
KasQ an Epimerase Primes the Biosynthesis of Aminoglycoside Antibiotic Kasugamycin and KasF/H Acetyltransferases Inactivate Its Activity

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cloning, Expression, and Purification
2.2. Crystallization and Data Collection
2.3. Structure Determination and Refinement
2.4. Site-Directed Mutagenesis
2.5. Isothermal Titration Calorimetry (ITC)
2.6. Synthesis of 2-Acetamidoglucal (AAG)
2.6.1. General Information
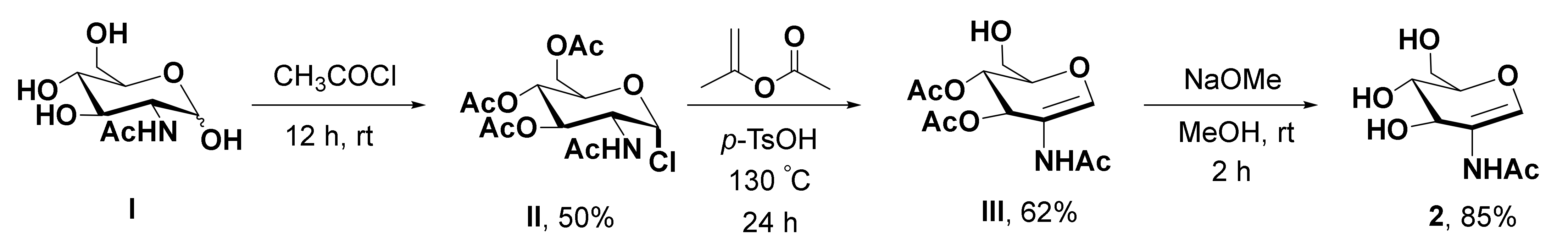
2.6.2. Procedure for the Synthesis of 2-Acetamidoglucal (AAG)
2.7. Construction of pMKBAC08-KAS and pWZC07-PrpsJ-kasT
2.8. In Vivo Isotope Incorporation Assay
- HPLC method A:
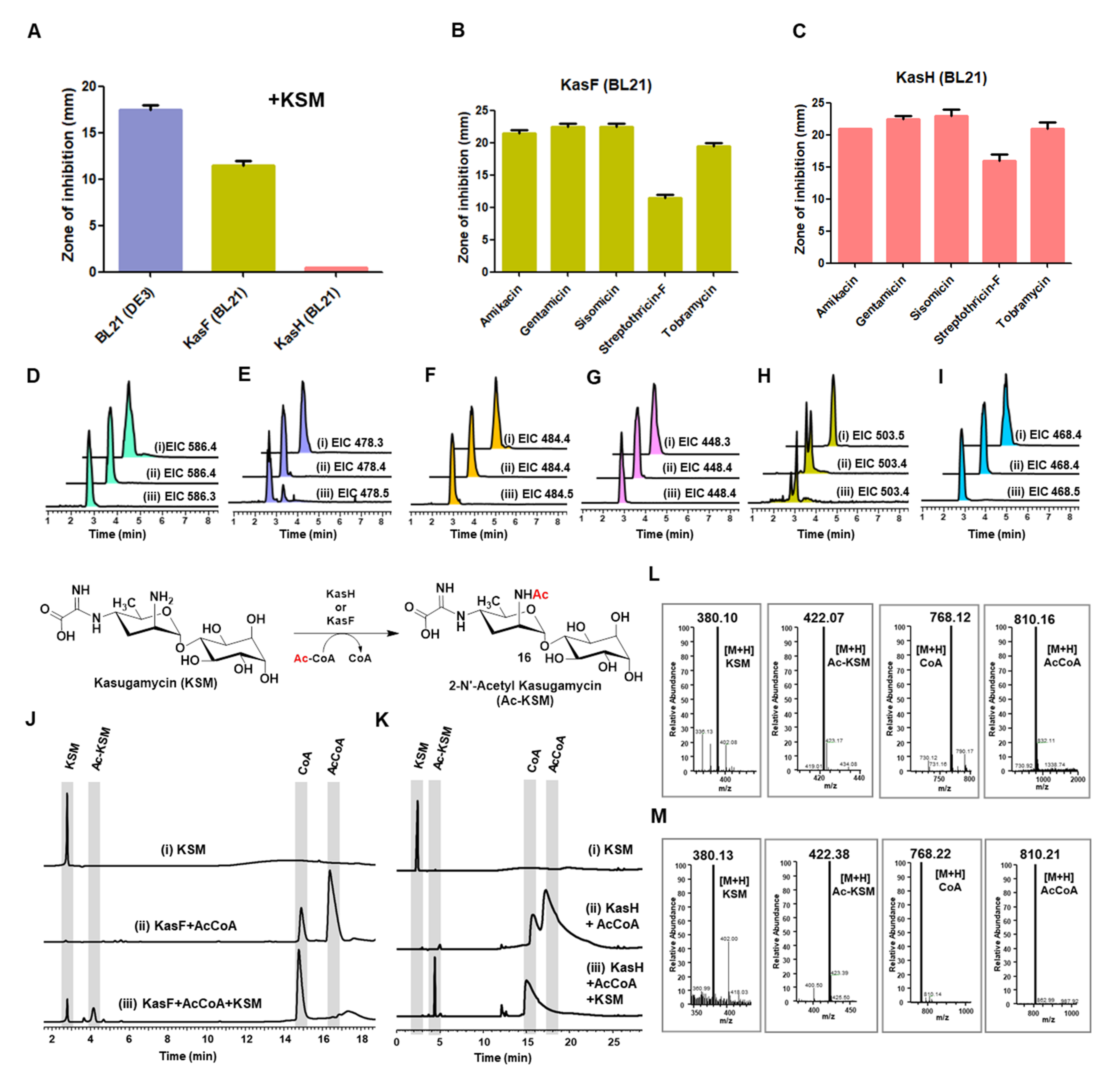
2.9. Disc Diffusion Assay
2.10. HPLC Activity Assay
- HPLC method B:
- HPLC method C:
2.11. Thin Layer Chromatography (TLC) Analysis
2.12. HPLC Kinetic Analysis
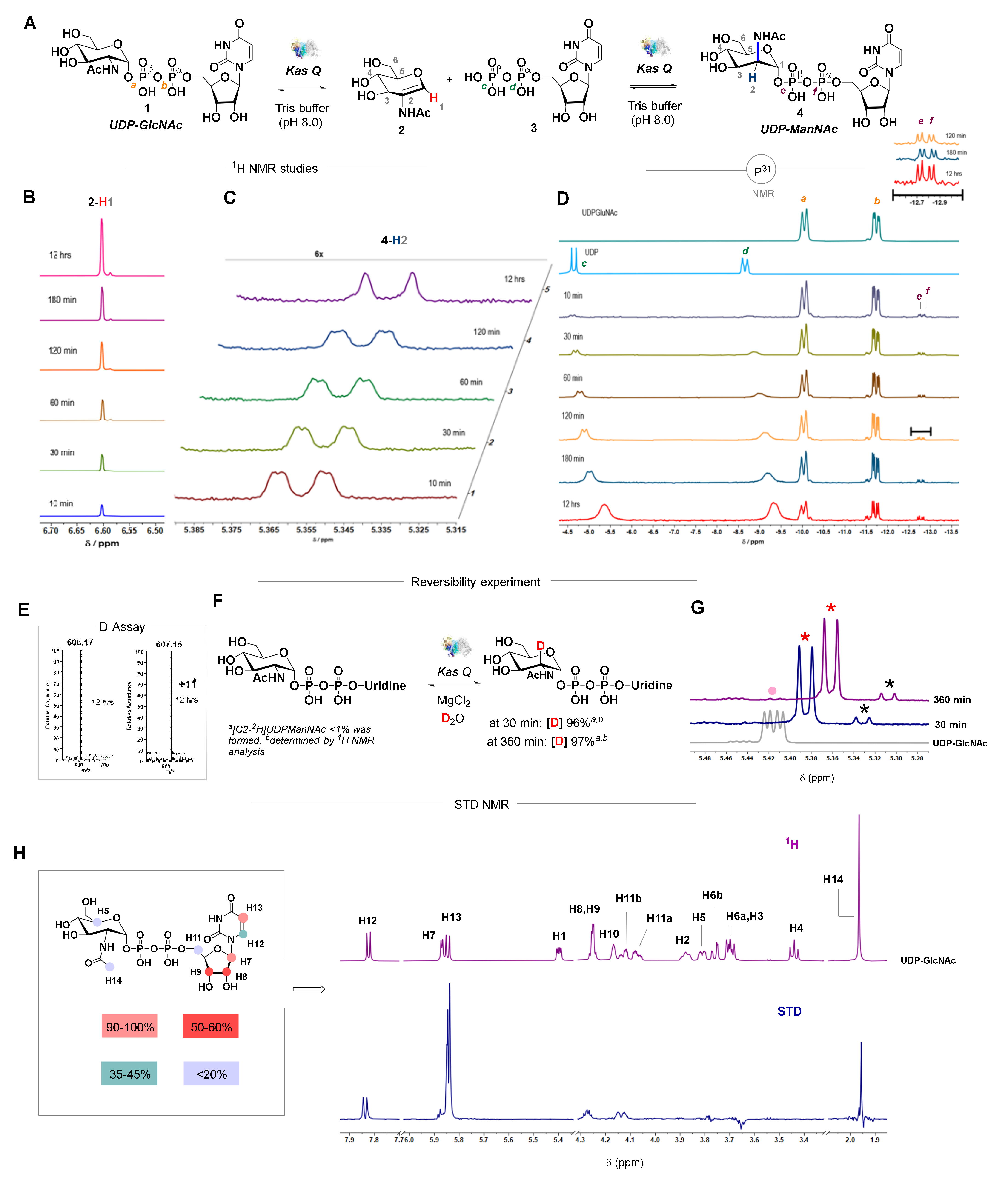
2.13. NMR Activity Assay
2.14. Deuterium Incorporation Assay
- HPLC method D:
