
Cooperation between Angiogenesis, Vasculogenesis, Chemotaxis, and Coagulation in Breast Cancer Metastases Development: Pathophysiological Point of View



Abstract
:Simple Summary
Abstract
1. Introduction
1.1. General Information Related to Breast Cancer
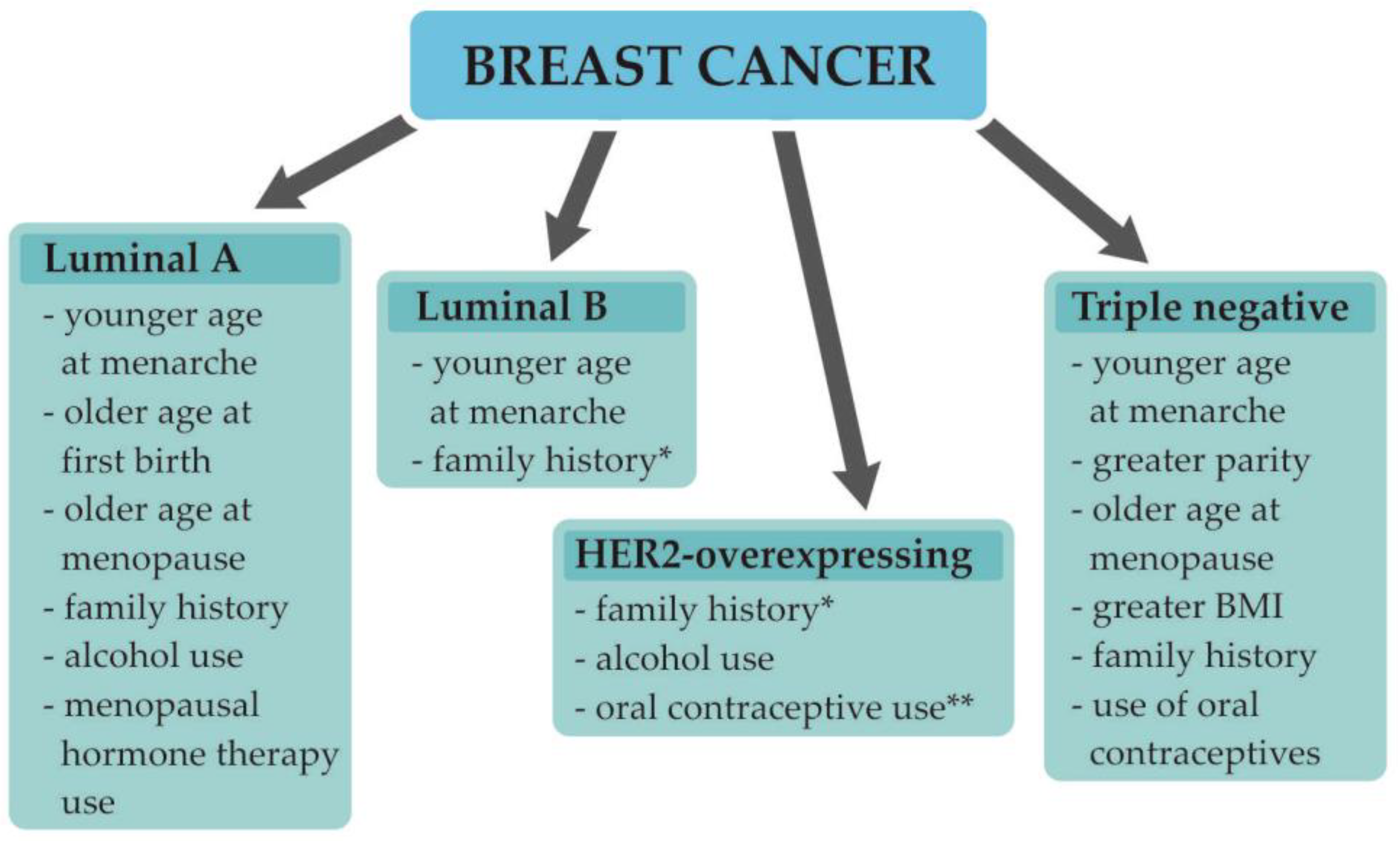
1.2. Understanding Breast Cancer Heterogeneity
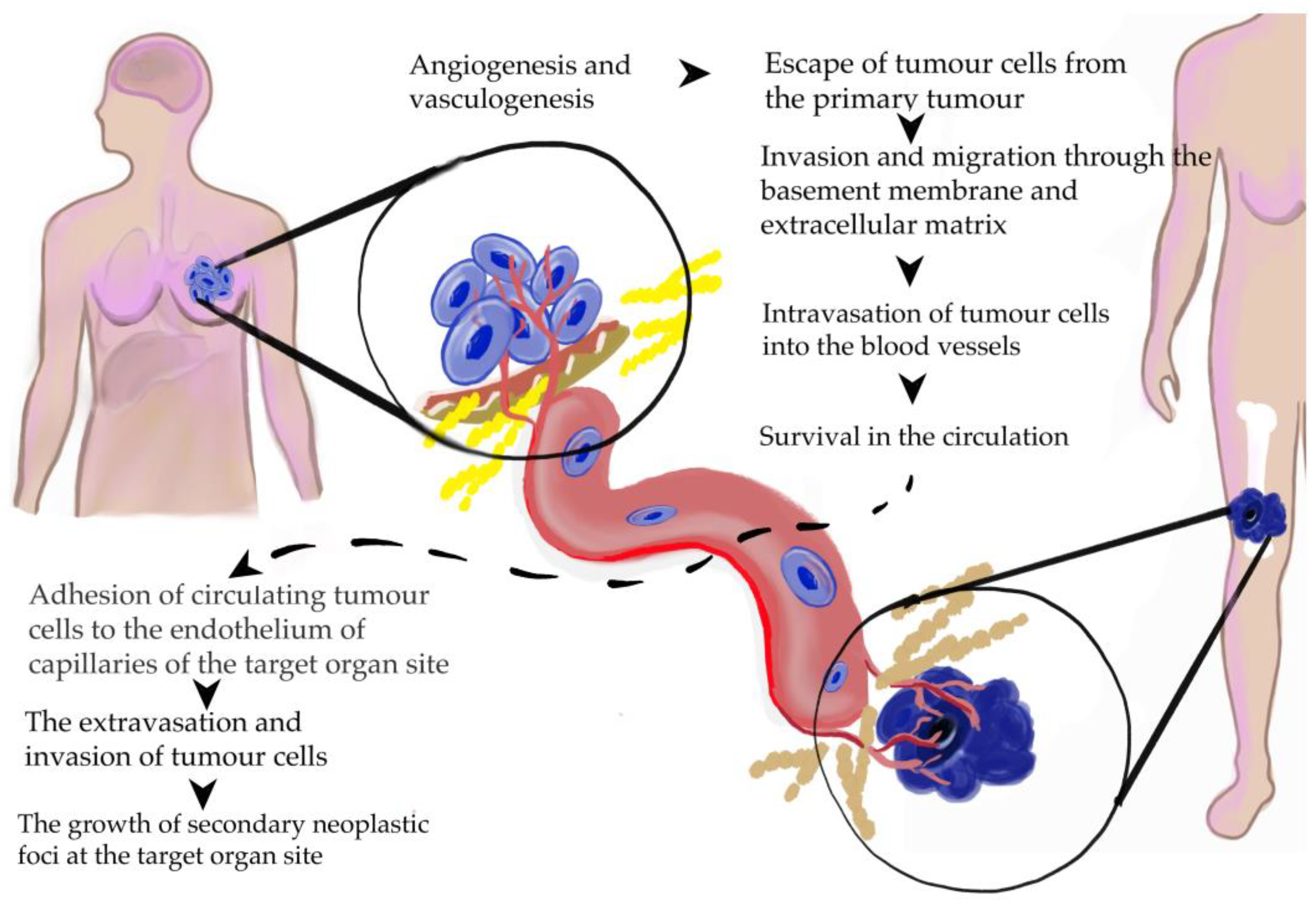
1.3. Breast Cancer Development, Progression, and Metastasis Formation
1.4. The Aim of This Review
2. Angiogenesis
2.1. The Role of Angiogenesis in Tumor Invasion
2.2. The Role of Angiogenesis in BC
2.3. Interactions of VEGF with Tumor and Tumor Microenviroment
2.4. Contribution of VEGF to the Metastatic Spread of BC
2.5. Structure of Tumoral Vessels
3. Vasculogenesis
3.1. Alternative Method of Tumor Vessel Formation
3.2. Role of EPCs in Breast Cancer
3.3. Role of Hypoxia and Inflammation in EPCs Mobilization and Homing
3.4. Interactions of EPCs with Tumor and Tumor Microenviroment
4. Chemotaxis
4.1. The Role of Chemotaxis in Tumor Invasion
4.2. The Role of Chemotaxis in BC
4.3. Interactions of CXCL12 with Tumor and Tumor Microenviroment
4.4. Contribution of CXCL12 to the Metastatic Spread of BC
5. Coagulation
5.1. Tissue Factor (TF)—A Potent Activator of the Coagulation Cascade
5.2. The Role of Coagulation in BC
5.3. Interactions of TF with Tumor and Tumor Microenviroment
5.4. Link between Coagulation and Angiogenesis
5.5. Contribution of TF to the Metastatic Spread of BC
6. Metastatic Disease
6.1. Advanced Breast Cancer—The Formation of Metastases in Distant Organs
6.2. Concepts of Tumor Spread
6.3. Systemic Dissemination of Cancer Cells
6.4. Homing of Circulating Tumor Cells in Target Organs
6.5. Bones
6.6. Lungs
6.7. Brain
6.8. Liver
7. Future Perspective
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 9 November 2021).
- Qiao, A.; Gu, F.; Guo, X.; Zhang, X.; Fu, L. Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front. Med. 2016, 10, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Barnard, M.E.; Boeke, C.E.; Tamimi, R.M. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim. Biophys. Acta. 2015, 1856, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, R.; Hayes, M.M.; Gelmon, K.A. Breast carcinoma—Rare types: Review of the literature. Ann. Oncol. 2009, 20, 1763–1770. [Google Scholar] [CrossRef]
- Rakha, E.A.; Green, A.R. Molecular classification of breast cancer: What the pathologist needs to know. Pathology 2017, 49, 111–119. [Google Scholar] [CrossRef]
- Sachdev, J.C.; Sandoval, A.C.; Jahanzeb, M. Update on Precision Medicine in Breast Cancer. Cancer Treat. Res. 2019, 178, 45–80. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Tosello, G.; Torloni, M.R.; Mota, B.S.; Neeman, T.; Riera, R. Breast surgery for metastatic breast cancer. Cochrane Database Syst. Rev. 2018, 3, CD011276. [Google Scholar] [CrossRef]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Januškevičienė, I.; Petrikaitė, V. Heterogeneity of breast cancer: The importance of interaction between different tumor cell populations. Life Sci. 2019, 239, 117009. [Google Scholar] [CrossRef] [PubMed]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Brown, N.J.; Holen, I. The breast tumor microenvironment: Role in cancer development, progression and response to therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.H. Palliation and breast cancer. J. Surg Oncol. 2017, 115, 538–543. [Google Scholar] [CrossRef]
