabara, M.; Ortiz, A.; Covic, A.; Kanbay, M. Renin-angiotensin system and cancer: Epidemiology, cell signaling, genetics and epigenetics. Clin. Transl Oncol. 2021, 23, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Zouein, F.A.; Deris Zayeri, Z.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care. Spec. Pharm. 2007, 13 (8 Suppl B), 9–20. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef]
- Iwai, M.; Horiuchi, M. Devil and angel in the renin–angiotensin system: ACE–angiotensin II–AT1 receptor axis vs. ACE2–angiotensin-(1–7)–Mas receptor axis. Hypertens Res. 2009, 32, 533–536. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Fan, J.; Wu, F.; Huang, Q.; Guo, M.; Lv, Z.; Han, J.; Duan, L.; Hu, G.; Chen, L. The ACE2/angiotensin-(1–7)/mas receptor axis: Pleiotropic roles in cancer. Front. Physiol. 2017, 8, 276. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.R.; Wickremesekera, A.C.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Glioblastoma multiforme cancer stem cells express components of the renin–angiotensin system. Front. Surg. 2016, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Siljee, S.; Buchanan, O.; Brasch, H.; Bockett, N.; Patel, J.; Paterson, E.; Purdie, G.; Davis, P.; Itinteang, T.; Tan, S. Cancer stem cells in metastatic head and neck cutaneous squamous cell carcinoma express components of the renin-angiotensin system. Cells 2021, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.S.; Brasch, H.D.; Dunne, J.C.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer stem cells in moderately differentiated lip squamous cell carcinoma express components of the renin–angiotensin system. Front Surg. 2017, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siljee, S.; Pilkington, T.; Brasch, H.D.; Bockett, N.; Patel, J.; Paterson, E.; Davis, P.F.; Tan, S.T. Cancer stem cells in head and neck metastatic malignant melanoma express components of the renin-angiotensin system. Life 2020, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Siljee, S.; Milne, B.; Brasch, H.D.; Bockett, N.; Patel, J.; Davis, P.F.; Kennedy-Smith, A.; Itinteang, T.; Tan, S.T. Expression of components of the renin-angiotensin system by cancer stem cells in renal clear cell carcinoma. Biomolecules 2021, 11, 537. [Google Scholar] [CrossRef]
- Narayanan, A.; Wickremesekera, S.K.; van Schaijik, B.; Marsh, R.W.; Brasch, H.D.; Tan, S.T.; Itinteang, T. Cancer stem cells in liver metastasis from colon adenocarcinoma express components of the renin-angiotensin system. J. Cancer Metastais Treat 2019, 5, 36. [Google Scholar] [CrossRef]
- Hashemzehi, M.; Beheshti, F.; Hassanian, S.M.; Ferns, G.A.; Khazaei, M.; Avan, A. Therapeutic potential of renin angiotensin system inhibitors in cancer cells metastasis. Pathol. Res. Pract. 2020, 216, 153010. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef] [Green Version]
- Ishikane, S.; Takahashi-Yanaga, F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem. Pharm. 2018, 151, 96–103. [Google Scholar] [CrossRef]
- Roth, I.; Wickremesekera, A.C.; Wickremesekera, S.K.; Davis, P.F.; Tan, S.T. Therapeutic targeting of cancer stem cells via modulation of the renin-angiotensin system. Front Oncol. 2019, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.P.; Wickremesekera, A.C.; Brasch, H.D.; Marsh, R.; Tan, S.T.; Itinteang, T. Expression of cathepsins B, D, and G in isocitrate dehydrogenase-wildtype glioblastoma. Front Surg. 2017, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Featherston, T.; Brasch, H.D.; Siljee, S.D.; van Schaijik, B.; Patel, J.; de Jongh, J.; Marsh, R.W.; Itinteang, T.; Tan, S.T. Cancer stem cells in head and neck cutaneous squamous cell carcinoma express cathepsins. Plast. Reconstr. Surg.-Glob. Open. 2020, 8, e3042. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Chang-McDonald, B.; Patel, J.; Bockett, N.; Paterson, E.; Davis, P.F.; Tan, S.T. Cathepsins B, D, and G Are Expressed in Metastatic Head and Neck Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 3697. [Google Scholar] [CrossRef] [PubMed]
