
Whole-Blood Transcriptional Profiles Enable Early Prediction of the Presence of Coronary Atherosclerosis and High-Risk Plaque Features at Coronary CT Angiography

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Coronary Computed Tomography Angiography
2.3. Blood Sample Collection
2.4. RNA Isolation and Sequencing
2.5. RNA-Seq Data Pre-Processing
2.6. Differential Expression Analysis
2.7. Gene Set Enrichment Analysis
2.8. Microarray Analysis
2.9. Statistical Analysis
3. Results
3.1. Study Population
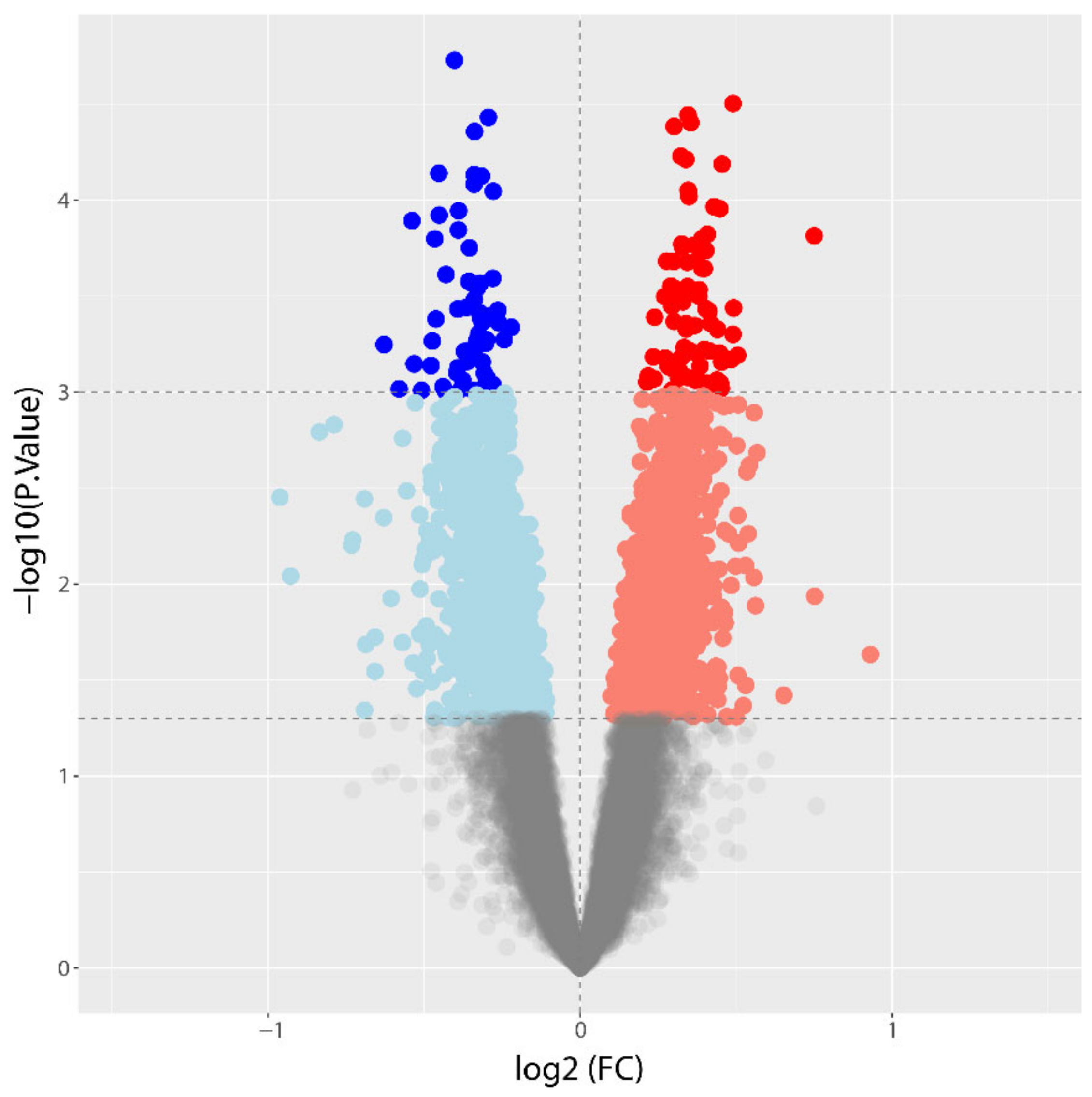
3.2. Differential Gene Expression between CAD and NoCAD Patients
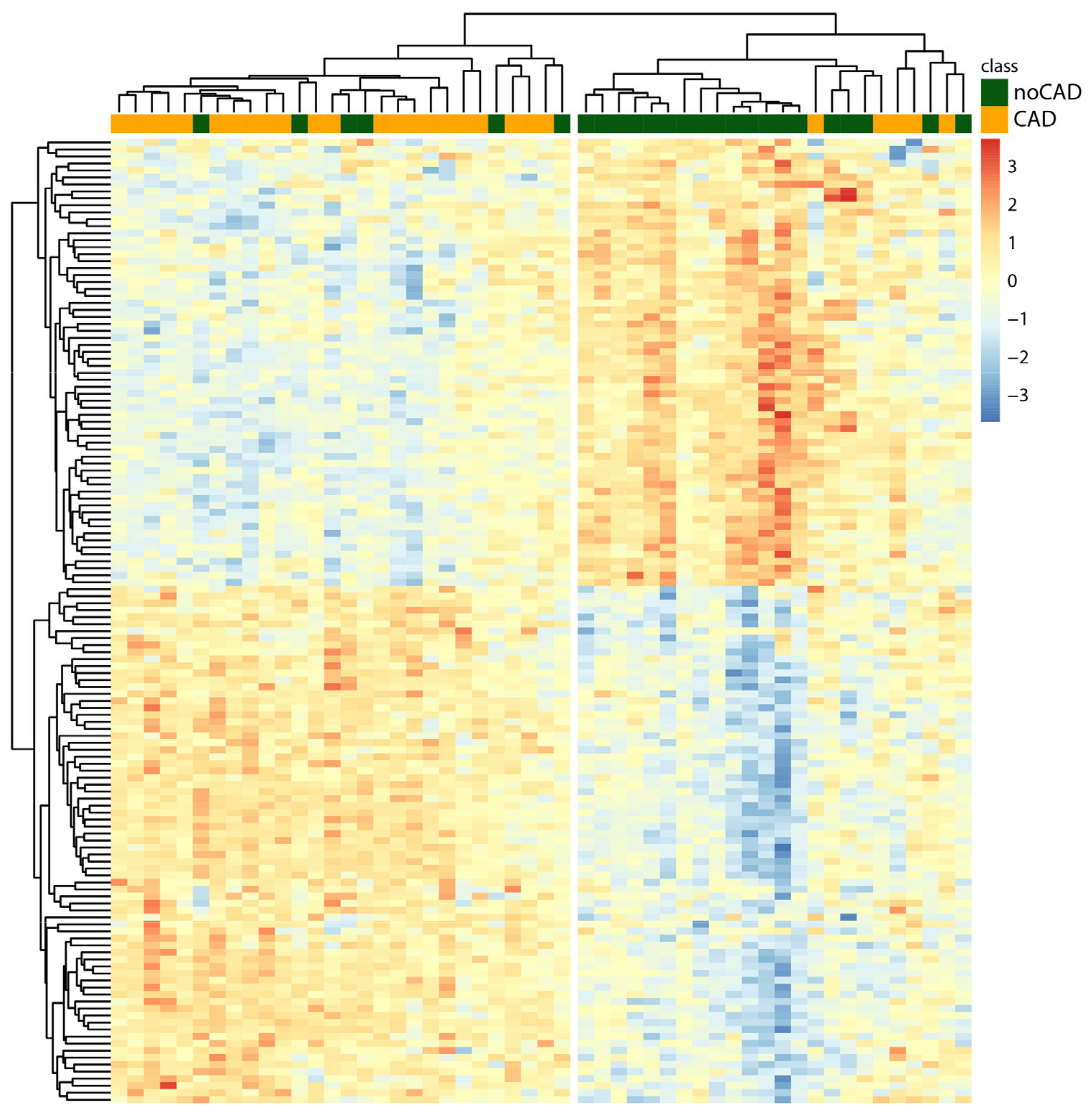
3.3. Hierarchical Clustering
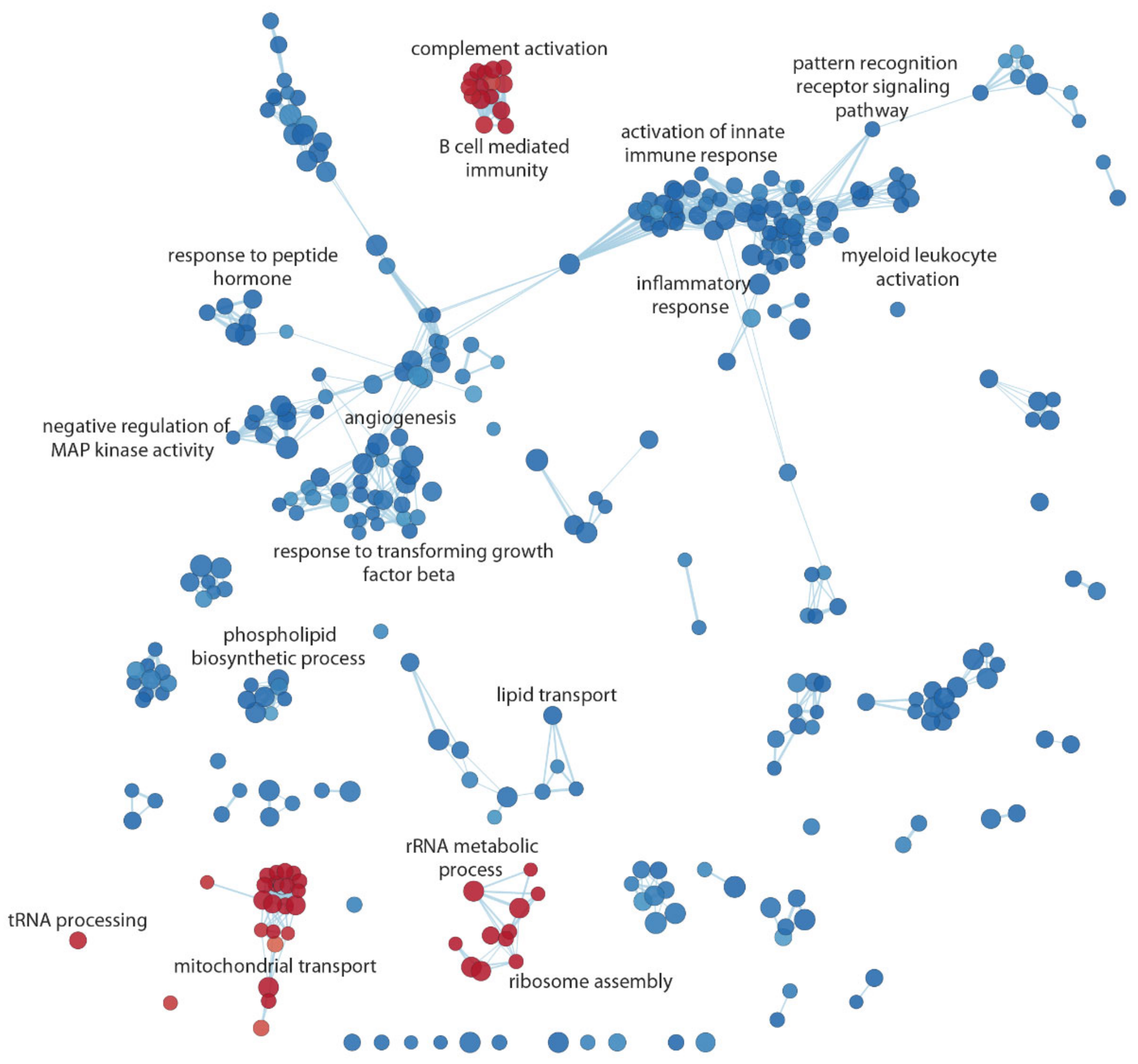
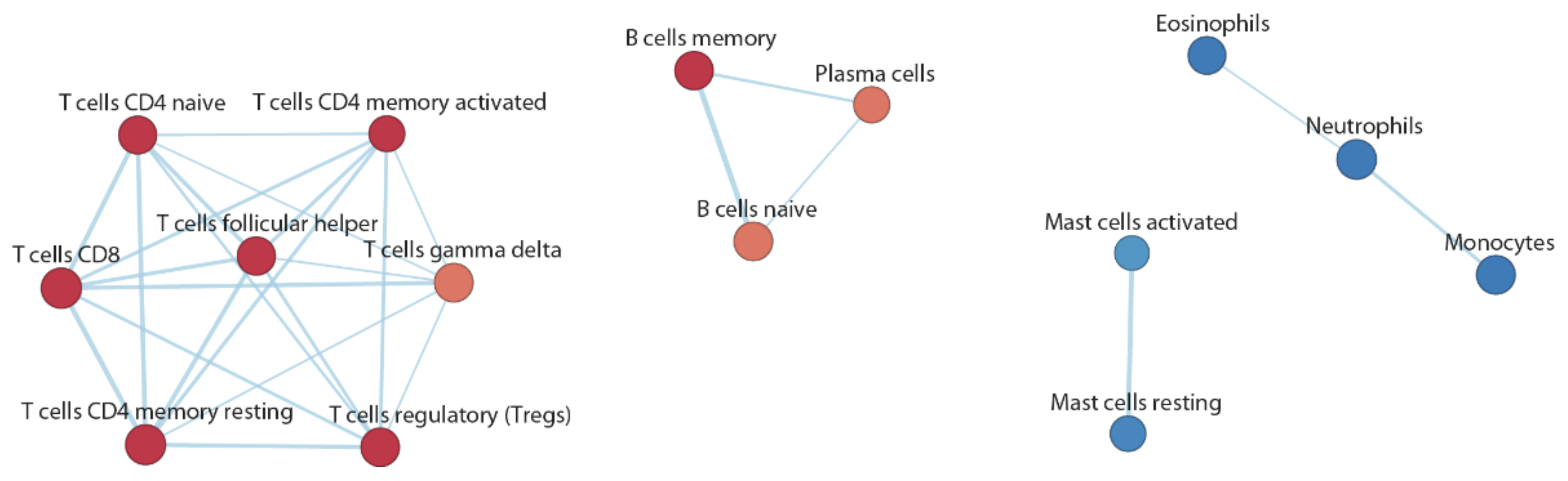
3.4. Functional Inference
3.5. Associations between Gene Expression and High-Risk Plaque Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 2022, 29, 5–115. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wang, T.J. Circulating, imaging, and genetic biomarkers in cardiovascular risk prediction. Trends Cardiovasc. Med. 2011, 21, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.M.; Rochitte, C.E.; Dewey, M.; Arbab-Zadeh, A.; Niinuma, H.; Gottlieb, I.; Paul, N.; Clouse, M.E.; Shapiro, E.P.; Hoe, J.; et al. Diagnostic performance of coronary angiography by 64-row CT. N. Engl. J. Med. 2008, 359, 2324–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoenacker, P.K.; Heijenbrok-Kal, M.H.; Van Heste, R.; Decramer, I.; Van Hoe, L.R.; Wijns, W.; Hunink, M.G. Diagnostic performance of multidetector CT angiography for assessment of coronary artery disease: Meta-analysis. Radiology 2007, 244, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, S.; Sarai, M.; Harigaya, H.; Anno, H.; Inoue, K.; Hara, T.; Naruse, H.; Ishii, J.; Hishida, H.; Wong, N.D.; et al. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J. Am. Coll. Cardiol. 