2.15. Computational Analysis
3. Results and Discussion
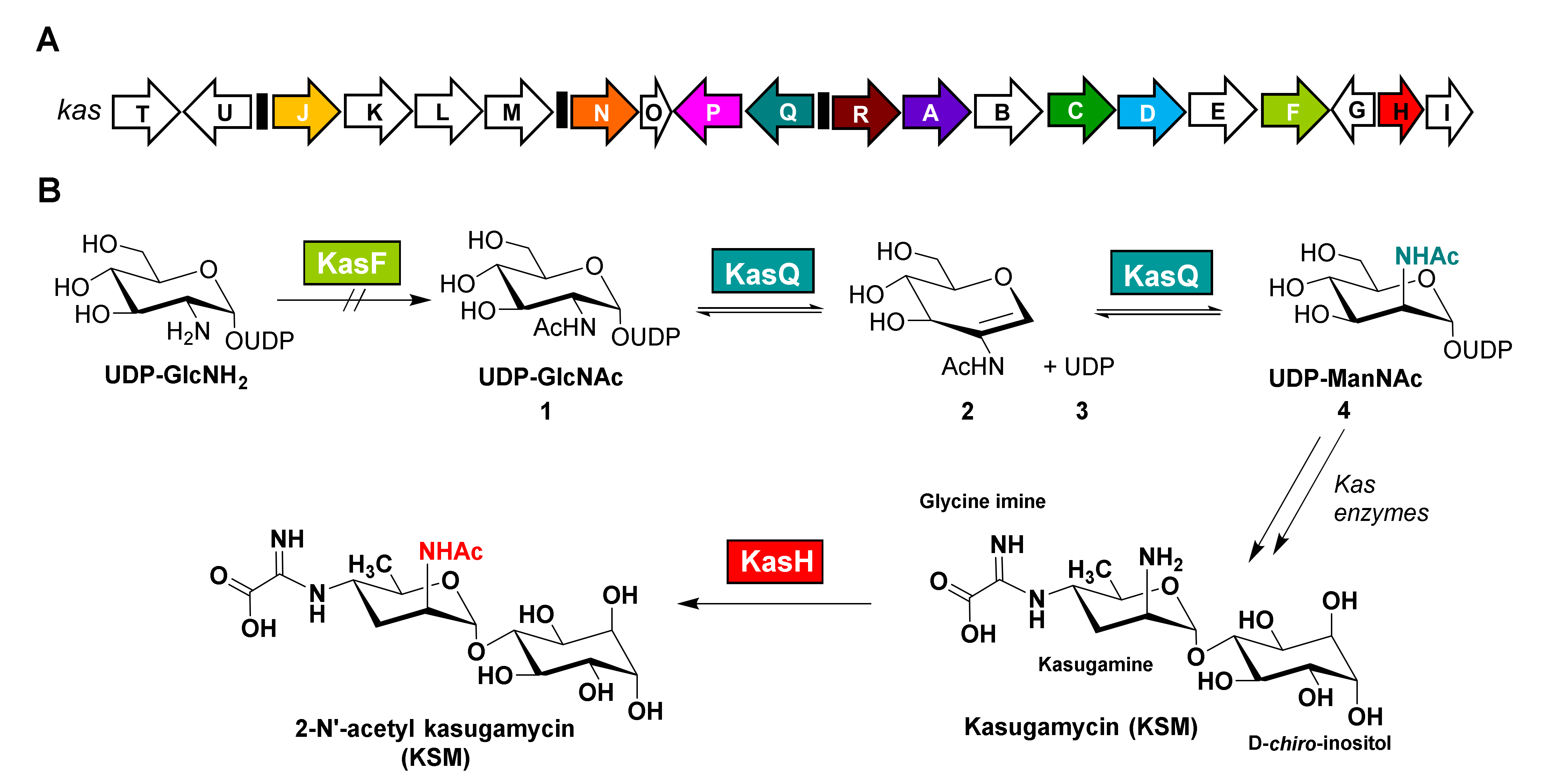
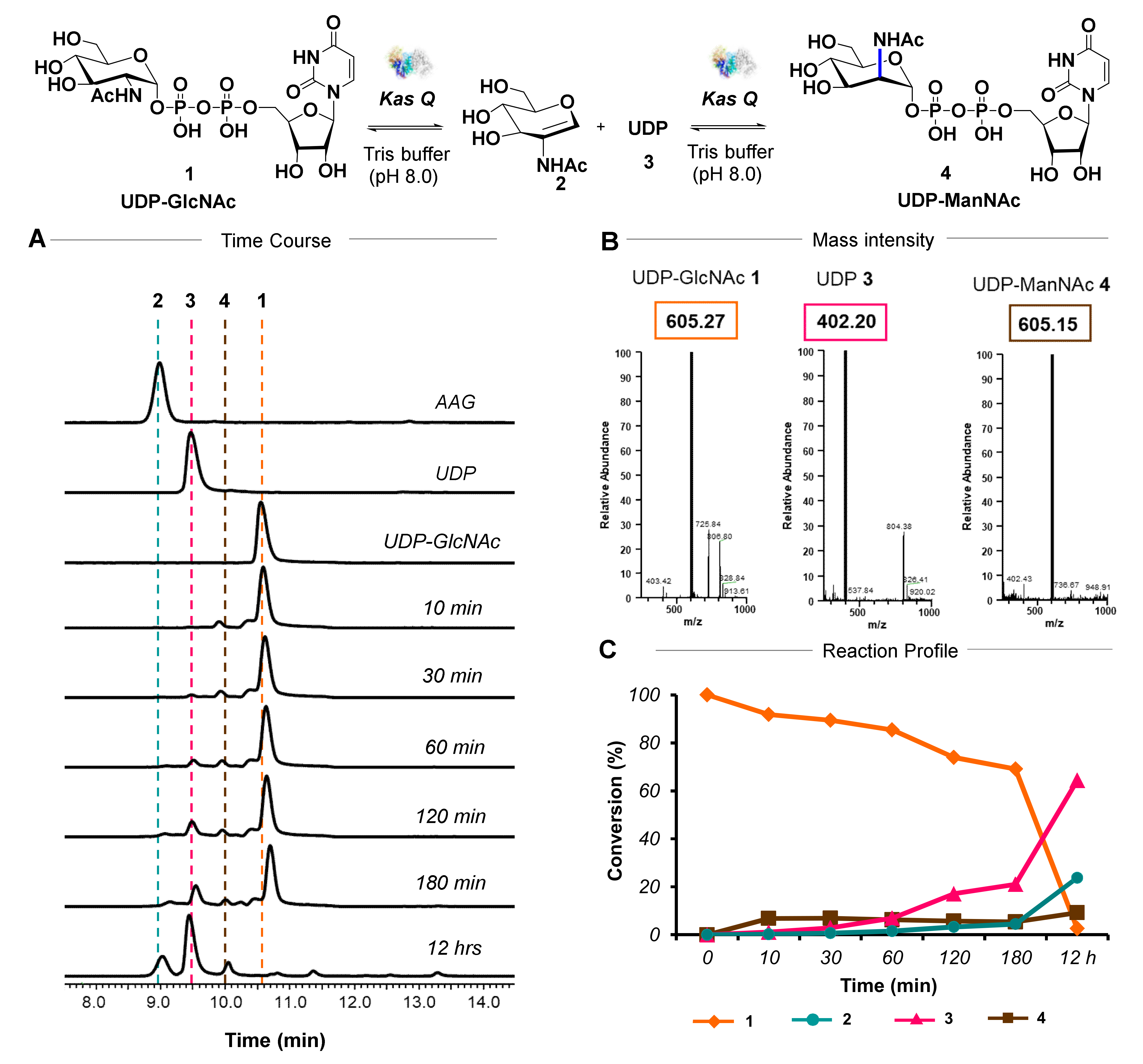
3.1. Biochemical Investigation of an Epimerase
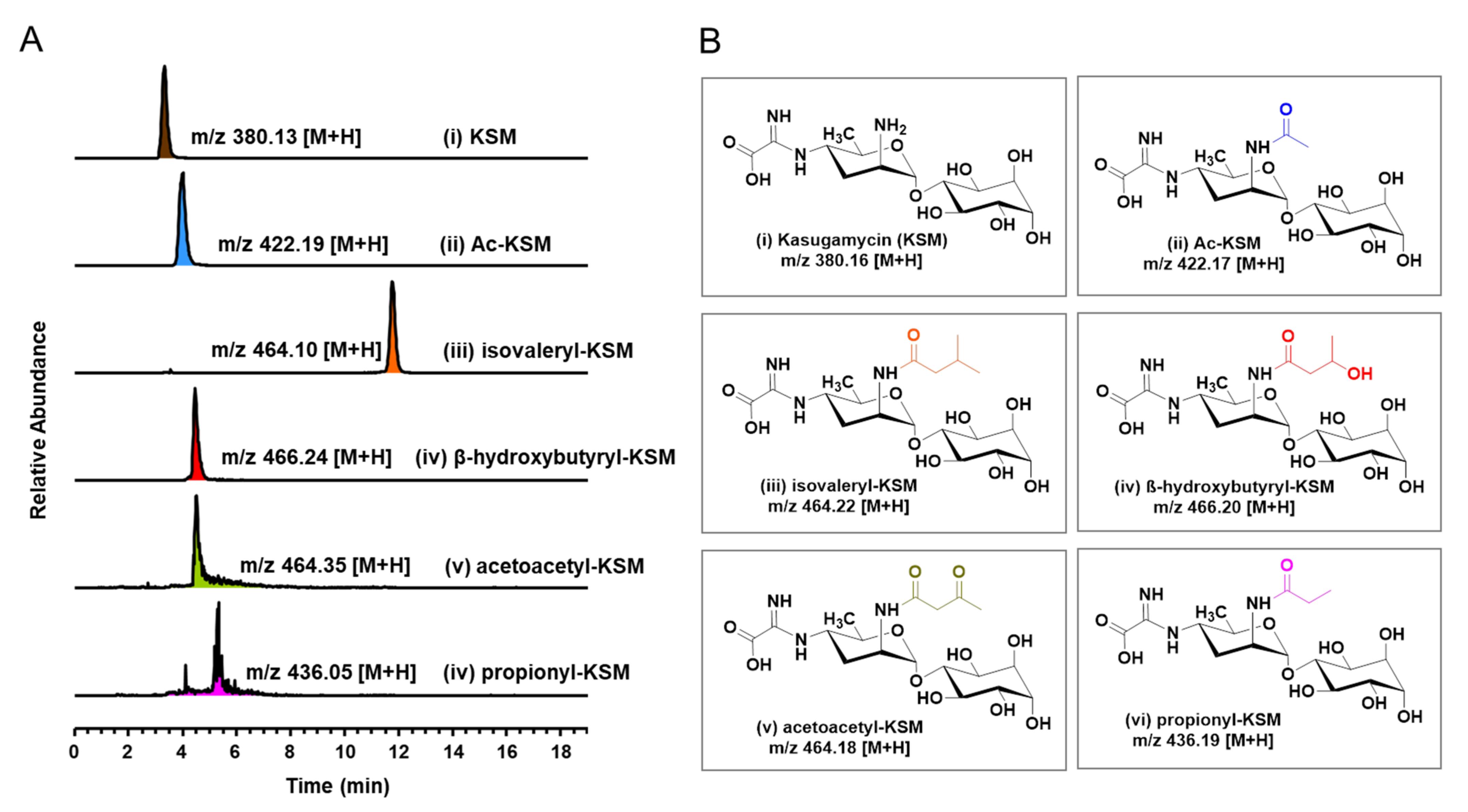
3.2. Biochemical Characterization of Acetyltransferases
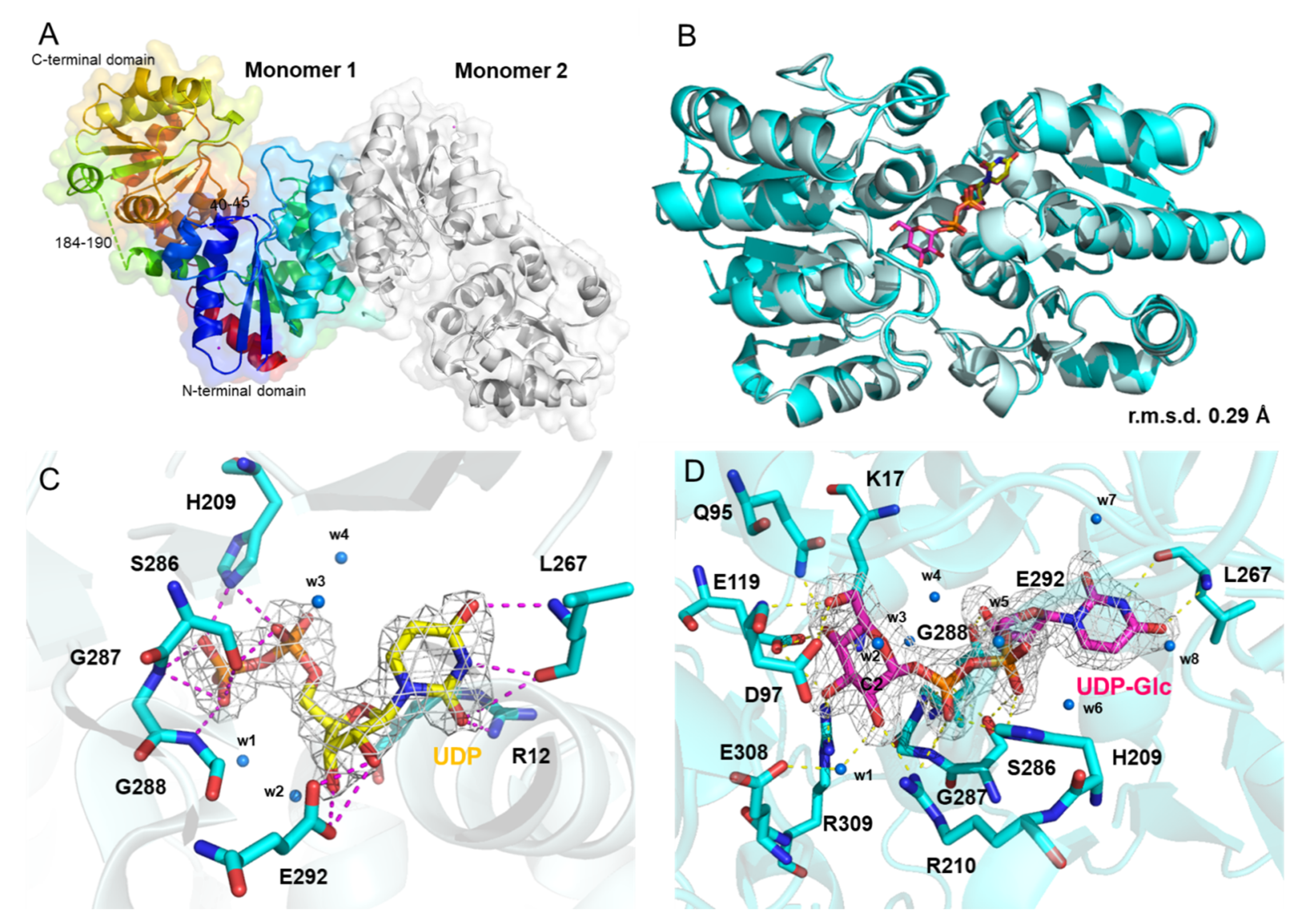
3.3. Crystal Structure of KasQ in Complex with UDP and UDP-Glc
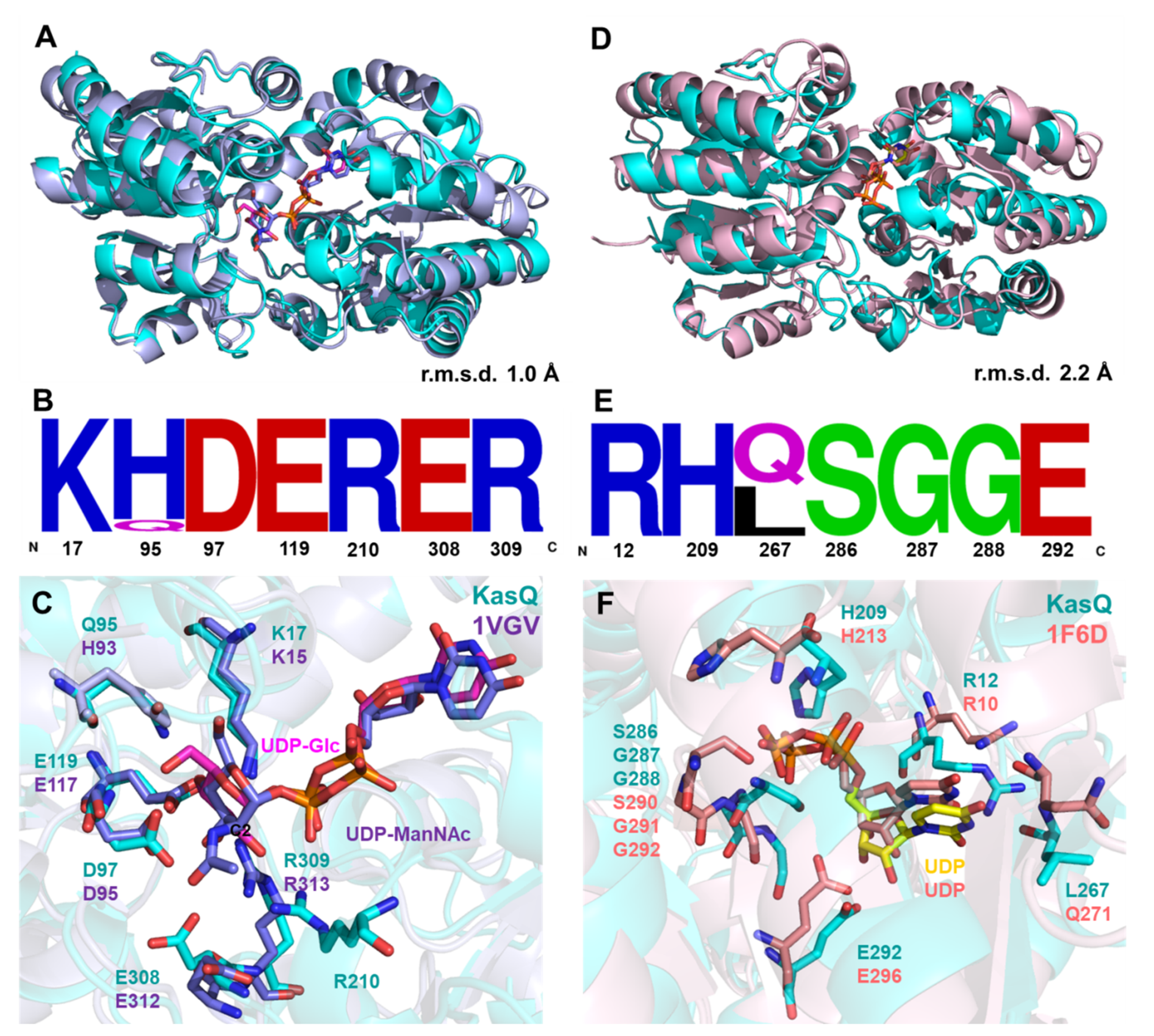
3.4. Structural Comparison
3.5. KasQ Substrate Specificity
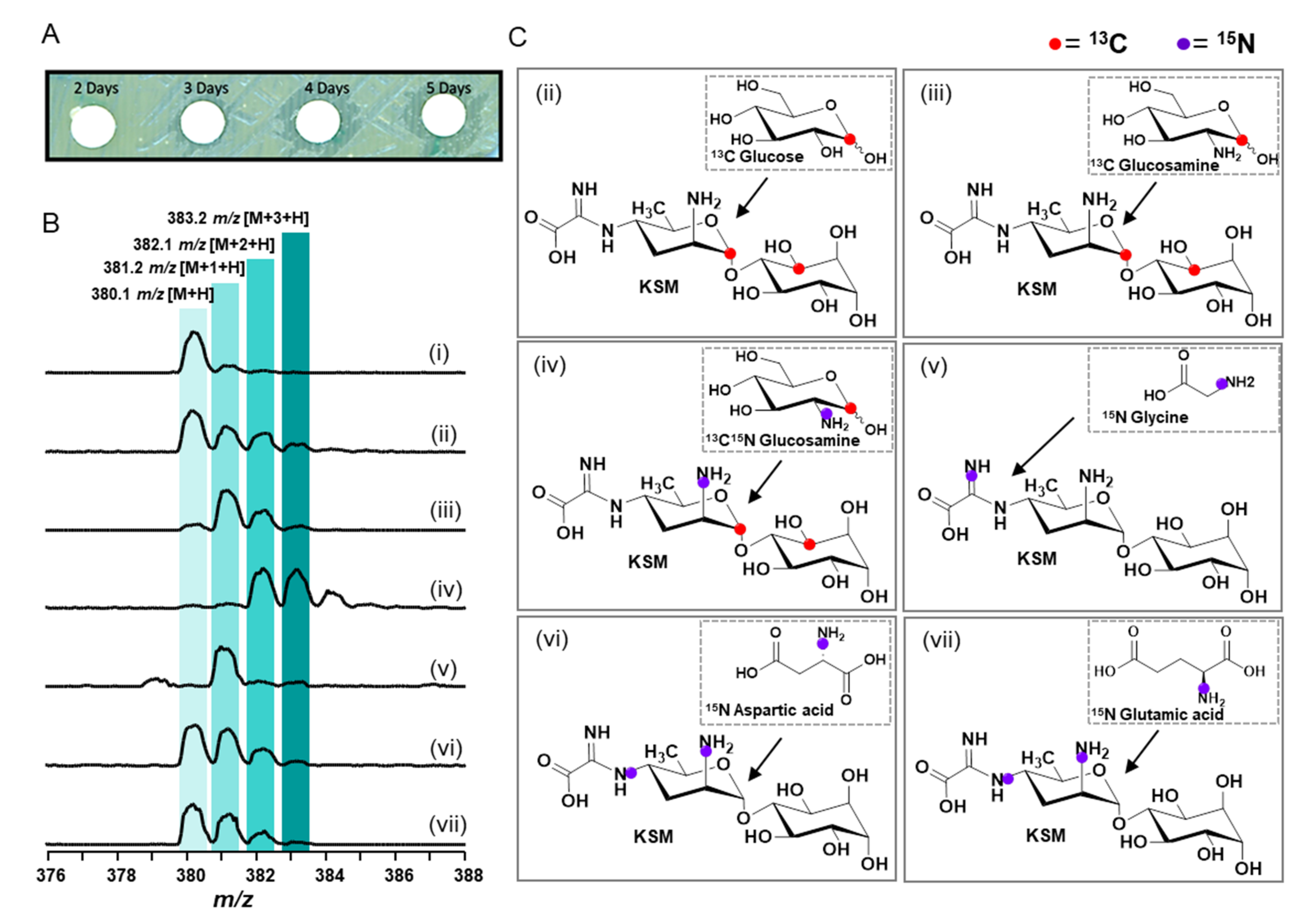
3.6. In Vivo Isotope Incorporation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ikekawa, T.; Umezawa, H.; Iitaka, Y. The structure of kasugamycin hydrobromide by x-ray crystallographic analysis. J. Antibiot. 1966, 19, 49–50. [Google Scholar]
- Umezawa, H.; Hamada, M.; Suhara, Y.; Hashimoto, T.; Ikekawa, T. Kasugamycin, a new antibiotic. Antimicrob. Agents Chemother. 1965, 5, 753–757. [Google Scholar] [PubMed]