- Sangster, A.B.; Chang-McDonald, B.; Patel, J.; Bockett, N.; Paterson, E.; Davis, P.F.; Tan, S.T. Expression of cathepsins B and D by cancer stem cells in head and neck metastatic malignant melanoma. Melanoma. Res. 2021, 31, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Featherston, T.; Marsh, R.W.; van Schaijik, B.; Brasch, H.D.; Tan, S.T.; Itinteang, T. Expression and localization of cathepsins B, D, and G in two cancer stem cell subpopulations in moderately differentiated oral tongue squamous cell carcinoma. Front. Med. 2017, 4, 100. [Google Scholar] [CrossRef]
- Mehrotra, S.; Wickremesekera, S.K.; Brasch, H.D.; Van Schaijik, B.; Marsh, R.W.; Tan, S.T.; Itinteang, T. Expression and localization of cathepsins B, D and G in cancer stem cells in liver metastasis from colon adenocarcinoma. Front. Surg. 2018, 5, 40. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity. Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Novellasdemunt, L.; Antas, P.; Li, V.S. Targeting Wnt signaling in colorectal cancer. A review in the theme: Cell signaling: Proteins, pathways and mechanisms. Am. J. Physiol. Cell 2015, 309, C511–C521. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J. Wnt signaling pathway in non–small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, DJT356. [Google Scholar] [CrossRef] [Green Version]
- Howe, L.R.; Brown, A.M. Wnt signaling and breast cancer. Cancer Biol. Ther. 2004, 3, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takigawa, Y.; Brown, A. Wnt signaling in liver cancer. Curr. Drug. Targets 2008, 9, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- O’Rawe, M.; Kilmister, E.J.; Mantamadiotis, T.; Kaye, A.H.; Tan, S.T.; Wickremesekera, A.C. The renin–angiotensin system in the tumor microenvironment of glioblastoma. Cancers 2021, 13, 4004. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Ahmad, A.; Azmi, A.S.; Banerjee, S.; Kong, D.; Sarkar, F.H. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim. Biophys. Acta. Rev. Cancer 2010, 1806, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Panelos, J.; Massi, D. Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol. Ther. 2009, 8, 1986–1993. [Google Scholar] [CrossRef]
- Sikandar, S.S.; Pate, K.T.; Anderson, S.; Dizon, D.; Edwards, R.A.; Waterman, M.L.; Lipkin, S.M. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010, 70, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Banerjee, S.; Li, Y.; Rahman, K.W.; Zhang, Y.; Sarkar, F.H. Down-regulation of Notch-1 inhibits invasion by inactivation of nuclear factor-κB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 2006, 66, 2778–2784. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, Y.; Li, Y.; Banerjee, S.; Liao, J.; Sarkar, F.H. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol. Cancer Ther. 2006, 5, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Y.; Banerjee, S.; Kong, D.; Ahmad, A.; Nogueira, V.; Hay, N.; Sarkar, F.H. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-κB signaling pathways. J. Cell Biochem. 2010, 109, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Rizzo, P.; Rajan, P.; Albain, K.; Rychlik, K.; Shah, S.; Miele, L. Notch-1 and notch-4 receptors as prognostic markers in breast cancer. Int. J. Surg. Pathol. 2011, 19, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef]