2009, 54, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Obaid, D.R.; Calvert, P.A.; Brown, A.; Gopalan, D.; West, N.E.J.; Rudd, J.H.F.; Bennett, M.R. Coronary CT angiography features of ruptured and high-risk atherosclerotic plaques: Correlation with intra-vascular ultrasound. J. Cardiovasc. Comput. Tomogr. 2017, 11, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Takamura, K.; Fujimoto, S.; Kondo, T.; Hiki, M.; Kawaguchi, Y.; Kato, E.; Daida, H. Incremental prognostic value of coronary computed tomography angiography: High-risk plaque characteristics in asymptomatic patients. J. Atheroscler. Thromb. 2017, 24, 1174–1185. [Google Scholar] [CrossRef] [Green Version]
- Navas-Carrillo, D.; Marin, F.; Valdes, M.; Orenes-Pinero, E. Deciphering acute coronary syndrome biomarkers: High-resolution proteomics in platelets, thrombi and microparticles. Crit. Rev. Clin. Lab. Sci. 2017, 54, 49–58. [Google Scholar] [CrossRef]
- Alvarez-Llamas, G.; de la Cuesta, F.; Barderas, M.E.; Darde, V.; Padial, L.R.; Vivanco, F. Recent advances in atherosclerosis-based proteomics: New biomarkers and a future perspective. Expert. Rev. Proteom. 2008, 5, 679–691. [Google Scholar] [CrossRef]
- Aziz, H.; Zaas, A.; Ginsburg, G.S. Peripheral blood gene expression profiling for cardiovascular disease assessment. Genom. Med. 2007, 1, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Joehanes, R.; Johnson, A.D.; Barb, J.J.; Raghavachari, N.; Liu, P.; Woodhouse, K.A.; O’Donnell, C.J.; Munson, P.J.; Levy, D. Gene expression analysis of whole blood, peripheral blood mononuclear cells, and lymphoblastoid cell lines from the Framingham Heart Study. Physiol. Genom. 2012, 44, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.J.; Lee, A.H.; Smilowitz, N.R.; Hausvater, A.; Fishman, G.I.; Hochman, J.S.; Reynolds, H.R.; Berger, J.S. Whole-blood transcriptome profiling identifies women with myocardial infarction with nonobstructive coronary artery disease. Circ. Genom. Precis. Med. 2018, 