
Engineering a Therapeutic Protein to Enhance the Study of Anti-Drug Immunity

 ,
, 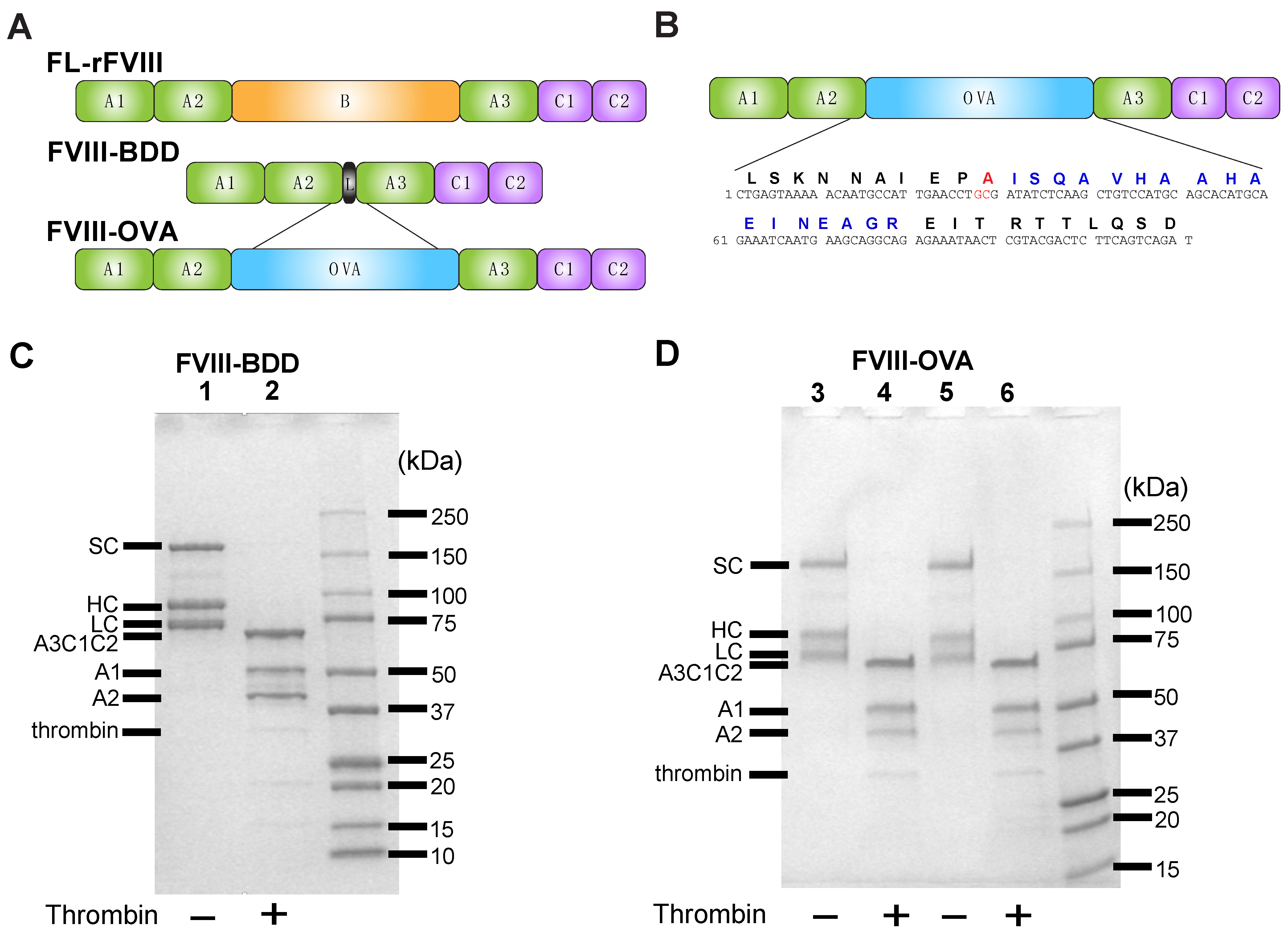{kind=link}
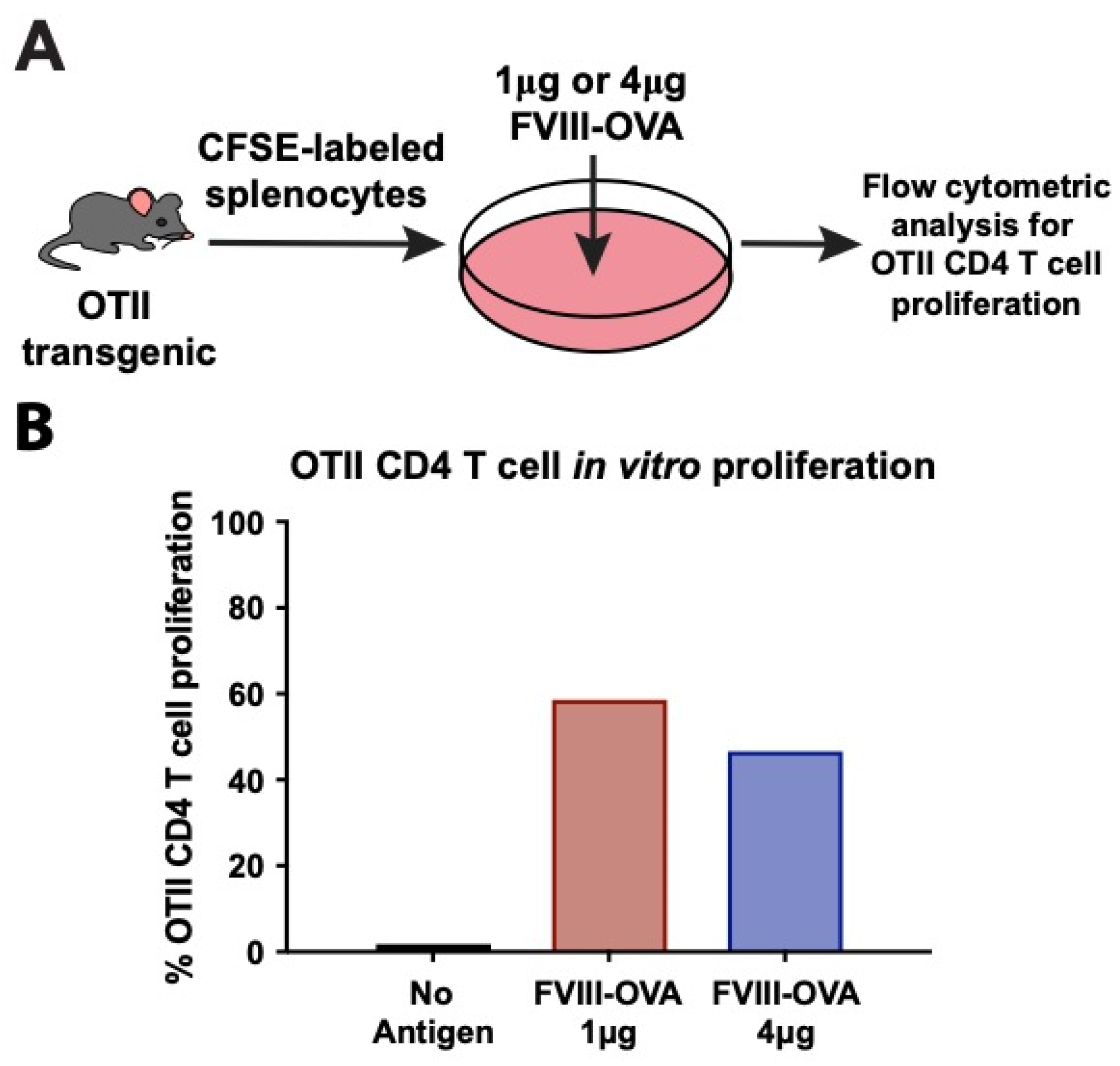{kind=link}
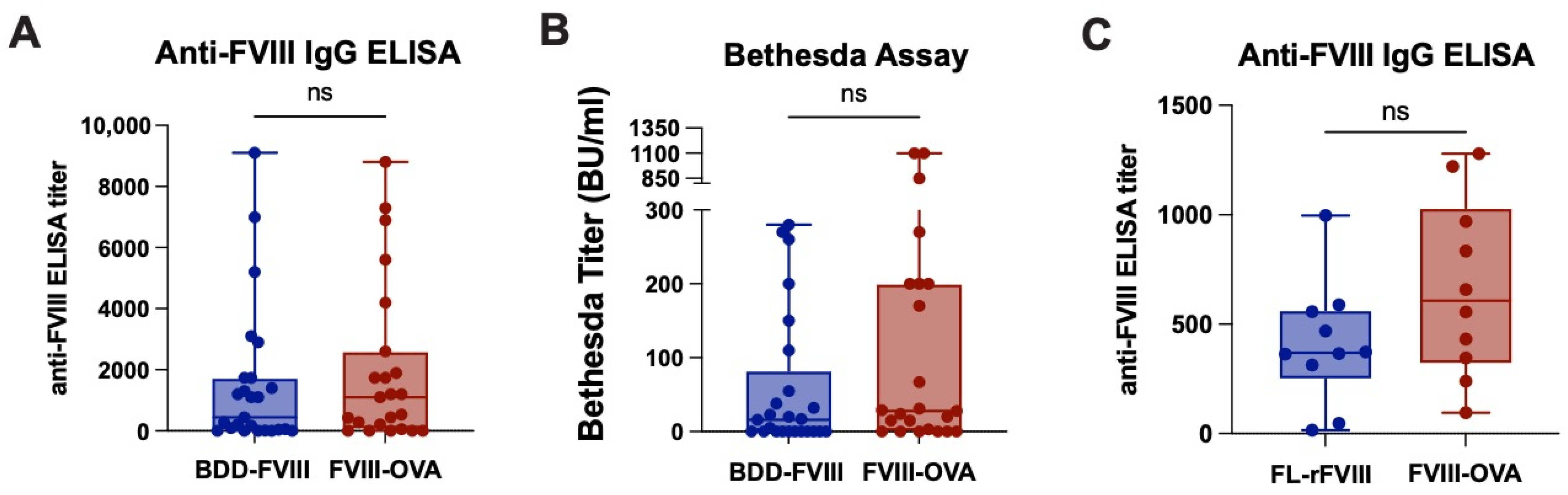{kind=link}
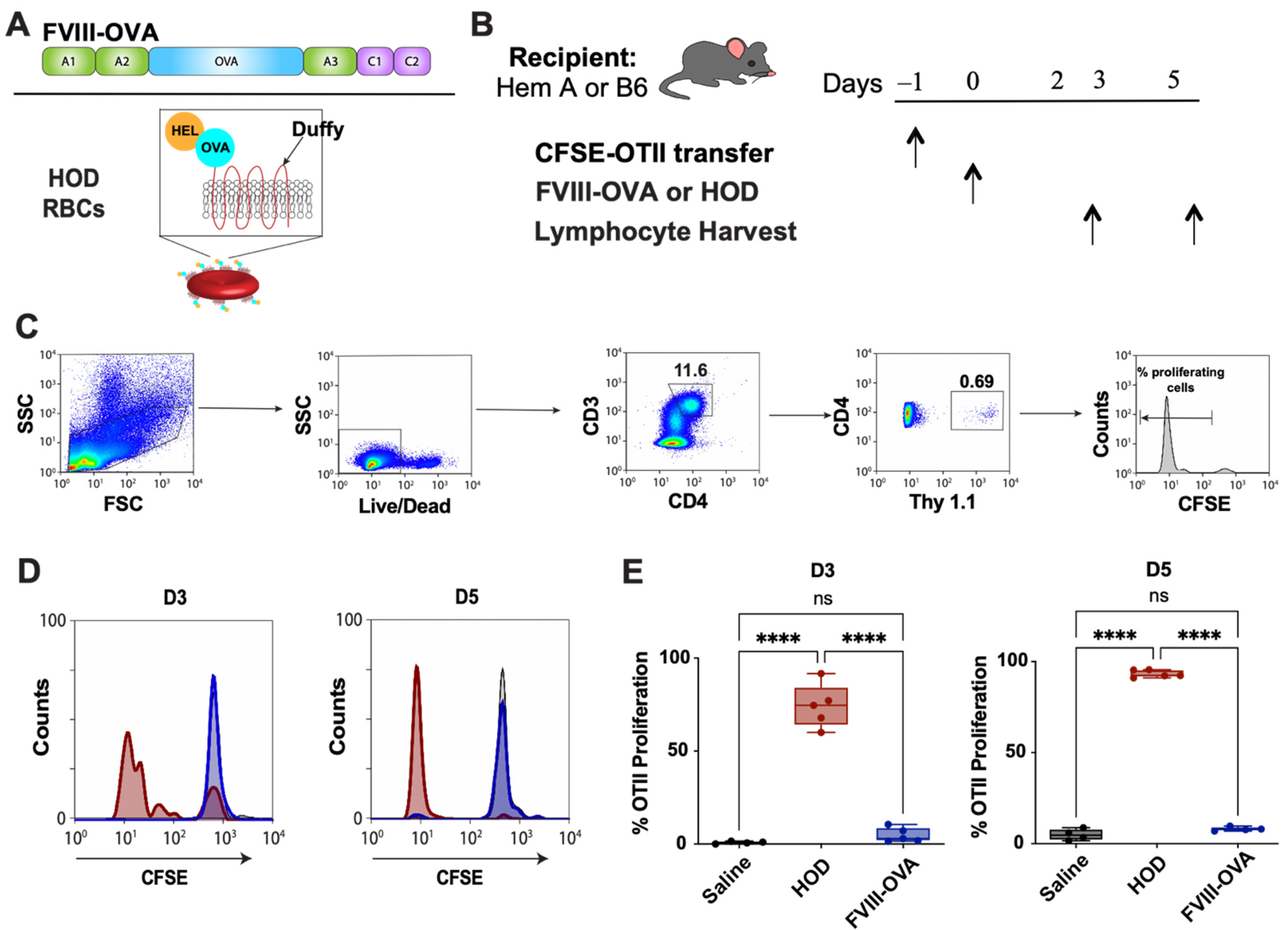{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction, Expression and Purification of FVIII-OVA (RENeo FVIII with R740A Mutation)
2.2. Activation of FVIII by Thrombin and SDS-PAGE of FVIII
2.3. Coagulation Assay
2.4. Mice
2.5. OTII Splenocyte Labeling, Adoptive Transfer and Analysis of CD4 T Cell Proliferation In Vitro and In Vivo
2.6. RBC Isolation and Transfusion
2.7. FVIII Administration, Plasma Collection and Analysis of Anti-FVIII Antibodies
2.8. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brunelli, M.J.; Atallah, A.N.; da Silva, E.M. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis type VI. Cochrane Database Syst. Rev. 2021, 9, CD009806. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, L.; Quan, S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst. Rev. 2017, 11, CD011539. [Google Scholar] [CrossRef] [PubMed]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, e191–e197. [Google Scholar] [CrossRef]
- Yari, M.; Ghoshoon, M.B.; Vakili, B.; Ghasemi, Y. Therapeutic Enzymes: Applications and Approaches to Pharmacological Improvement. Curr. Pharm. Biotechnol. 2017, 18, 531–540. [Google Scholar] [CrossRef]
- Cafuir, L.A.; Kempton, C.L. Current and emerging factor VIII replacement products for hemophilia A. Ther. Adv. Hematol. 2017, 8, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Fernandez-Pereira, C.; San Millan-Tejado, B.; Gallardo-Gomez, M.; Perez-Marquez, T.; Alves-Villar, M.; Melcon-Crespo, C.; Fernandez-Martin, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef]
- Kinoshita, S.; Yoshioka, A.; Park, Y.D.; Ishizashi, H.; Konno, M.; Funato, M.; Matsui, T.; Titani, K.; Yagi, H.; Matsumoto, M.; et al. Upshaw-Schulman syndrome revisited: A concept of congenital thrombotic thrombocytopenic purpura. Int. J. Hematol. 2001, 74, 101–108. [Google Scholar] [CrossRef]
- Abolhassani, H.; Sadaghiani, M.S.; Aghamohammadi, A.; Ochs, H.D.; Rezaei, N. Home-based subcutaneous immunoglobulin versus hospital-based intravenous immunoglobulin in treatment of primary antibody deficiencies: Systematic review and meta analysis. J. Clin. Immunol. 2012, 32, 1180–1192. [Google Scholar] [CrossRef]
- Sykes, M. Immune tolerance: Mechanisms and application in clinical transplantation. J. Intern. Med. 2007, 262, 288–310. [Google Scholar] [CrossRef]