- Schuwirth, B.S.; Day, J.M.; Hau, C.W.; Janssen, G.R.; Dahlberg, A.E.; Cate, J.H.; Vila-Sanjurjo, A. Structural analysis of kasugamycin inhibition of translation. Nat. Struct. Mol. Biol. 2006, 13, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, T.; Hara, I.; Matsuoka, M.; Sato, K.; Shimada, S.; Izawa, R.; Hashimoto, T.; Hamada, M.; Okami, Y.; Takeuchi, T.; et al. Studies on the preventive effect of kasugamycin on rice blast. J. Antibiot. 1965, 18, 115–119. [Google Scholar]
- Chaudhuri, S.; Li, L.; Zimmerman, M.; Chen, Y.; Chen, Y.X.; Toosky, M.N.; Gardner, M.; Pan, M.; Li, Y.Y.; Kawaji, Q.; et al. Kasugamycin potentiates rifampicin and limits emergence of resistance in Mycobacterium tuberculosis by specifically decreasing mycobacterial mistranslation. elife 2018, 7, e36782. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Kim, M.V.; Rakib, T.; Wong, P.W.; van Zandt, M.; Barry, N.A.; Kaisho, T.; Goodman, A.L.; Iwasaki, A. Topical application of aminoglycoside antibiotics enhances host resistance to viral infections in a microbiota-independent manner. Nat. Microbiol. 2018, 3, 611–621. [Google Scholar] [CrossRef]