- Mitsuhashi, Y.; Horiuchi, A.; Miyamoto, T.; Kashima, H.; Suzuki, A.; Shiozawa, T. Prognostic significance of Notch signalling molecules and their involvement in the invasiveness of endometrial carcinoma cells. Histopathology 2012, 60, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeasmin, S.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Ishikawa, M.; Iida, K.; Otsuki, Y.; Kobayashi, H.; Nakayama, S.; Miyazaki, K. Expression of nuclear Notch3 in cervical squamous cell carcinomas and its association with adverse clinical outcomes. Gynecol. Oncol. 2010, 117, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Xu, J.; Sun, Y. Notch1 activation is a poor prognostic factor in patients with gastric cancer. Br. J. Cancer 2014, 110, 2283–2290. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Wen, J.; Ning, Y.; Li, Y. Higher notch expression implies poor survival in pancreatic ductal adenocarcinoma: A systematic review and meta-analysis. Pancreatology 2018, 18, 954–961. [Google Scholar] [CrossRef]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar]
- Osipo, C.; Patel, P.; Rizzo, P.; Clementz, A.; Hao, L.; Golde, T.; Miele, L. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene 2008, 27, 5019–5032. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Singh, P.K.; Hollingsworth, M.A. Cancer metastasis facilitated by developmental pathways: Sonic hedgehog, Notch, and bone morphogenic proteins. J. Cell Biochem. 2007, 102, 829–839. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Ray, H. Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 2019, 42, 263–279. [Google Scholar] [CrossRef]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 2011, 6, e17687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Daya-Grosjean, L.; Couvé-Privat, S. Sonic hedgehog signaling in basal cell carcinomas. Cancer Lett. 2005, 225, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Kallassy, M.; Toftgård, R.; Ueda, M.; Nakazawa, K.; Vŏrechovský, I.; Yamasaki, H.; Nakazawa, H. Patched (ptch)-associated preferential expression of smoothened (smoh) in human basal cell carcinoma of the skin. Cancer Res. 1997, 57, 4731–4735. [Google Scholar] [PubMed]
- Bonifas, J.M.; Epstein Jr, E.H.; Pennypacker, S.; Chuang, P.-T.; McMahon, A.P.; Williams, M.; Rosenthal, A.; de Sauvage, F.J. Activation of expression of hedgehog target genes in basal cell carcinomas. J. Investig. Dermatol. 2001, 116, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. Cancer 2012, 131, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Nakamura, M.; Yamaguchi, H.; Yamanaka, N.; Akiyoshi, T.; Koga, K.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Nuclear factor-κB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006, 66, 7041–7049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, P.; Hernández, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta. Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotto, R.; Mishra, V.; Herz, E.; Yaacov, A.; Solomon, O.; Rauch, R.; Mondshine, A.; Motin, M.; Leibovich-Rivkin, T.; Davis, M.; et al. AL101, a gamma-secretase inhibitor, has potent antitumor activity against adenoid cystic carcinoma with activated NOTCH signaling. Cell Death Dis. 2022, 13, 678. [Google Scholar] [CrossRef] [PubMed]
- Deangelo, D.; Stone, R.; Silverman, L.; Stock, W.; Attar, E.; Fearen, I.; Dallob, A.; Matthews, C.; Stone, J.; Freedman, S. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J. Clin. Oncol. 2006, 24 (Suppl. S18), 6585. [Google Scholar] [CrossRef]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- McKeage, M.J.; Kotasek, D.; Markman, B.; Hidalgo, M.; Millward, M.J.; Jameson, M.B.; Harris, D.L.; Stagg, R.J.; Kapoun, A.M.; Xu, L. Phase IB trial of the anti-cancer stem cell DLL4-binding agent demcizumab with pemetrexed and carboplatin as first-line treatment of metastatic non-squamous NSCLC. Target. Oncol. 2018, 13, 89–98. [Google Scholar] [CrossRef]