- Turgay Yagmur, I.; Unal Uzun, O.; Kucukcongar Yavas, A.; Kulhas Celik, I.; Toyran, M.; Gunduz, M.; Civelek, E.; Dibek Misirlioglu, E. Management of hypersensitivity reactions to enzyme replacement therapy in children with lysosomal storage diseases. Ann. Allergy Asthma Immunol. 2020, 125, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Drost, J.B.; Lusher, J.M.; Warrier, I.; Shapiro, A.; Koerper, M.A.; Dimichele, D.; Westman, J.; Key, N.S.; Sommer, S.S. Anaphylactic response to factor IX replacement therapy in haemophilia B patients: Complete gene deletions confer the highest risk. Haemophilia 1999, 5, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment With Monoclonal Antibodies. Front. Immunol. 2020, 11, 1951. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.D.; Lillicrap, D. Factor VIII inhibitors: Advances in basic and translational science. Int. J. Lab. Hematol. 2017, 39 (Suppl. S1), 6–13. [Google Scholar] [CrossRef] [Green Version]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef]
- Franchini, M.; Marano, G.; Pati, I.; Candura, F.; Profili, S.; Veropalumbo, E.; Masiello, F.; Catalano, L.; Piccinini, V.; Vaglio, S.; et al. Emicizumab for the treatment of haemophilia A: A narrative review. Blood Transfus. 2019, 17, 223–228. [Google Scholar] [CrossRef]
- Knoebl, P.; Thaler, J.; Jilma, P.; Quehenberger, P.; Gleixner, K.; Sperr, W.R. Emicizumab for the treatment of acquired hemophilia A. Blood 2021, 137, 410–419. [Google Scholar] [CrossRef]
- Kempton, C.L.; Meeks, S.L. Toward optimal therapy for inhibitors in hemophilia. Blood 2014, 124, 3365–3372. [Google Scholar] [CrossRef] [Green Version]
- Castaman, G.; Santoro, C.; Coppola, A.; Mancuso, M.E.; Santoro, R.C.; Bernardini, S.; Pugliese, F.R.; Lubrano, R.; Golato, M.; Tripodi, A.; et al. Emergency management in patients with haemophilia A and inhibitors on prophylaxis with emicizumab: AICE practical guidance in collaboration with SIBioC, SIMEU, SIMEUP, SIPMeL and SISET. Blood Transfus. 2020, 18, 143–151. [Google Scholar] [CrossRef]
- Okaygoun, D.; Oliveira, D.D.; Soman, S.; Williams, R. Advances in the management of haemophilia: Emerging treatments and their mechanisms. J. Biomed. Sci. 2021, 28, 64. [Google Scholar] [CrossRef]
- Sorensen, P.S. Antidrug Antibodies Against Biological Treatments for Multiple Sclerosis. CNS Drugs 2022, 36, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Lacroix-Desmazes, S.; Reding, M.T. Inhibitor development. Haemophilia 2008, 14 (Suppl. S3), 36–42. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, D.; Fijnvandraat, K.; Young, G.; Mancuso, M.E. Patients with hemophilia A and inhibitors: Prevention and evolving treatment paradigms. Expert Rev. Hematol. 2020, 13, 313–321. [Google Scholar] [CrossRef]
- Kevane, B.; O’Connell, N. The current and future role of plasma-derived clotting factor concentrate in the treatment of haemophilia A. Transfus. Apher. Sci. 2018, 57, 502–506. [Google Scholar] [CrossRef]