- Giussani, M.; Merlino, G.; Cappelletti, V.; Tagliabue, E.; Daidone, M.G. Tumor-extracellular matrix interactions: Identification of tools associated with breast cancer progression. Semin. Cancer Biol. 2015, 35, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Xie, H.Y.; Shao, Z.M.; Li, D.Q. Tumor microenvironment: Driving forces and potential therapeutic targets for breast cancer metastasis. Chin. J Cancer 2017, 36, 36. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.J.; Gray, J.W. Chemokine signaling in cancer-stroma communications. J. Cell Commun. Signal 2021, 15, 361–381. [Google Scholar] [CrossRef]
- Zarychta, E.; Ruszkowska-Ciastek, B.; Bielawski, K.; Rhone, P. Stromal Cell-Derived Factor 1α (SDF-1α) in Invasive Breast Cancer: Associations with Vasculo-Angiogenic Factors and Prognostic Significance. Cancers 2021, 13, 1952. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [Green Version]
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J Clin. Invest. 2020, 130, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Koutras, A.; Kotoula, V.; Fountzilas, G. Prognostic and predictive role of vascular endothelial growth factor polymorphisms in breast cancer. Pharmacogenomics 2015, 16, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madu, C.O.; Wang, S.; Madu, C.O.; Lu, Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J. Cancer. 2020, 11, 4474–4494. [Google Scholar] [CrossRef] [PubMed]
- Aalders, K.C.; Tryfonidis, K.; Senkus, E.; Cardoso, F. Anti-angiogenic treatment in breast cancer: Facts, successes, failures and future perspectives. Cancer Treat Rev. 2017, 53, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Bando, H. Vascular endothelial growth factor and bevacitumab in breast cancer. Breast Cancer 2007, 14, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, Y.S.; Yao, Y.D.; Chen, J.Q.; Chen, J.N.; Huang, S.Y.; Zeng, Y.J.; Yao, H.R.; Zeng, S.H.; Fu, Y.S.; et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [Green Version]
- Farzaneh Behelgardi, M.; Zahri, S.; Gholami Shahvir, Z.; Mashayekhi, F.; Mirzanejad, L.; Asghari, S.M. Targeting signaling pathways of VEGFR1 and VEGFR2 as a potential target in the treatment of breast cancer. Mol. Biol. Rep. 2020, 47, 2061–2071. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Devel. Ther. 2015, 9, 4953–4964. [Google Scholar] [CrossRef] [Green Version]