- Tomar, P.P.S.; Krugliak, M.; Arkin, I.T. Blockers of the SARS-CoV-2 3a Channel Identified by Targeted Drug Repurposing. Viruses 2021, 13, 532. [Google Scholar] [CrossRef]
- Kamle, S.; Ma, B.; He, C.H.; Akosman, B.; Zhou, Y.; Lee, C.M.; El-Deiry, W.S.; Huntington, K.; Liang, O.; Machan, J.T.; et al. Chitinase 3-like-1 is a therapeutic target that mediates the effects of aging in COVID-19. JCI Insight 2021, 6, e148749. [Google Scholar] [CrossRef] [PubMed]
- Flatt, P.M.; Mahmud, T. Biosynthesis of aminocyclitol-aminoglycoside antibiotics and related compounds. Nat. Prod. Rep. 2007, 24, 358–392. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, S.; Aoki, D.; Hamada, M.; Hori, M.; Tsuchiya, K.S. DNA sequencing and transcriptional analysis of the kasugamycin biosynthetic gene cluster from Streptomyces kasugaensis M338-M1. J. Antibiot. 2006, 59, 18–28. [Google Scholar] [CrossRef]
- Kasuga, K.; Sasaki, A.; Matsuo, T.; Yamamoto, C.; Minato, Y.; Kuwahara, N.; Fujii, C.; Kobayashi, M.; Agematu, H.; Tamura, T.; et al. Heterologous production of kasugamycin, an aminoglycoside antibiotic from Streptomyces kasugaensis, in Streptomyces lividans and Rhodococcus erythropolis L-88 by constitutive expression of the biosynthetic gene cluster. Appl. Microbiol. Biotechnol. 2017, 101, 4259–4268. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, S.; Tsuji, T.; Higashide, K.; Kinoshita, N.; Hamada, M.; Hori, M. A 7.6 kb DNA region from Streptomyces kasugaensis M338-M1 includes some genes responsible for kasugamycin biosynthesis. J. Antibiot. 1998, 51, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Kudo, F. 2.22—Biosynthesis of Aminoglycoside Antibiotics. In Comprehensive Natural Products III; Liu, H.-W., Begley, T.P., Eds.; Elsevier: Oxford, UK, 2020; pp. 588–612. [Google Scholar] [CrossRef]
- Morgan, P.M.; Sala, R.F.; Tanner, M.E. Eliminations in the Reactions Catalyzed by UDP-N-Acetylglucosamine 2-Epimerase. J. Am. Chem. Soc. 1997, 119, 10269–10277. [Google Scholar] [CrossRef]
- Swartley, J.S.; Liu, L.J.; Miller, Y.K.; Martin, L.E.; Edupuganti, S.; Stephens, D.S. Characterization of the gene cassette required for biosynthesis of the (alpha1-->6)-linked N-acetyl-D-mannosamine-1-phosphate capsule of serogroup A Neisseria meningitidis. J. Bacteriol. 1998, 180, 1533–1539. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Muthana, M.M.; Yu, H.; McArthur, J.B.; Qu, J.; Chen, X. Characterizing non-hydrolyzing Neisseria meningitidis serogroup A UDP-N-acetylglucosamine (UDP-GlcNAc) 2-epimerase using UDP-N-acetylmannosamine (UDP-ManNAc) and derivatives. Carbohydr. Res. 2016, 419, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badger, J.; Sauder, J.M.; Adams, J.M.; Antonysamy, S.; Bain, K.; Bergseid, M.G.; Buchanan, S.G.; Buchanan, M.D.; Batiyenko, Y.; Christopher, J.A.; et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 2005, 60, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 18 November 2021).