- Covington, M.L.; Voma, C.; Stowell, S.R. Shortage of plasma-derived products: A looming crisis? Blood 2022, 139, 3222–3225. [Google Scholar] [CrossRef] [PubMed]
- Verkerke, H.; Saeedi, B.J.; Boyer, D.; Allen, J.W.; Owens, J.; Shin, S.; Horwath, M.; Patel, K.; Paul, A.; Wu, S.; et al. Are we forgetting about IgA? A re-examination of COVID-19 convalescent plasma. Transfusion 2021, 61, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Verkerke, H.; Horwath, M.; Saeedi, B.; Boyer, D.; Allen, J.W.; Owens, J.; Arthur, C.M.; Nakahara, H.; Rha, J.; Patel, K.; et al. Comparison of Antibody Class Specific SARS-CoV-2 Serology for the Diagnosis of Acute COVID-19. J. Clin. Microbiol. 2021, 59, e02026-20. [Google Scholar] [CrossRef]
- Ehrenforth, S.; Kreuz, W.; Scharrer, I.; Kornhuber, B. Factor VIII inhibitors in haemophiliacs. Lancet 1992, 340, 253. [Google Scholar] [CrossRef]
- Kempton, C.L.; White, G.C., 2nd. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood 2009, 113, 11–17. [Google Scholar] [CrossRef]
- Ehrenforth, S.; Kreuz, W.; Scharrer, I.; Linde, R.; Funk, M.; Gungor, T.; Krackhardt, B.; Kornhuber, B. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992, 339, 594–598. [Google Scholar] [CrossRef]
- Meeks, S.L.; Healey, J.F.; Parker, E.T.; Barrow, R.T.; Lollar, P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood 2007, 110, 4234–4242. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.M. Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activity. J. Thromb. Haemost. 2004, 2, 1700–1708. [Google Scholar] [CrossRef] [PubMed]
- Lollar, P. Pathogenic antibodies to coagulation factors. Part one: Factor VIII and factor IX. J. Thromb. Haemost. 2004, 2, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- White, G.C., 2nd; Kempton, C.L.; Grimsley, A.; Nielsen, B.; Roberts, H.R. Cellular immune responses in hemophilia: Why do inhibitors develop in some, but not all hemophiliacs? J. Thromb. Haemost. 2005, 3, 1676–1681. [Google Scholar] [CrossRef]
- Lorenzo, J.I.; Lopez, A.; Altisent, C.; Aznar, J.A. Incidence of factor VIII inhibitors in severe haemophilia: The importance of patient age. Br. J. Haematol. 2001, 113, 600–603. [Google Scholar] [CrossRef]
- Lusher, J.M.; Arkin, S.; Abildgaard, C.F.; Schwartz, R.S. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N. Engl. J. Med. 1993, 328, 453–459. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E.C.; Valentino, L.A. Emicizumab: Review of the literature and critical appraisal. Haemophilia 2019, 25, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Yada, K.; Nogami, K. Spotlight on emicizumab in the management of hemophilia A: Patient selection and special considerations. J. Blood Med. 2019, 10, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Aledort, L.; Mannucci, P.M.; Schramm, W.; Tarantino, M. Factor VIII replacement is still the standard of care in haemophilia A. Blood Transfus. 2019, 17, 479–486. [Google Scholar] [CrossRef]