- Sasi, S.P.; Yan, X.; Enderling, H.; Park, D.; Gilbert, H.Y.; Curry, C.; Coleman, C.; Hlatky, L.; Qin, G.; Kishore, R.; et al. Breaking the ‘harmony’ of TNF-α signaling for cancer treatment. Oncogene 2012, 31, 4117–4127. [Google Scholar] [CrossRef] [Green Version]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.; Elizalde, P.V.; Schillaci, R. Harnessing Tumor Necrosis Factor Alpha to Achieve Effective Cancer Immunotherapy. Cancers 2021, 13, 564. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Chen, I.C.; Lin, P.Y.; Lung, J.H.; Li, Y.C.; Lin, Y.C.; Yang, C.T.; Tsai, Y.H. Epidermal growth factor receptor mutation enhances expression of vascular endothelial growth factor in lung cancer. Oncol. Lett. 2016, 12, 4598–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. 2017, 28, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Vazquez Rodriguez, G.; Abrahamsson, A.; Jensen, L.D.E.; Dabrosin, C. Adipocytes Promote Early Steps of Breast Cancer Cell Dissemination via Interleukin-8. Front. Immunol. 2018, 9, 1767. [Google Scholar] [CrossRef] [Green Version]
- Zarychta, E.; Rhone, P.; Bielawski, K.; Michalska, M.; Rość, D.; Ruszkowska-Ciastek, B. Anti-angiogenic efficacy in invasive breast carcinoma patients depends on clinicopathological determinants. Adv. Med. Sci. 2019, 64, 216–223. [Google Scholar] [CrossRef]
- Chung, A.; Cui, X.; Audeh, W.; Giuliano, A. Current status of anti-human epidermal growth factor receptor 2 therapies: Predicting and overcoming herceptin resistance. Clin. Breast Cancer 2013, 13, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Meng, Y.G.; Untch, M.; Wang, H.J.; Bauerfeind, I.; Epstein, M.; Stieber, P.; Vernes., J.-M.; Gutierrez, J.; Hong, K.; et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin. Cancer Res. 2004, 10, 1706–1716. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Yao, H.; Hu, H. Disrupting Tumor Angiogenesis and “the Hunger Games” for Breast Cancer. Adv. Exp. Med. Biol. 2017, 1026, 171–195. [Google Scholar] [CrossRef]
- Salimifard, S.; Masjedi, A.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Irandoust, M.; Azizi, G.; Mohammadi, H.; Keramati, M.R.; Jadidi-Niaragh, F. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathol. Res. Pract. 2020, 216, 152915. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Liu, C.; Hou, L.; Meng, M.; Zhang, X.; Luo, M.; Shao, S.; Zuo, X.; Zhao, X. Vascular endothelial growth factor receptor-1 activation promotes migration and invasion of breast cancer cells through epithelial-mesenchymal transition. PLoS ONE 2013, 8, e65217. [Google Scholar] [CrossRef] [PubMed]
- Sadremomtaz, A.; Kobarfard, F.; Mansouri, K.; Mirzanejad, L.; Asghari, S.M. Suppression of migratory and metastatic pathways via blocking VEGFR1 and VEGFR2. J. Recept. Signal Transduct. Res. 2018, 38, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Perrot-Applanat, M.; Di Benedetto, M. Autocrine functions of VEGF in breast tumor cells: Adhesion, survival, migration and invasion. Cell Adh. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markiewski, M.M.; Daugherity, E.; Reese, B.; Karbowniczek, M. The Role of Complement in Angiogenesis. Antibodies 2020, 9, 67. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N. Abnormalities of tumor endothelial cells and cancer progression. Oral. Sci. Int. 2018, 15, 1–6. [Google Scholar] [CrossRef]