- Elder, D.; Halton, D.E.; Hague, A.; Paraskeva, C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: Independence from COX-2 protein expression. Clin. Cancer Res. 1997, 3, 1679–1683. [Google Scholar]
- Maier, T.J.; Janssen, A.; Schmidt, R.; Geisslinger, G.; Grösch, S. Targeting the beta-catenin/APC pathway: A novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005, 19, 1353–1355. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA. 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickström, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjörnsson, B.; Sandén, E.; Darabi, A.; Siesjö, P.; Kool, M. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2013, 2501. [Google Scholar] [CrossRef]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34, e15271. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.; Licitra, L.; Dutriaux, C.; Thomas, L. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L. A Randomized Phase II Trial of Vismodegib versus Placebo with FOLFOX or FOLFIRI and Bevacizumab in Patients with Previously Untreated Metastatic Colorectal CancerVismodegib with Chemotherapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro. Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung. Cancer. 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012–12 (EDALINE) study. Investig. New. Drugs. 2019, 37, 98–108. [Google Scholar] [CrossRef]
- Ross, A.E.; Hughes, R.M.; Glavaris, S.; Ghabili, K.; He, P.; Anders, N.M.; Harb, R.; Tosoian, J.J.; Marchionni, L.; Schaeffer, E.M. Pharmacodynamic and pharmacokinetic neoadjuvant study of hedgehog pathway inhibitor Sonidegib (LDE-225) in men with high-risk localized prostate cancer undergoing prostatectomy. Oncotarget 2017, 8, 104182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudary, N.; Pintilie, M.; Hedley, D.; Hill, R.P.; Milosevic, M.; Mackay, H. Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. Br. J. Cancer 2017, 116, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Coon, V.; Laukert, T.; Pedone, C.A.; Laterra, J.; Kim, K.J.; Fults, D.W. Molecular Therapy Targeting Sonic Hedgehog and Hepatocyte Growth Factor Signaling in a Mouse Model of MedulloblastomaShh-and HGF-Targeted Therapy in Medulloblastoma. Mol. Cancer Ther. 2010, 9, 2627–2636. [Google Scholar] [CrossRef] [Green Version]
- Dinić, J.; Podolski-Renić, A.; Jovanović, M.; Musso, L.; Tsakovska, I.; Pajeva, I.; Dallavalle, S.; Pešić, M. Novel heat shock protein 90 inhibitors suppress P-glycoprotein activity and overcome multidrug resistance in cancer cells. Int. J. Mol. Sci. 2019, 20, 4575. [Google Scholar] [CrossRef] [Green Version]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targetingt tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Wang, T.; Wu, X.; Guo, C.; Zhang, K.; Xu, J.; Li, Z.; Jiang, S. Development of inhibitors of the programmed cell death-1/programmed cell death-ligand 1 signaling pathway. J. Med. Chem. 2019, 62, 1715–1730. [Google Scholar] [CrossRef]
- Castelli, V.; Giordano, A.; Benedetti, E.; Giansanti, F.; Quintiliani, M.; Cimini, A.; d’Angelo, M. The great escape: The power of cancer stem cells to evade programmed cell death. Cancers 2021, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in therapeutic targeting of cancer stem cells within the tumor microenvironment: An updated review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Li, M.; Luo, T.; Yin, Y.; Jiang, Y. Matrix metalloproteinases in tumorigenesis: An evolving paradigm. Cell Mol. Life Sci. 2011, 68, 3853–3868. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Giaccone, G.; Seymour, L.; Debruyne, C.; Bezjak, A.; Hirsh, V.; Smylie, M.; Rubin, S.; Martins, H.; Lamont, A.; et al. Prospective, randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients with small-cell lung cancer: A trial of the National Cancer Institute of Canada-Clinical Trials Group and the European Organization for Research and Treatment of Cancer. J. Clin. Oncol. 2002, 20, 4434–4439. [Google Scholar] [PubMed]