- Navarrete, A.; Dasgupta, S.; Delignat, S.; Caligiuri, G.; Christophe, O.D.; Bayry, J.; Nicoletti, A.; Kaveri, S.V.; Lacroix-Desmazes, S. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J. Thromb. Haemost. 2009, 7, 1816–1823. [Google Scholar] [CrossRef]
- Zerra, P.E.; Cox, C.; Baldwin, W.H.; Patel, S.R.; Arthur, C.M.; Lollar, P.; Meeks, S.L.; Stowell, S.R. Marginal zone B cells are critical to factor VIII inhibitor formation in mice with hemophilia A. Blood 2017, 130, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Ing, M.; Delignat, S.; Peyron, I.; Gilardin, L.; Vogel, C.W.; Fritzinger, D.C.; Fremeaux-Bacchi, V.; Kaveri, S.V.; Roumenina, L.T.; et al. Complement C3 is a novel modulator of the anti-factor VIII immune response. Haematologica 2018, 103, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, K.; Scott, D.W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018, 2, 2332–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, R.W.; Kuteyeva, V.; Saboungi, R.; Terhorst, C.; Biswas, M. Reprogrammed CD4(+) T Cells That Express FoxP3(+) Control Inhibitory Antibody Formation in Hemophilia A Mice. Front. Immunol. 2019, 10, 274. [Google Scholar] [CrossRef]
- Gupta, N.; Culina, S.; Meslier, Y.; Dimitrov, J.; Arnoult, C.; Delignat, S.; Gangadharan, B.; Lecerf, M.; Justesen, S.; Gouilleux-Gruart, V.; et al. Regulation of immune responses to protein therapeutics by transplacental induction of T cell tolerance. Sci. Transl. Med. 2015, 7, 275ra221. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered antigen-specific human regulatory T cells: Immunosuppression of FVIII-specific T- and B-cell responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, R.A.; James, E.A.; Kwok, W.W.; Thompson, A.R.; Pratt, K.P. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood 2009, 114, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.; Rogers, G.L.; Sherman, A.; Byrne, B.J.; Markusic, D.M.; Jiang, H.; Herzog, R.W. Combination therapy for inhibitor reversal in haemophilia A using monoclonal anti-CD20 and rapamycin. Thromb. Haemost. 2017, 117, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Jing, W.; Chen, J.; Cai, Y.; Chen, Y.; Schroeder, J.A.; Johnson, B.D.; Cui, W.; Shi, Q. Induction of activated T follicular helper cells is critical for anti-FVIII inhibitor development in hemophilia A mice. Blood Adv. 2019, 3, 3099–3110. [Google Scholar] [CrossRef] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4(+)T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10, 2138. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Heise, M.T. Mouse Models as Resources for Studying Infectious Diseases. Clin. Ther. 2019, 41, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herati, R.S.; Wherry, E.J. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Consideration of Strategies to Improve the Value of Animal Models. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, J.L.; Pai, C.C.; Murphy, W.J. Preclinical modeling of hematopoietic stem cell transplantation—Advantages and limitations. FEBS J. 2016, 283, 1595–1606. [Google Scholar] [CrossRef]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895. [Google Scholar] [CrossRef]
- Cho, Y.B.; Lee, I.G.; Joo, Y.H.; Hong, S.H.; Seo, Y.J. TCR Transgenic Mice: A Valuable Tool for Studying Viral Immunopathogenesis Mechanisms. Int. J. Mol. Sci. 2020, 21, 9690. [Google Scholar] [CrossRef]
- Alsughayyir, J.; Chhabra, M.; Qureshi, M.S.; Mallik, M.; Ali, J.M.; Gamper, I.; Moseley, E.L.; Peacock, S.; Kosmoliaptsis, V.; Goddard, M.J.; et al. Relative Frequencies of Alloantigen-Specific Helper CD4 T Cells and B Cells Determine Mode of Antibody-Mediated Allograft Rejection. Front. Immunol. 2018, 9, 3039. [Google Scholar] [CrossRef] [Green Version]
- Reding, M.T.; Wu, H.; Krampf, M.; Okita, D.K.; Diethelm-Okita, B.M.; Key, N.S.; Conti-Fine, B.M. CD4+ T cell response to factor VIII in hemophilia A, acquired hemophilia, and healthy subjects. Thromb. Haemost. 1999, 82, 509–515. [Google Scholar] [CrossRef]
- Bray, G.L.; Kroner, B.L.; Arkin, S.; Aledort, L.W.; Hilgartner, M.W.; Eyster, M.E.; Ragni, M.V.; Goedert, J.J. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: A report from the Multi-Center Hemophilia Cohort Study. Am. J. Hematol. 1993, 42, 375–379. [Google Scholar] [CrossRef]
- Zerra, P.E.; Patel, S.R.; Jajosky, R.P.; Arthur, C.M.; McCoy, J.W.; Allen, J.W.L.; Chonat, S.; Fasano, R.M.; Roback, J.D.; Josephson, C.D.; et al. Marginal zone B cells mediate a CD4 T-cell-dependent extrafollicular antibody response following RBC transfusion in mice. Blood 2021, 138, 706–721. [Google Scholar] [CrossRef]
- Meuer, S.C.; Schlossman, S.F.; Reinherz, E.L. Clonal analysis of human cytotoxic T lymphocytes: T4+ and T8+ effector T cells recognize products of different major histocompatibility complex regions. Proc. Natl. Acad. Sci. USA 1982, 79, 4395–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellino, F.; Zhong, G.; Germain, R.N. Antigen presentation by MHC class II molecules: Invariant chain function, protein trafficking, and the molecular basis of diverse determinant capture. Hum. Immunol. 1997, 54, 159–169. [Google Scholar] [CrossRef]