- Debatin, K.M.; Wei, J.; Beltinger, C. Endothelial progenitor cells for cancer gene therapy. Gene Ther. 2008, 15, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Flamini, V.; Jiang, W.G.; Lane, J.; Cui, Y.X. Significance and therapeutic implications of endothelial progenitor cells in angiogenic-mediated tumour metastasis. Crit. Rev. Oncol. Hematol. 2016, 100, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into Endothelial Progenitor Cells: Origin, Classification, Potentials, and Prospects. Stem Cells Int. 2018, 2018, 9847015. [Google Scholar] [CrossRef]
- Botelho, M.C.; Alves, H. Endothelial Progenitor Cells in Breast Cancer. Int. J. Immunother. Cancer Res. 2016, 2, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, H.Q.; Li, J.; Liu, X.L. Endothelial progenitor cells promote tumor growth and progression by enhancing new vessel formation. Oncol. Lett. 2016, 12, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, M.; Dudek, A.; Jahagirdar, B.; Koodie, L.; Marker, P.H.; Verfaillie, C.M. Origin of endothelial progenitors in human postnatal bone marrow. J Clin. Investig. 2002, 109, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.P.; Jin, D.; Chuang, E.; Gold, E.G.; Tousimis, E.A.; Moore, A.L.; Christos, P.J.; de Dalmas, T.; Donovan, D.; Rafii, S.; et al. Circulating endothelial progenitor cells correlate to stage in patients with invasive breast cancer. Breast Cancer Res. Treat. 2008, 107, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Rhone, P.; Bielawski, K.; Ziołkowska, K.; Rość, D.; Ruszkowska-Ciastek, B. Low Pre-Treatment Count of Circulating Endothelial Pro-genitors as a Prognostic Biomarker of the High Risk of Breast Cancer Recurrence. J. Clin. Med. 2019, 8, 1984. [Google Scholar] [CrossRef] [Green Version]
- Dome, B.; Timar, J.; Dobos, J.; Meszaros, L.; Raso, E.; Paku, S.; Kenessey, I.; Ostoros, G.; Magyar, M.; Ladanyi, A.; et al. Identification and clinical significance of circulating endothelial progenitor cells in human non-small cell lung cancer. Cancer Res. 2006, 66, 7341–7347. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.H.; Lin, C.H.; Shau, W.Y.; Chen, T.J.; Yang, S.H.; Huang, S.M.; Hsu, C.; Lu, Y.S.; Cheng, A.L. Dynamics of circulating endothelial cells and endothelial progenitor cells in breast cancer patients receiving cytotoxic chemotherapy. BMC Cancer 2012, 12, 620. [Google Scholar] [CrossRef] [Green Version]
- Shaked, Y.; Henke, E.; Roodhart, J.M.; Mancuso, P.; Langenberg, M.H.; Colleoni, M.; Daenen, L.G.; Man, S.; Xu, P.; Emmenegger, U.; et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: Implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 2008, 14, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Fürstenberger, G.; von Moos, R.; Lucas, R.; Thürlimann, B.; Senn, H.J.; Hamacher, J.; Boneberg, E.M. Circulating endothelial cells and angiogenic serum factors during neoadjuvant chemotherapy of primary breast cancer. Br. J. Cancer 2006, 94, 524–531. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; Mishima, Y.; Sahin, I.; Manier, S.; Glavey, S.; Vacca, A.; Roccaro, A.M.; Ghobrial, I.M. Role of endothelial progenitor cells in cancer progression. Biochim. Biophys. Acta 2014, 1846, 26–39. [Google Scholar] [CrossRef]