- Díaz, N.; Suárez, D. Molecular dynamics studies of matrix metalloproteases. Methods Mol. Biol. 2017, 1579, 111–134. [Google Scholar] [PubMed]
- Hira, V.V.; Breznik, B.; Vittori, M.; Loncq de Jong, A.; Mlakar, J.; Oostra, R.-J.; Khurshed, M.; Molenaar, R.J.; Lah, T.; Van Noorden, C.J. Similarities between stem cell niches in glioblastoma and bone marrow: Rays of hope for novel treatment strategies. J. Histochem. Cytochem. 2020, 68, 33–57. [Google Scholar] [CrossRef]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.; Van Noorden, C.J.; Molenaar, R.J. CXCR4 antagonists as stem cell mobilizers and therapy sensitizers for acute myeloid leukemia and glioblastoma? Biology 2020, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Bernasconi, P.; Borsani, O. Targeting leukemia stem cell-niche dynamics: A new challenge in AML treatment. J. Oncol. 2019, 2019, 8323592. [Google Scholar] [CrossRef] [Green Version]
- Lever, A.F.; Hole, D.J.; Gillis, C.R.; McCallum, I.R.; McInnes, G.T.; MacKinnon, P.L.; Meredith, P.A.; Murray, L.S.; Reid, J.L.; Robertson, J.W. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 1998, 352, 179–184. [Google Scholar] [CrossRef]
- Kong, A.P.; Yang, X.; So, W.-Y.; Luk, A.; Ma, R.C.; Ozaki, R.; Cheung, K.K.; Lee, H.-M.; Yu, L.; Xu, G. Additive effects of blood glucose lowering drugs, statins and renin-angiotensin system blockers on all-site cancer risk in patients with type 2 diabetes. BMC Med. 2014, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, Y.Y.; Chen, K.B.; Tsai, T.H.; Tsai, W.C. Lowered cancer risk with ACE inhibitors/ARBs: A population-based cohort study. J. Clin. Hypertens. (Greenwich) 2014, 16, 27–33. [Google Scholar] [CrossRef]
- Wang, K.-L.; Liu, C.-J.; Chao, T.-F.; Huang, C.-M.; Wu, C.-H.; Chen, T.-J.; Chiang, C.-E. Long-term use of angiotensin II receptor blockers and risk of cancer: A population-based cohort analysis. Int. J. Cardiol. 2012, 167, 2162–2166. [Google Scholar] [CrossRef] [PubMed]
- Christian, J.B.; Lapane, K.L.; Hume, A.L.; Eaton, C.B.; Weinstock, M.A. Association of ACE inhibitors and angiotensin receptor blockers with keratinocyte cancer prevention in the randomized VATTC trial. J. Natl. Cancer Inst. 2008, 100, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.-P.; Yuan, J.-M.; Sun, C.-L.; van den Berg, D.; Seow, A.; Lee, H.-P.; Mimi, C.Y. Angiotensin I-converting enzyme (ACE) gene polymorphism and breast cancer risk among Chinese women in Singapore. Cancer Res. 2003, 63, 573–578. [Google Scholar]
- Chae, Y.K.; Valsecchi, M.E.; Kim, J.; Bianchi, A.L.; Khemasuwan, D.; Desai, A.; Tester, W. Reduced risk of breast cancer recurrence in patients using ACE inhibitors, ARBs, and/or statins. Cancer Investig. 2011, 29, 585–593. [Google Scholar] [CrossRef]
- Santala, E.E.; Murto, M.O.; Artama, M.; Pukkala, E.; Visvanathan, K.; Murtola, T.J. Angiotensin Receptor Blockers Associated with Improved Breast Cancer Survival—A Nationwide Cohort Study from Finland. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2376–2382. [Google Scholar] [CrossRef] [PubMed]