- Oxenius, A.; Bachmann, M.F.; Zinkernagel, R.M.; Hengartner, H. Virus-specific MHC-class II-restricted TCR-transgenic mice: Effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 1998, 28, 390–400. [Google Scholar] [CrossRef]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Wingerath, J.; Ostroumov, D.; Woller, N.; Manns, M.P.; Pinschewer, D.D.; Orlinger, K.; Berka, U.; Kuhnel, F.; Wirth, T.C. Recombinant LCMV Vectors Induce Protective Immunity following Homologous and Heterologous Vaccinations. Mol. Ther. 2017, 25, 2533–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehst, B.D.; Ingulli, E.; Jenkins, M.K. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am. J. Transplant. 2003, 3, 1355–1362. [Google Scholar] [CrossRef]
- Khan, S.H.; Badovinac, V.P. Listeria monocytogenes: A model pathogen to study antigen-specific memory CD8 T cell responses. Semin. Immunopathol. 2015, 37, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Desmarets, M.; Cadwell, C.M.; Peterson, K.R.; Neades, R.; Zimring, J.C. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood 2009, 114, 2315–2322. [Google Scholar] [CrossRef] [Green Version]
- Lubin, I.M.; Healey, J.F.; Scandella, D.; Runge, M.S.; Lollar, P. Elimination of a major inhibitor epitope in factor VIII. J. Biol. Chem. 1994, 269, 8639–8641. [Google Scholar] [CrossRef]
- Barrow, R.T.; Healey, J.F.; Gailani, D.; Scandella, D.; Lollar, P. Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood 2000, 95, 564–568. [Google Scholar] [CrossRef]
- Healey, J.F.; Barrow, R.T.; Tamim, H.M.; Lubin, I.M.; Shima, M.; Scandella, D.; Lollar, P. Residues Glu2181-Val2243 contain a major determinant of the inhibitory epitope in the C2 domain of human factor VIII. Blood 1998, 92, 3701–3709. [Google Scholar] [CrossRef] [PubMed]
- Grushin, K.; Miller, J.; Dalm, D.; Parker, E.T.; Healey, J.F.; Lollar, P.; Stoilova-McPhie, S. Lack of recombinant factor VIII B-domain induces phospholipid vesicle aggregation: Implications for the immunogenicity of factor VIII. Haemophilia 2014, 20, 723–731. [Google Scholar] [CrossRef]
- Doering, C.; Parker, E.T.; Healey, J.F.; Craddock, H.N.; Barrow, R.T.; Lollar, P. Expression and characterization of recombinant murine factor VIII. Thromb. Haemost. 2002, 88, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Gao, G.P.; Chirmule, N.; Tazelaar, J.; Kazazian, H.H., Jr. Partial correction of murine hemophilia A with neo-antigenic murine factor VIII. Hum. Gene Ther. 2000, 11, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Chao, B.N.; Baldwin, W.H.; Healey, J.F.; Parker, E.T.; Shafer-Weaver, K.; Cox, C.; Jiang, P.; Kanellopoulou, C.; Lollar, P.; Meeks, S.L.; et al. Characterization of a genetically engineered mouse model of hemophilia A with complete deletion of the F8 gene. J. Thromb. Haemost. 2016, 14, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, C.M.; Zerra, P.E.; Shin, S.; Wang, J.; Song, X.; Doering, C.B.; Lollar, P.; Meeks, S.; Stowell, S.R. Nonhuman glycans can regulate anti-factor VIII antibody formation in mice. Blood 2022, 139, 1312–1317. [Google Scholar] [CrossRef]