11, e002387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.X.; He, S.; Wang, Y.J.; Gan, X.K.; Zhou, Y.Q.; Hua, L.; Hou, C.; Zhang, S.; Zhou, H.X.; Jia, E.Z. Comprehensive analysis of mRNA expression profiling and identification of potential diagnostic biomarkers in coronary artery disease. ACS Omega 2021, 6, 24016–24026. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Toma, I.; Yang, Z.; Katz, R.; Reiner, J.; Mazhari, R.; Shah, P.; Tackett, M.; Jones, D.; Jepson, T.; et al. RNA sequencing of blood in coronary artery disease: Involvement of regulatory T cell imbalance. BMC Med. Genom. 2021, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; van Rooij, J.G.J.; Wakabayashi, Y.; Hwang, S.J.; Yang, Y.; Ghanbari, M.; Bos, D.; Consortium, B.; Levy, D.; Johnson, A.D.; et al. Genome-wide transcriptome study using deep RNA sequencing for myocardial infarction and coronary artery calcification. BMC Med. Genom. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Andreini, D.; Magnoni, M.; Masson, S.; Mushtaq, S.; Berti, S.; Canestrari, M.; Casolo, G.; Gabrielli, D.; Latini, R.; et al. Association of high-risk coronary atherosclerosis at CCTA with clinical and circulating biomarkers: Insight from CAPIRE study. J. Cardiovasc. Comput. Tomogr. 2021, 15, 73–80. [Google Scholar] [CrossRef]
- Conte, E.; Mushtaq, S.; Pontone, G.; Li Piani, L.; Ravagnani, P.; Galli, S.; Collet, C.; Sonck, J.; Di Odoardo, L.; Guglielmo, M.; et al. Plaque quantification by coronary computed tomography angiography using intravascular ultrasound as a reference standard: A comparison between standard and last generation computed tomography scanners. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 191–201. [Google Scholar] [CrossRef]