- Makar, G.A.; Holmes, J.H.; Yang, Y.X. Angiotensin-converting enzyme inhibitor therapy and colorectal cancer risk. J. Natl. Cancer Inst. 2014, 106, djt374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engineer, D.R.; Burney, B.O.; Hayes, T.G.; Garcia, J.M. Exposure to ACEI/ARB and beta-Blockers Is Associated with Improved Survival and Decreased Tumor Progression and Hospitalizations in Patients with Advanced Colon Cancer. Transl Oncol. 2013, 6, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Sasahira, N.; Hirano, K.; Kogure, H.; Kawakubo, K.; Yagioka, H.; Yashima, Y.; et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br. J. Cancer 2010, 103, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Sasaki, T.; Takahara, N.; Saito, K.; Ishigaki, K.; Hamada, T.; Mizuno, S.; Miyabayashi, K.; Yamamoto, K.; et al. The inhibition of renin-angiotensin system in advanced pancreatic cancer: An exploratory analysis in 349 patients. J. Cancer Res. Clin. Oncol. 2015, 141, 933–939. [Google Scholar] [CrossRef]
- Tanaka, N.; Miyajima, A.; Kikuchi, E.; Matsumoto, K.; Hagiwara, M.; Ide, H.; Kosaka, T.; Masuda, T.; Nakamura, S.; Oya, M. Prognonstic impact of renin-angiotensin system blockade in localised upper-tract urothelial carcinoma. Br. J. Cancer 2012, 106, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Huang, C.H.; Lu, H.I.; Chen, C.H.; Huang, W.T.; Hsieh, M.J.; Rau, K.M.; Chang, A.Y.; Lin, W.C.; Li, S.H. Prognostic impact of renin-angiotensin system blockade in esophageal squamous cell carcinoma. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Wilop, S.; von Hobe, S.; Crysandt, M.; Esser, A.; Osieka, R.; Jost, E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J. Cancer Res. Clin. Oncol. 2009, 135, 1429–1435. [Google Scholar] [CrossRef]
- Wei, J.; Zhou, Z.; Xu, Z.; Zeng, S.; Chen, X.; Wang, X.; Liu, W.; Liu, M.; Gong, Z.; Yan, Y. Retrospective clinical study of renin-angiotensin system blockers in lung cancer patients with hypertension. PeerJ 2019, 7, e8188. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.A.; Mann, J.R.; Shoaibi, A.; Pai, S.G.; Bottai, M.; Sutton, S.S.; Haddock, K.S.; Bennett, C.L.; Hebert, J.R. Angiotensin receptor blockers: Are they related to lung cancer? J. Hypertens. 2013, 31, 1669. [Google Scholar] [CrossRef] [Green Version]
- Rao, G.A.; Mann, J.R.; Bottai, M.; Uemura, H.; Burch, J.B.; Bennett, C.L.; Haddock, K.S.; Hebert, J.R. Angiotensin receptor blockers and risk of prostate cancer among United States veterans. J. Clin. Pharm. 2013, 53, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C.; Chan, W.-L.; Chen, Y.-C.; Chen, T.-J.; Lin, S.-J.; Chen, J.-W.; Leu, H.-B. Angiotensin II receptor blockers and risk of cancer in patients with systemic hypertension. Am. J. Oncol. 2011, 107, 1028–1033. [Google Scholar] [CrossRef]
- Algazi, M.; Plu-Bureau, G.; Flahault, A.; Dondon, M.; Le, M. Is beta-blocker treatment associated with a decrease in the risk of cancer. Lett. Drug Des. Discov. 2006, 3, 653–661. [Google Scholar] [CrossRef]
- Lin, C.-S.; Lin, W.-S.; Lin, C.-L.; Kao, C.-H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-Y.; Huang, W.-Y.; Lin, C.-L.; Huang, T.-C.; Wu, Y.-Y.; Chen, J.-H.; Kao, C.-H. Propranolol reduces cancer risk: A population-based cohort study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Grazzini, M.; Gandini, S.; Benemei, S.; Lotti, T.; Marchionni, N.; Geppetti, P. Treatment with β-blockers and reduced disease progression in patients with thick melanoma. Arch. Intern. Med. 2011, 171, 779–781. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Gandini, S.; Grazzini, M.; Benemei, S.; Marchionni, N.; Geppetti, P. Effect of β-blockers and other antihypertensive drugs on the risk of melanoma recurrence and death. Mayo. Clin. Proc 2013, 88, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Geppetti, P.; Gandini, S. β-Blocker use and reduced disease progression in patients with thick melanoma: 8 years of follow-up. Melanoma Res. 2017, 27, 268–270. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018, 7, e1405205. [Google Scholar] [CrossRef] [Green Version]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta blockers and breast cancer mortality: A population-based study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef] [Green Version]