- Mener, A.; Patel, S.R.; Arthur, C.M.; Chonat, S.; Wieland, A.; Santhanakrishnan, M.; Liu, J.; Maier, C.L.; Jajosky, R.P.; Girard-Pierce, K.; et al. Complement serves as a switch between CD4+ T cell-independent and -dependent RBC antibody responses. JCI Insight 2018, 3, e121631. [Google Scholar] [CrossRef]
- Patel, S.R.; Bennett, A.; Girard-Pierce, K.; Maier, C.L.; Chonat, S.; Arthur, C.M.; Zerra, P.E.; Mener, A.; Stowell, S.R. Recipient priming to one RBC alloantigen directly enhances subsequent alloimmunization in mice. Blood Adv. 2018, 2, 105–115. [Google Scholar] [CrossRef]
- Mener, A.; Patel, S.R.; Arthur, C.M.; Stowell, S.R. Antibody-mediated immunosuppression can result from RBC antigen loss independent of Fcgamma receptors in mice. Transfusion 2019, 59, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Zerra, P.E.; Arthur, C.M.; Chonat, S.; Maier, C.L.; Mener, A.; Shin, S.; Allen, J.W.L.; Baldwin, W.H.; Cox, C.; Verkerke, H.; et al. Fc Gamma Receptors and Complement Component 3 Facilitate Anti-fVIII Antibody Formation. Front. Immunol. 2020, 11, 905. [Google Scholar] [CrossRef]
- Maier, C.L.; Mener, A.; Patel, S.R.; Jajosky, R.P.; Bennett, A.L.; Arthur, C.M.; Hendrickson, J.E.; Stowell, S.R. Antibody-mediated immune suppression by antigen modulation is antigen-specific. Blood Adv. 2018, 2, 2986–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowell, S.R.; Liepkalns, J.S.; Hendrickson, J.E.; Girard-Pierce, K.R.; Smith, N.H.; Arthur, C.M.; Zimring, J.C. Antigen modulation confers protection to red blood cells from antibody through Fcgamma receptor ligation. J. Immunol. 2013, 191, 5013–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.R.; Gibb, D.R.; Girard-Pierce, K.; Zhou, X.; Rodrigues, L.C.; Arthur, C.M.; Bennett, A.L.; Jajosky, R.P.; Fuller, M.; Maier, C.L.; et al. Marginal Zone B Cells Induce Alloantibody Formation Following RBC Transfusion. Front. Immunol. 2018, 9, 2516. [Google Scholar] [CrossRef] [PubMed]
- Mener, A.; Arthur, C.M.; Patel, S.R.; Liu, J.; Hendrickson, J.E.; Stowell, S.R. Complement Component 3 Negatively Regulates Antibody Response by Modulation of Red Blood Cell Antigen. Front. Immunol. 2018, 9, 676. [Google Scholar] [CrossRef] [Green Version]
- Calabro, S.; Gallman, A.; Gowthaman, U.; Liu, D.; Chen, P.; Liu, J.; Krishnaswamy, J.K.; Nascimento, M.S.; Xu, L.; Patel, S.R.; et al. Bridging channel dendritic cells induce immunity to transfused red blood cells. J. Exp. Med. 2016, 213, 887–896. [Google Scholar] [CrossRef]
- Soldatenko, A.; Hoyt, L.R.; Xu, L.; Calabro, S.; Lewis, S.M.; Gallman, A.E.; Hudson, K.E.; Stowell, S.R.; Luckey, C.J.; Zimring, J.C.; et al. Innate and Adaptive Immunity to Transfused Allogeneic RBCs in Mice Requires MyD88. J. Immunol. 2022, 208, 991–997. [Google Scholar] [CrossRef]