- Cury, R.C.; Abbara, S.; Achenbach, S.; Agatston, A.; Berman, D.S.; Budoff, M.J.; Dill, K.E.; Jacobs, J.E.; Maroules, C.D.; Rubin, G.D.; et al. CAD-RADSTM Coronary Artery Disease—Reporting and Data System. An expert consensus document of the Society of Cardiovascular Computed Tomography (SCCT), the American College of Radiology (ACR) and the North American Society for Cardiovascular Imaging (NASCI). Endorsed by the American College of Cardiology. J. Cardiovasc. Comput. Tomogr. 2016, 10, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Pang, T.; Shi, R.; Qian, W.L.; Yan, W.F.; Li, Y.; Yang, Z.G. Effect of smoking on coronary artery plaques in type 2 diabetes mellitus: Evaluation with coronary computed tomography angiography. Front. Endocrinol. 2021, 12, 750773. [Google Scholar] [CrossRef]
- Mushtaq, S.; De Araujo Goncalves, P.; Garcia-Garcia, H.M.; Pontone, G.; Bartorelli, A.L.; Bertella, E.; Campos, C.M.; Pepi, M.; Serruys, P.W.; Andreini, D. Long-term prognostic effect of coronary atherosclerotic burden: Validation of the computed tomography-Leaman score. Circ. Cardiovasc. Imaging 2015, 8, e002332. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Storey, J.D. A general framework for multiple testing dependence. Proc. Natl. Acad. Sci. USA 2008, 105, 18718–18723. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, M.; Colombo, G.I.; Piacentini, L. DaMiRseq-an R/Bioconductor package for data mining of RNA-Seq data: Normalization, feature selection and classification. Bioinformatics 2018, 34, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Wingrove, J.A.; Daniels, S.E.; Sehnert, A.J.; Tingley, W.; Elashoff, M.R.; Rosenberg, S.; Buellesfeld, L.; Grube, E.; Newby, L.K.; Ginsburg, G.S.; et al. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circ. Cardiovasc. Genet. 2008, 1, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnaeve, P.R.; Donahue, M.P.; Grass, P.; Seo, D.; Vonderscher, J.; Chibout, S.D.; Kraus, W.E.; Sketch, M., Jr.; Nelson, C.; Ginsburg, G.S.; et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS ONE 2009, 4, e7037. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of lipid accumulation and inflammation in atherosclerosis: Focus on molecular and cellular mechanisms. Front. Cardiovasc. Med. 2021, 8, 707529. [Google Scholar] [CrossRef]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA damage in the context of pathogenesis of atherosclerosis. Biomedicines 2020, 8, 166. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Chiesa, M.; Piacentini, L.; Bono, E.; Milazzo, V.; Campodonico, J.; Marenzi, G.; Colombo, G.I. Whole blood transcriptome profile at hospital admission discriminates between patients with ST-segment elevation and non-ST-segment elevation acute myocardial infarction. Sci. Rep. 2020, 10, 8731. [Google Scholar] [CrossRef]
- Andreini, D.; Magnoni, M.; Conte, E.; Masson, S.; Mushtaq, S.; Berti, S.; Canestrari, M.; Casolo, G.; Gabrielli, D.; Latini, R.; et al. Coronary plaque features on CTA can identify patients at increased risk of cardiovascular events. JACC Cardiovasc. Imaging 2020, 13, 1704–1717. [Google Scholar] [CrossRef]
- Chang, H.J.; Lin, F.Y.; Lee, S.E.; Andreini, D.; Bax, J.; Cademartiri, F.; Chinnaiyan, K.; Chow, B.J.W.; Conte, E.; Cury, R.C.; et al. Coronary atherosclerotic precursors of acute coronary syndromes. J. Am. Coll. Cardiol. 2018, 71, 2511–2522. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Hulten, E.; Belur, P.; Smith, R.; Voros, S.; Villines, T.C. Coronary CT angiography versus intravascular ultrasound for estimation of coronary stenosis and atherosclerotic plaque burden: A meta-analysis. J. Cardiovasc. Comput. Tomogr. 2013, 7, 256–266. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | CAD (n = 27) | noCAD (n = 25) | p-Value |
|---|---|---|---|
| Males | 21 (77.8%) | 20 (80%) | 0.84 |
| Age (years) | 63 ± 8 | 62 ± 7 | 0.65 |
| BMI (kg/m3) | 26.2 ± 3 | 26 ± 2.7 | 0.8 |
| Smokers | 16 (59.2%) | 13 (52%) | 0.60 |
| Hypertension | 19 (70%) | 10 (40%) | 0.045 |
| Hypercholesterolemia | 19 (70%) | 16 (64%) | 0.64 |
| Use of statin | 15 (55%) | 7 (28%) | 0.06 |
| Diabetes mellitus | 4 (14.8%) | 3 (12%) | 0.76 |