- Horbal, L.; Fedorenko, V.; Luzhetskyy, A. Novel and tightly regulated resorcinol and cumate-inducible expression systems for Streptomyces and other actinobacteria. Appl. Microbiol. Biot. 2014, 98, 8641–8655. [Google Scholar] [CrossRef] [PubMed]
- Kieser, T.; Hopwood, D.A. Genetic Manipulation of Streptomyces—Integrating Vectors and Gene Replacement. Methods Enzymol. 1991, 204, 430–458. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salo, W.L. The incorporation of tritium from tritium-enriched water into UDP-N-Acetylglucosamine and UDP-N-Acetyl Mannosamine catalyzed by UDP-N-Acetylglucosamine 2-Epimerase from Escherichia coli. Biochim. Biophys. Acta (BBA) Enzymol. 1976, 452, 625–628. [Google Scholar] [CrossRef]
- Bellucci, M.C.; Volonterio, A. Aminoglycosides: From Antibiotics to Building Blocks for the Synthesis and Development of Gene Delivery Vehicles. Antibiotics 2020, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Litovchick, A.; Lapidot, A.; Eisenstein, M.; Kalinkovich, A.; Borkow, G. Neomycin B−Arginine Conjugate, a Novel HIV-1 Tat Antagonist: Synthesis and Anti-HIV Activities. Biochemistry 2001, 40, 15612–15623. [Google Scholar] [CrossRef]
- Llewellyn, N.M.; Spencer, J.B. Chemoenzymatic acylation of aminoglycoside antibiotics. Chem. Commun. 2008, 32, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Thamban Chandrika, N.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar] [CrossRef]
- Ban, Y.H.; Song, M.C.; Kim, H.J.; Lee, H.; Wi, J.B.; Park, J.W.; Lee, D.G.; Yoon, Y.J. Development of 6′-N-Acylated Isepamicin Analogs with Improved Antibacterial Activity Against Isepamicin-Resistant Pathogens. Biomolecules 2020, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Samuel, J.; Tanner, M.E. Active site mutants of the “non-hydrolyzing” UDP-N-acetylglucosamine 2-epimerase from Escherichia coli. Biochim. Biophys. Acta 2004, 1700, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, G.E.; Macauley, M.S.; Stubbs, K.A.; Dennis, R.J.; Taylor, E.J.; Davies, G.J.; Greig, I.R.; Vocadlo, D.J. Analysis of PUGNAc and NAG-thiazoline as transition state analogues for human O-GlcNAcase: Mechanistic and structural insights into inhibitor selectivity and transition state poise. J. Am. Chem. Soc. 2007, 129, 635–644. [Google Scholar] [CrossRef]
- Hurlburt, N.K.; Guan, J.; Ong, H.; Yu, H.; Chen, X.; Fisher, A.J. Structural characterization of a nonhydrolyzing UDP-GlcNAc 2-epimerase from Neisseria meningitidis serogroup A. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Mosimann, S.C.; Tanner, M.E.; Strynadka, N.C. The structure of UDP-N-acetylglucosamine 2-epimerase reveals homology to phosphoglycosyl transferases. Biochemistry 2000, 39, 14993–15001. [Google Scholar] [CrossRef]
- Huang, C.M.; Lyu, S.Y.; Lin, K.H.; Chen, C.L.; Chen, M.H.; Shih, H.W.; Hsu, N.S.; Lo, I.W.; Wang, Y.L.; Li, Y.S.; et al. Teicoplanin Reprogrammed with the N-Acyl-Glucosamine Pharmacophore at the Penultimate Residue of Aglycone Acquires Broad-Spectrum Antimicrobial Activities Effectively Killing Gram-Positive and -Negative Pathogens. ACS Infect. Dis. 2019, 5, 430–442. [Google Scholar] [CrossRef]
- Fukagawa, Y.; Sawa, T.; Takeuchi, T.; Umezawa, H. Studies on biosynthesis of kasugamycin. I. Biosynthesis of kasugamycin and the kasugamine moiety. J. Antibiot. 1968, 21, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Fukagawa, Y.; Sawa, T.; Takeuchi, T.; Umezawa, H. Biosynthesis of kasugamycin. II. Biosynthesis of the two-carbon-side chain of kasugamycin. J. Antibiot. 1968, 21, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Fukagawa, Y.; Sawa, T.; Takeuchi, T.; Umezawa, H. Studies on biosynthesis of kasugamycin. 3. Biosynthesis of the d-inositol moiety. J. Antibiot. 1968, 21, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, T.; Fukagawa, Y.; Homma, I.; Takeuchi, T.; Umezawa, H. Studies on biosynthesis of kasugamycin. VI. Some relationships between the incorporation of 14C-compounds and the production of kasugamycin. J. Antibiot. 1968, 21, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukagawa, Y.; Sawa, T.; Homma, I.; Takeuchi, T.; Umezawa, H. Studies on biosynthesis of kasugamycin. V. Biosynthesis of the amidine group. J. Antibiot. 1968, 21, 410–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.-W.; Kim, J.; Cho, H.-S.; Shin, J.-S. Active Site Engineering of ω-Transaminase Guided by Docking Orientation Analysis and Virtual Activity Screening. ACS Catal. 2017, 7, 3752–3762. [Google Scholar] [CrossRef]
- Dairi, T.; Yamaguchi, K.; Hasegawa, M. N-formimidoyl fortimicin A synthase, a unique oxidase involved in fortimicin A biosynthesis: Purification, characterization and gene cloning. Mol. Gen. Genet. MGG 1992, 236, 49–59. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KasQ–WT | KasQ–UDP | KasQ–UDP-Glc | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PDB code | 7VYY | 7VZA | 7VZ6 | ||||||