- Botteri, E.; Munzone, E.; Rotmensz, N.; Cipolla, C.; De Giorgi, V.; Santillo, B.; Zanelotti, A.; Adamoli, L.; Colleoni, M.; Viale, G. Therapeutic effect of β-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res. Treat 2013, 140, 567–575. [Google Scholar] [CrossRef]
- Powe, D.G.; Voss, M.J.; Zänker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628. [Google Scholar] [CrossRef] [Green Version]
- Melhem-Bertrandt, A.; Chavez-MacGregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.-M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A.; Håheim, L.L. Use of β-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate 2013, 73, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A. Association between use of β-blockers and prostate cancer–specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2013, 65, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.S.; Karlan, B.Y.; Li, A.J. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol. Oncol. 2012, 127, 375–378. [Google Scholar] [CrossRef]
- Herrera, I.; Pascual, S.; Zapater, P.; Carnicer, F.; Bellot, P.; Maria Palazon, J. The use of β-blockers is associated with a lower risk of developing hepatocellular carcinoma in patients with cirrhosis. Eur. J. Gastroenterol. Hepatol. 2016, 28, 1194–1197. [Google Scholar] [CrossRef]
- Ahl, R.; Matthiessen, P.; Sjölin, G.; Cao, Y.; Wallin, G.; Ljungqvist, O.; Mohseni, S. Effects of beta-blocker therapy on mortality after elective colon cancer surgery: A Swedish nationwide cohort study. BMJ Open 2020, 10, e036164. [Google Scholar] [CrossRef]
- Cardwell, C.R.; Mc Menamin, Ú.C.; Hicks, B.M.; Hughes, C.; Cantwell, M.M.; Murray, L.J. Drugs affecting the renin-angiotensin system and survival from cancer: A population based study of breast, colorectal and prostate cancer patient cohorts. BMC Med. 2014, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mandilaras, V.; Bouganim, N.; Yin, H.; Asselah, J.; Azoulay, L. The use of drugs acting on the renin-angiotensin system and the incidence of pancreatic cancer. Br. J. Cancer 2017, 116, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, G.; Rodríguez, L.A.G.; Ruigómez, A.; Johansson, S.; Wallander, M.A.; Frithz, G.; Svärdsudd, K. Association between captopril, other antihypertensive drugs and risk of prostate cancer. Prostate 2004, 58, 50–56. [Google Scholar] [CrossRef]
- Livingstone, E.; Hollestein, L.; van Herk-Sukel, M.; van de Poll-Franse, L.; Nijsten, T.; Schadendorf, D.; de Vries, E. β-Blocker use and all-cause mortality of melanoma patients: Results from a population-based Dutch cohort study. Eur. J. Cancer 2013, 49, 3863–3871. [Google Scholar] [CrossRef]
- McCourt, C.; Coleman, H.; Murray, L.; Cantwell, M.; Dolan, O.; Powe, D.; Cardwell, C. Beta-blocker usage after malignant melanoma diagnosis and survival: A population-based nested case–control study. Br. J. Dermatol. 2014, 170, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Sakellakis, M.; Kostaki, A.; Starakis, I.; Koutras, A. β-Blocker use and risk of recurrence in patients with early breast cancer. Chemotherapy 2014, 60, 288–289. [Google Scholar] [CrossRef]