| Peripheral artery disease | 5 (19%) | 2 (8%) | 0.42 |
| Clinical presentation | |||
| Any of the symptoms below | 15 (55.5%) | 13 (52%) | 0.80 |
| Angina pectoris | 3 (11%) | 2 (8%) | 0.71 |
| Atypical chest pain | 6 (22%) | 4 (16%) | 0.58 |
| Dyspnea | 3 (11%) | 2 (8%) | 0.71 |
| Arrhythmias | 4 (14%) | 7 (28%) | 0.21 |
| Laboratory | |||
| Erythrocytes (106/µL) | 4.88 ± 0.5 | 4.91 ± 0.41 | 0.81 |
| Leucocytes (103/µL) | 8.32 ± 1.67 | 7.67 ± 1.84 | 0.19 |
| Hemoglobin (g/dL) | 14.83 ± 1.42 | 14.85 ± 1.12 | 0.95 |
| Hematocrit (%) | 43.04 ± 3.91 | 43.50 ± 2.67 | 0.62 |
| Platelets (103/µL) | 239.88 ± 50.38 | 255.48 ± 55.82 | 0.33 |
| Glycemia (mg/dL) | 100.33 ± 10.9 | 102 ± 27.27 | 0.77 |
| Uric acid (mg/dL) | 5.34 ± 1.05 | 5.2 ± 1.40 | 0.68 |
| γ-GT (UI/L) | 35.34 ± 22.35 | 34.08 ± 23.9 | 0.84 |
| Total bilirubin (mg/dL) | 0.62 ± 0.27 | 0.71 ± 0.34 | 0.31 |
| Troponin I (ng/L) | 5.07 ± 11.2 | 2.81 ± 2.23 | 0.31 |
| Triglycerides (mg/dL) | 109 ± 65.82 | 94.44 ± 33.64 | 0.31 |
| Total cholesterol (mg/dL) | 200.48 ± 49.26 | 195.84 ± 40.42 | 0.71 |
| HLD-c (mg/dL) | 58.22 ± 13.37 | 66.76 ± 18.05 | 0.06 |
| LDL-c (mg/dL) | 120.40 ± 41.59 | 110.08 ± 33.99 | 0.33 |
| CRP (mg/dL) | 2.12 ± 2.59 | 1.46 ± 1.90 | 0.30 |
| NAME | Gene Ontology ID | NES | q-Value |
|---|---|---|---|
| Positively associated | |||
| rRNA metabolic process | GO:0016072 | 4.706 | 0 |
| Aerobic respiration | GO:0009060 | 3.535 | 0 |
| Ribosome assembly | GO:0042255 | 3.732 | 0 |
| Complement activation | GO:0006956 | 2.956 | 0.00007 |
| B cell-mediated immunity | GO:0019724 | 2.686 | 0.00053 |
| Mitochondrial transport | GO:0006839 | 2.490 | 0.00185 |
| tRNA processing | GO:0008033 | 2.070 | 0.02193 |
| Negatively associated | |||
| Pattern recognition receptor signaling pathway | GO:0002221 | −3.129 | 0 |
| Negative regulation of MAP kinase activity | GO:0043407 | −2.894 | 0.00058 |
| Response to peptide hormone | GO:0043434 | −2.719 | 0.00206 |