| Wavelength (Å) | 1.0 | 1.0 | 1.0 | ||||||
| Space group | C2 | P1 | P1 | ||||||
| Cell dimensions | |||||||||
| a, b, c (Å) | 112.4 | 106.1 | 94.3 | 81.8 | 104.7 | 106.3 | 82.8 | 104.9 | 106.8 |
| α, β, γ (°) | 90 | 124.9 | 90 | 74.5 | 73.0 | 68.4 | 74.9 | 72.9 | 68.1 |
| Resolution (Å) | 25.45–2.44 (2.55–2.46) | 19.89–2.60 (2.68–2.60) | 21.56–2.10 (2.18–2.10) | ||||||
| Rmerge | 0.031 (0.387) | 0.023 (0.365) | 0.035 (0.296) | ||||||
| I/I | 26.6 (2.4) | 23.8 (1.9) | 19.7 (2.7) | ||||||
| Completeness (%) | 85.9 (86) | 93.9 (94.1) | 95.2 (95.1) | ||||||
| Redundancy | 3.6 (3.9) | 2.4 (2.5) | 2.2 (2.1) | ||||||
| Refinement | |||||||||
| Resolution (Å) | 25.45–2.44 (2.55–2.44) | 19.9–2.58 (2.68–2.58) | 21.5–2.09 (2.14–2.09) | ||||||
| No. reflections | 28,957 | 91,395 | 176,474 | ||||||
| Rwork/Rfree | 0.22/0.27 | 0.21/0.24 | 0.20/0.22 | ||||||
| R.m.s deviations | |||||||||
| Bond lengths (Å) | 0.66 | 0.66 | 0.70 | ||||||
| Bond angles (°) | 0.77 | 0.79 | 0.84 | ||||||
| Mutants | Primers |
|---|---|
| Q95A | Forward 5′-TGGTGTCTCCCGCTACCACGGCGACGTCC-3′ Reverse 5′-GGACGTCGCCGTGGTAGCGGGAGACACCA-3′ |
| Q95E | Forward 5′-TGTCTCCCTCTACCACGGCGACGTCCA-3′ Reverse 5′-TGGACGTCGCCGTGGTAGAGGGAGACA-3′ |
| E308A | Forward 5′-CCTCGGGGCGCGCCGTGCGGTCC-3′ Reverse 5′-GGACCGCACGGCGCGCCCCGAGG-3′ |
| E308Q | Forward 5′-CTCGGGGCGCTGCGTGCGGTCCC-3′ Reverse 5′-GGGACCGCACGCAGCGCCCCGAG-3′ |
| Primers | Sequence |
|---|---|
| P303 P304 P115 P116 P117 P118 | TAACCGTTTAAACTTAATTAAGAAGATCCTTTGATCTTTTCTACGG AAGCCCTGGATCCAATTCCCCAATGTCAAGCACTT ATTATTATTCATATGCGCGTCATCGACGGGATGCACCGGCT ACCTGTGTTCAACCAAGGAATTTCCTGGGTTGAACACCGC AAGATCAGATCCCCGCGGTGACGTAGTACGAGAT ACAGCGTCATGACGTGAAGTTCAGCGACCCGT |
| P119 P120 P121 P122 P123 P124 | ATTATTATTCAATTGCACTACAAGTTCCGCTGGGTGTACCTGAATTT ATTATTATTTTAATTAAGCTTATTCGCAGCAGTCCTCGATGACGAAGACA CAGGAAATTCGAGGCGGAATTCGCCGAATCCTT TGCCTTGTCGTCGTAGGTGTGGAGGCTGTT TTCGACATCGGCATGATCACCCACGGTCTGCTCT ATTATTATTAAGCTTAACAACGTGACGGGTTCAGCCAGTTACCGCTTCATGCTT |
| S.No | KasQ with | n | Kd (µM) | ∆G (kcal mol−1) | ∆H (kcal mol−1) | ∆S (cal mol−1 K−1) |
|---|---|---|---|---|---|---|
| 1 | AMP | NBD | NBD | NBD | NBD | NBD |
| 2 | ADP | NBD | NBD | NBD | NBD | NBD |
| 3 | ATP | NBD | NBD | NBD | NBD | NBD |
| 4 | GMP | NBD | NBD | NBD | NBD | NBD |
| 5 | GDP | NBD | NBD | NBD | NBD | NBD |
| 6 | GTP | NBD | NBD | NBD | NBD | NBD |
| 7 | TTP | 1.1 ± 0.1 | 174.5 ± 20.2 | −5.128 ± 1.221 | −7.612 ± 1.221 | −8.33 |
| 8 | UMP | 1.0 ± 0.0 | 123.9 ± 70.9 | −13.19 ± 1.245 | −9.254 ± 1.245 | 13.2 |
| 9 | UDP 3 | 1.0 ± 0.0 | 32.2 ± 6.8 | −6.128 ± 0.5523 | −4.130 ± 0.5523 | 6.70 |
| 10 | UTP | 1.0 ± 0.0 | 71.4 ± 5.7 | −5.645 ± 1.340 | −9.104 ± 1.340 | −11.6 |
| 11 | UDP-GlcNAc 1 | NBD | NBD | NBD | NBD | NBD |
| 12 | UDP-GlcNAc/Mg2+ | NBD | NBD | NBD | NBD | NBD |
| 13 | UDP-GalNAc | 1.8 ± 0.4 | 334.4 ± 133. 7 | −4.742 ± 1.700 | −3.418 ± 1.700 | 4.44 |
| 14 | UDP-Glc | 0.5 ± 0.0 | 9.5 ± 0.3 | −6.834 ± 0.0375 | −0.3944 ± 0.0375 | 21.6 |
| 15 | TDP-Glc | 0.3 ± 0.0 | 25.1 ± 0.2 | −6.273 ± 0.2773 | −4.568 ± 0.2773 | 5.72 |
| 16 | GDP-Glc | NBD | NBD | NBD | NBD | NBD |
| 17 | UDP-Gal | 1.0 ± 0.2 | 273.3 ± 54.6 | −4.862 ± 1.926 | −6.636 ± 1.926 | −5.95 |
| 18 | UDP-GlcA | 1.5 ± 0.2 | 76.3 ± 15.2 | −5.61 ± 0.5751 | −1.376 ± 0.5751 | 14.2 |
| 19 | GDP-Man | NBD | NBD | NBD | NBD | NBD |
| 20 | UDP-GlcNH2 | NBD | NBD | NBD | NBD | NBD |
| 21 | GlcNAc | NBD | NBD | NBD | NBD | NBD |
| 22 | GalNAc | NBD | NBD | NBD | NBD | NBD |
| 23 | GlcNH2 | NBD | NBD | NBD | NBD | NBD |
| 24 | GlcA | NBD | NBD | NBD | NBD | NBD |
| 25 | GalA | NBD | NBD | NBD | NBD | NBD |
| 26 | D-Glucose | NBD | NBD | NBD | NBD | NBD |
| 27 | D-Mannose | NBD | NBD | NBD | NBD | NBD |
| 28 | D-Galactose | NBD | NBD | NBD | NBD | NBD |
| 29 | AAG 2 | NBD | NBD | NBD | NBD | NBD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattinam, R.; Basha, R.S.; Wang, Y.-L.; Wang, Z.-C.; Hsu, N.-S.; Lin, K.-H.; Zadeh, S.M.; Adhikari, K.; Lin, J.-P.; Li, T.-L. KasQ an Epimerase Primes the Biosynthesis of Aminoglycoside Antibiotic Kasugamycin and KasF/H Acetyltransferases Inactivate Its Activity. Biomedicines 2022, 10, 212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020212
Rattinam R, Basha RS, Wang Y-L, Wang Z-C, Hsu N-S, Lin K-H, Zadeh SM, Adhikari K, Lin J-P, Li T-L. KasQ an Epimerase Primes the Biosynthesis of Aminoglycoside Antibiotic Kasugamycin and KasF/H Acetyltransferases Inactivate Its Activity. Biomedicines. 2022; 10(2):212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020212
Chicago/Turabian StyleRattinam, Rajesh, R. Sidick Basha, Yung-Lin Wang, Zhe-Chong Wang, Ning-Shian Hsu, Kuan-Hung Lin, Saeid Malek Zadeh, Kamal Adhikari, Jin-Ping Lin, and Tsung-Lin Li. 2022. "KasQ an Epimerase Primes the Biosynthesis of Aminoglycoside Antibiotic Kasugamycin and KasF/H Acetyltransferases Inactivate Its Activity" Biomedicines 10, no. 2: 212. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020212