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071. [Google Scholar] [CrossRef]
- Kong, L.; Guo, S.; Liu, C.; Zhao, Y.; Feng, C.; Liu, Y.; Wang, T.; Li, C. Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int. J. Oncol. 2016, 48, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, Z.; Deng, M.; Liu, Q.; Zhang, T.; Guo, W.; Li, P.; Qiao, W. Prognostic and clinicopathological value of CXCL12/SDF1 expression in breast cancer: A meta-analysis. Clin. Chim. Acta 2018, 484, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, E.; Przylipiak, A.; Zajkowska, M.; Piskor, B.M.; Sidorkiewicz, I.; Szmitkowski, M.; Lawicki, S. Possible Diagnostic Application of CXCL12 and CXCR4 as Tumor Markers in Breast Cancer Patients. Anticancer Res. 2020, 40, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Qian, L.; Chen, X.; Ding, B. Prognostic significance of CXCL12, CXCR4, and CXCR7 in patients with breast cancer. Int J. Clin. Exp. Pathol. 2015, 8, 13217–13224. [Google Scholar]
- Samarendra, H.; Jones, K.; Petrinic, T.; Silva, M.A.; Reddy, S.; Soonawalla, Z.; Gordon-Weeks, A. A meta-analysis of CXCL12 expression for cancer prognosis. Br. J. Cancer 2017, 117, 124–135. [Google Scholar] [CrossRef]
- Janssens, R.; Struyf, S.; Proost, P. Pathological roles of the homeostatic chemokine CXCL12. Cytokine Growth Factor Rev. 2018, 44, 51–68. [Google Scholar] [CrossRef]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.E.; Walenkamp, A.M.E. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.M.; Elbaz, M.; Cuitiño, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef]
- Coussens, L.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The Role of CXC Chemokine Receptors 1–4 on Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Chang, W.; Fang, B.; Qin, J.; Qu, X.; Cheng, F. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci. Rep. 2016, 6, 39541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mego, M.; Cholujova, D.; Minarik, G.; Sedlackova, T.; Gronesova, P.; Karaba, M.; Benca, J.; Cingelova, S.; Cierna, Z.; Manasova, D.; et al. CXCR4-SDF-1 interaction potentially mediates trafficking of circulating tumor cells in primary breast cancer. BMC Cancer 2016, 16, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.; Esmaeili, A.A.; Gopalakrishna-Pillai, S.; Murad, J.P.; Andersen, E.S.; Reddy, N.K.; Srinivasan, G.; Armstrong, B.; Chu, C.; Kim, Y.; et al. Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12. NPJ Breast Cancer 2017, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Endo, M.; Yamamoto, Y.; Odagiri, H.; Kadomatsu, T.; Nakamura, T.; Tanoue, H.; Ito, H.; Yugami, M.; Miyata, K.; et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci. Rep. 2015, 5, 9170. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Peng, Z.; Lin, J.; Ren, X.; Zhang, G.; Cui, Y. Forkhead box C1 boosts triple-negative breast cancer metastasis through activating the transcription of chemokine receptor-4. Cancer Sci. 2018, 109, 3794–3804. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ordoñez, A.; Seoane, S.; Cabezas, P.; Eiro, N.; Sendon-Lago, J.; Macia, M.; García-Caballero, T.; Gonzalez, L.O.; Sanchez, L.; Vizoso, F.; et al. Breast cancer metastasis to liver and lung is facilitated by Pit-1-CXCL12-CXCR4 ax-is. Oncogene 2018, 37, 1430–1444. [Google Scholar] [CrossRef]