- Cardwell, C.R.; Coleman, H.G.; Murray, L.J.; Entschladen, F.; Powe, D.G. Beta-blocker usage and breast cancer survival: A nested case-control study within a UK clinical practice research datalink cohort. Int. J. Epidemiol. 2013, 42, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, P.A.; Habel, L.A.; Weltzien, E.K.; Caan, B.J.; Cole, S.W. Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: Results from the LACE cohort. Breast Cancer Res. Treat 2011, 129, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, L.; Below, J.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M. Beta blocker use and colorectal cancer risk: Population-based case-control study. Cancer 2012, 118, 3911–3919. [Google Scholar] [CrossRef]
- Hicks, B.; Murray, L.; Powe, D.; Hughes, C.; Cardwell, C. β-Blocker usage and colorectal cancer mortality: A nested case–control study in the UK Clinical Practice Research Datalink cohort. Ann. Oncol. 2013, 24, 3100–3106. [Google Scholar] [CrossRef]
- Cata, J.P.; Villarreal, J.; Keerty, D.; Thakar, D.R.; Liu, D.D.; Sood, A.K.; Gottumukkala, V. Perioperative beta-blocker use and survival in lung cancer patients. J. Clin. Anesth. 2014, 26, 106–117. [Google Scholar] [CrossRef]
- Johannesdottir, S.A.; Schmidt, M.; Phillips, G.; Glaser, R.; Yang, E.V.; Blumenfeld, M.; Lemeshow, S. Use of B-blockers and mortality following ovarian cancer diagnosis: A population-based cohort study. BMC Cancer 2013, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Assayag, J.; Pollak, M.N.; Azoulay, L. Post-diagnostic use of beta-blockers and the risk of death in patients with prostate cancer. Eur. J. Cancer 2014, 50, 2838–2845. [Google Scholar] [CrossRef]
- Friis, S.; Sørensen, H.T.; Mellemkjær, L.; McLaughlin, J.K.; Nielsen, G.L.; Blot, W.J.; Olsen, J.H. Angiotensin-converting enzyme inhibitors and the risk of cancer: A population-based cohort study in Denmark. Cancer 2001, 92, 2462–2470. [Google Scholar] [CrossRef]
- Kim, S.-A.; Moon, H.; Roh, J.-L.; Kim, S.-B.; Choi, S.-H.; Nam, S.; Kim, S. Postdiagnostic use of β-blockers and other antihypertensive drugs and the risk of recurrence and mortality in head and neck cancer patients: An observational study of 10,414 person–years of follow-up. Clin. Transl Oncol. 2017, 19, 826–833. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef] [PubMed]
- O’Rawe, M.; Wickremesekera, A.C.; Pandey, R.; Young, D.; Sim, D.; FitzJohn, T.; Burgess, C.; Kaye, A.H.; Tan, S.T. Treatment of glioblastoma with re-purposed renin-angiotensin system modulators: Results of a phase I clinical trial. J. Clin. Neurosci. 2022, 95, 48–54. [Google Scholar] [CrossRef]
- Kast, R.E.; Boockvar, J.A.; Brüning, A.; Cappello, F.; Chang, W.W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013, 4, 502–530. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef] [Green Version]
- Skaga, E.; Skaga, I.Ø.; Grieg, Z.; Sandberg, C.J.; Langmoen, I.A.; Vik-Mo, E.O. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J. Cancer Res. Clin. Oncol. 2019, 145, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Kast, R.E.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. A phase Ib/IIa trial of 9 repurposed drugs combined with temozolomide for the treatment of recurrent glioblastoma: CUSP9v3. Neurooncol Adv. 2021, 3, vdab075. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilmister, E.J.; Koh, S.P.; Weth, F.R.; Gray, C.; Tan, S.T. Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment. Biomedicines 2022, 10, 2988. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112988
Kilmister EJ, Koh SP, Weth FR, Gray C, Tan ST. Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment. Biomedicines. 2022; 10(11):2988. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112988
Chicago/Turabian StyleKilmister, Ethan J., Sabrina P. Koh, Freya R. Weth, Clint Gray, and Swee T. Tan. 2022. "Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment" Biomedicines 10, no. 11: 2988. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10112988