| Myeloid leukocyte activation | GO:0002274 | −2.644 | 0.00243 |
| Inflammatory response | GO:0006954 | −2.544 | 0.00399 |
| Activation of innate immune response | GO:0002218 | −2.529 | 0.00422 |
| Response to transforming growth factor-beta | GO:0071559 | −2.384 | 0.00694 |
| Angiogenesis | GO:0001525 | −2.316 | 0.00956 |
| Phospholipid biosynthetic process | GO:0008654 | −2.197 | 0.01545 |
| Lipid transport | GO:0006869 | −2.032 | 0.02875 |
| Variable | Significantly Associated Genes | Positively Associated Genes | Negatively Associated Genes |
|---|---|---|---|
| Leaman Score | 19 | 11 | 8 |
| SIS | 34 | 17 | 17 |
| SSS | 17 | 7 | 10 |
| Plaque density < 150 HU | 58 | 42 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreini, D.; Melotti, E.; Vavassori, C.; Chiesa, M.; Piacentini, L.; Conte, E.; Mushtaq, S.; Manzoni, M.; Cipriani, E.; Ravagnani, P.M.; et al. Whole-Blood Transcriptional Profiles Enable Early Prediction of the Presence of Coronary Atherosclerosis and High-Risk Plaque Features at Coronary CT Angiography. Biomedicines 2022, 10, 1309. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061309
Andreini D, Melotti E, Vavassori C, Chiesa M, Piacentini L, Conte E, Mushtaq S, Manzoni M, Cipriani E, Ravagnani PM, et al. Whole-Blood Transcriptional Profiles Enable Early Prediction of the Presence of Coronary Atherosclerosis and High-Risk Plaque Features at Coronary CT Angiography. Biomedicines. 2022; 10(6):1309. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061309
Chicago/Turabian StyleAndreini, Daniele, Eleonora Melotti, Chiara Vavassori, Mattia Chiesa, Luca Piacentini, Edoardo Conte, Saima Mushtaq, Martina Manzoni, Eleonora Cipriani, Paolo M. Ravagnani, and et al. 2022. "Whole-Blood Transcriptional Profiles Enable Early Prediction of the Presence of Coronary Atherosclerosis and High-Risk Plaque Features at Coronary CT Angiography" Biomedicines 10, no. 6: 1309. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061309