- Unruh, D.; Horbinski, C. Beyond thrombosis: The impact of tissue factor signaling in cancer. J. Hematol. Oncol. 2020, 13, 93. [Google Scholar] [CrossRef]
- Morrissey, J.H. Tissue factor: An enzyme cofactor and a true receptor. Thromb. Haemost. 2001, 86, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.; Bromberg, M. Tissue factor as a novel target for treatment of breast cancer. Oncologist 2013, 18, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rak, J.; Milsom, C.; May, L.; Klement, P.; Yu, J. Tissue factor in cancer and angiogenesis: The molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin. Thromb. Hemost. 2006, 32, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Toi, M.; Koike, M.; Nakamura, S.; Tominaga, T. Tissue factor expression in breast cancer tissues: Its correlation with prognosis and plasma concentration. Br. J. Cancer 2000, 83, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Stämpfli, S.F.; Akhmedov, A.; Hausladen, S.; Varga, Z.; Dedes, K.J.; Hellermann, J.; Lüscher, T.F.; Kristiansen, G.; Tanner, F.C.; Breitenstein, A. Tissue Factor Expression Does Not Predict Mortality in Breast Cancer Patients. Anticancer Res. 2017, 37, 3259–3264. [Google Scholar] [CrossRef] [Green Version]
- Zarychta, E.; Rhone, P.; Bielawski, K.; Rosc, D.; Szot, K.; Zdunska, M.; Ruszkowska-Ciastek, B. Elevated plasma levels of tissue factor as a valuable diagnostic biomarker with relevant efficacy for prediction of breast cancer morbidity. J. Physiol. Pharmacol. 2018, 69, 10. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef]
- Shaker, H.; Bundred, N.J.; Landberg, G.; Pritchard, S.A.; Albadry, H.; Nicholson, S.L.; Harries, L.J.; Heah, J.Y.E.; Castle, J.; Kirwan, C.C. Breast cancer stromal clotting activation (Tissue Factor and thrombin): A pre-invasive phenomena that is prognostic in invasion. Cancer Med. 2020, 9, 1768–1778. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Shoji, M.; Chen, J.; Bierhaus, A.; Danave, I.; Micko, C.; Casper, K.; Dillehay, D.L.; Nawroth, P.P.; Rickles, F.R. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc. Natl. Acad. Sci. USA 1999, 96, 8663–8668. [Google Scholar] [CrossRef] [Green Version]
- Ollivier, V.; Chabbat, J.; Herbert, J.M.; Hakim, J.; de Prost, D. Vascular endothelial growth factor production by fibroblasts in response to factor VIIa binding to tissue factor involves thrombin and factor Xa. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- McInnes, L.M.; Jacobson, N.; Redfern, A.; Dowling, A.; Thompson, E.W.; Saunders, C.M. Clinical implications of circulating tumor cells of breast cancer patients: Role of epithelial-mesenchymal plasticity. Front. Oncol. 2015, 5, 42. [Google Scholar] [CrossRef]
- Krawczyk, N.; Meier-Stiegen, F.; Banys, M.; Neubauer, H.; Ruckhaeberle, E.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in circulating tumor cells of breast cancer patients. Biomed. Res. Int. 2014, 2014, 415721. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Karaba, M.; Minarik, G.; Benca, J.; Sedlácková, T.; Tothova, L.; Vlkova, B.; Cierna, Z.; Janega, P.; Luha, J.; et al. Relationship between circulating tumor cells, blood coagulation, and urokinase-plasminogen-activator system in early breast cancer patients. Breast J. 2015, 21, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Bourcy, M.; Suarez-Carmona, M.; Lambert, J.; Francart, M.E.; Schroeder, H.; Delierneux, C.; Skrypek, N.; Thompson, E.W.; Jérusalem, G.; Berx, G.; et al. Tissue Factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State That Drives Metastasis of Circulating Tumor Cells. Cancer Res. 2016, 76, 4270–4282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esumi, N.; Fan, D.; Fidler, I.J. Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res. 1991, 51, 4549–4556. [Google Scholar] [PubMed]
- Hu, L.; Lee, M.; Campbell, W.; Perez-Soler, R.; Karpatkin, S. Role of endogenous thrombin in tumor implantation, seeding, and spontaneous metastasis. Blood 2004, 104, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Kast, K.; Link, T.; Friedrich, K.; Petzold, A.; Niedostatek, A.; Schoffer, O.; Werner, C.; Klug, S.J.; Werner, A.; Gatzweiler, A.; et al. Impact of breast cancer subtypes and patterns of metastasis on outcome. Breast Cancer Res. Treat. 2015, 150, 621–629. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [Green Version]
- Kozłowski, J.; Kozłowska, A.; Kocki, J. Breast cancer metastasis—Insight into selected molecular mechanisms of the phenomenon. Postepy Hig. Med. Dosw. 2015, 69, 447–451. [Google Scholar] [CrossRef]
- Méndez-García, L.A.; Nava-Castro, K.E.; Ochoa-Mercado, T.L.; Palacios-Arreola, M.I.; Ruiz-Manzano, R.A.; Segovia-Mendoza, M.; Solleiro-Villavicencio, H.; Cázarez-Martínez, C.; Morales-Montor, J. Breast Cancer Metastasis: Are Cytokines Important Players During Its Development and Progression? J. Interferon Cytokine Res. 2019, 39, 39–55. [Google Scholar] [CrossRef]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Siegel, P.M. The influence of the pre-metastatic niche on breast cancer metastasis. Cancer Lett. 2016, 380, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T. Bone metastasis: Interaction between cancer cells and bone microenvironment. J. Oral. Biosci. 2019, 61, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr. Relat Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brook, N.; Brook, E.; Dharmarajan, A.; Dass, C.R.; Chan, A. Breast cancer bone metastases: Pathogenesis and therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 96, 63–78. [Google Scholar] [CrossRef]
- Salvador, F.; Llorente, A.; Gomis, R.R. From latency to overt bone metastasis in breast cancer: Potential for treatment and prevention. J. Pathol. 2019, 249, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ren, S.; Wang, Z.; Wang, Z.; Zhu, N.; Cai, D.; Ye, Z.; Ruan, J. Chemokines in bone-metastatic breast cancer: Therapeutic opportunities. Int. Immunopharmacol. 2020, 87, 106815. [Google Scholar] [CrossRef]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Urooj, T.; Wasim, B.; Mushtaq, S.; Shah, S.N.N.; Shah, M. Cancer Cell-derived Secretory Factors in Breast Cancer-associated Lung Metastasis: Their Mechanism and Future Prospects. Curr. Cancer Drug Targets 2020, 20, 168–186. [Google Scholar] [CrossRef]
- Yousefi, M.; Nosrati, R.; Salmaninejad, A.; Dehghani, S.; Shahryari, A.; Saberi, A. Organ-specific metastasis of breast cancer: Molecular and cellular mechanisms underlying lung metastasis. Cell Oncol. 2018, 41, 123–140. [Google Scholar] [CrossRef]
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Hosonaga, M.; Saya, H.; Arima, Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer Metastasis Rev. 2020, 39, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Custódio-Santos, T.; Videira, M.; Brito, M.A. Brain metastasization of breast cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Bale, R.; Putzer, D.; Schullian, P. Local Treatment of Breast Cancer Liver Metastasis. Cancers 2019, 11, 1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Feng, Y.; Lin, S.; Chen, J.; Lin, H.; Liang, X.; Zheng, H.; Cai, X. Mechanisms involved in breast cancer liver metastasis. J. Transl. Med. 2015, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Tayyeb, B.; Parvin, M. Pathogenesis of Breast Cancer Metastasis to Brain: A Comprehensive Approach to the Signaling Network. Mol. Neurobiol. 2016, 53, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA A Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Luminal A-Like | Luminal B-Like | HER2- Overexpressing | Triple-Negative | |
|---|---|---|---|---|
| ER | + | + | - | - |
| PR | ≥20% | <20% 1 | - | - |
| HER2 | - | + 1 | + | - |
| Ki-67 | <20% | ≥20% 1 | all | all |
| Grade | Low | Low/High | High | High |
| Frequency | 30–40% | 20–30% | 12–20% | 15–20% |
| Local–Regional Recurrence | 0.8–8% | 1.5–8.7% | 1.7–9.4% | 3–17% |
| Prognosis | Favorable | Intermediate | Unfavorable | Unfavorable/Poor |
| Cytokines and Growth Factors Involved in Angiogenesis | Role/Action |
|---|---|
| EGFR |
|
| bFGF |
|
| IL-8 |
|
| VEGF-A |
|
| TNF- α |
|
| MMPs |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarychta, E.; Ruszkowska-Ciastek, B. Cooperation between Angiogenesis, Vasculogenesis, Chemotaxis, and Coagulation in Breast Cancer Metastases Development: Pathophysiological Point of View. Biomedicines 2022, 10, 300. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020300
Zarychta E, Ruszkowska-Ciastek B. Cooperation between Angiogenesis, Vasculogenesis, Chemotaxis, and Coagulation in Breast Cancer Metastases Development: Pathophysiological Point of View. Biomedicines. 2022; 10(2):300. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020300
Chicago/Turabian StyleZarychta, Elżbieta, and Barbara Ruszkowska-Ciastek. 2022. "Cooperation between Angiogenesis, Vasculogenesis, Chemotaxis, and Coagulation in Breast Cancer Metastases Development: Pathophysiological Point of View" Biomedicines 10, no. 2: 300. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020300