- Kasper, C.K.; Aledort, L.; Aronson, D.; Counts, R.; Edson, J.R.; van Eys, J.; Fratantoni, J.; Green, D.; Hampton, J.; Hilgartner, M.; et al. Proceedings: A more uniform measurement of factor VIII inhibitors. Thromb. Diath. Haemorrh. 1975, 34, 612. [Google Scholar]
- Meeks, S.L.; Cox, C.L.; Healey, J.F.; Parker, E.T.; Doshi, B.S.; Gangadharan, B.; Barrow, R.T.; Lollar, P. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood 2012, 120, 2512–2520. [Google Scholar] [CrossRef] [Green Version]
- Barrow, R.T.; Lollar, P. Neutralization of antifactor VIII inhibitors by recombinant porcine factor VIII. J. Thromb. Haemost. 2006, 4, 2223–2229. [Google Scholar] [CrossRef]
- Pipe, S.W.; Kaufman, R.J. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proc. Natl. Acad. Sci. USA 1997, 94, 11851–11856. [Google Scholar] [CrossRef] [Green Version]
- Pittman, D.D.; Kaufman, R.J. Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII). Proc. Natl. Acad. Sci. USA 1988, 85, 2429–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadharan, B.; Ing, M.; Delignat, S.; Peyron, I.; Teyssandier, M.; Kaveri, S.V.; Lacroix-Desmazes, S. The C1 and C2 domains of blood coagulation factor VIII mediate its endocytosis by dendritic cells. Haematologica 2017, 102, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsuli, G.; Patel, S.; Cox, C.; Baldwin, W.H.; Lollar, J.; Meeks, S. Epitope dependent augmentation of the immune response in hemophilia A mice immunized with factor VIII/antibody immune complexes. Blood 2019, 134, 2387. [Google Scholar] [CrossRef]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Erlandsson Harris, H.; Andersson, U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 2004, 34, 1503–1512. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Valiante, N.M. Recent advances in the discovery and delivery of vaccine adjuvants. Nat. Rev. Drug Discov. 2003, 2, 727–735. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54 Pt 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; van der Bom, J.G.; Ljung, R.; Escuriola, C.; Cid, A.R.; Claeyssens-Donadel, S.; van Geet, C.; Kenet, G.; Makipernaa, A.; Molinari, A.C.; et al. Factor VIII products and inhibitor development in severe hemophilia A. N. Engl. J. Med. 2013, 368, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Reipert, B.M.; Gangadharan, B.; Hofbauer, C.J.; Berg, V.; Schweiger, H.; Bowen, J.; Blatny, J.; Fijnvandraat, K.; Mullins, E.S.; Klintman, J.; et al. The prospective Hemophilia Inhibitor PUP Study reveals distinct antibody signatures prior to FVIII inhibitor development. Blood Adv. 2020, 4, 5785–5796. [Google Scholar] [CrossRef]
- Oldenburg, J.; Lacroix-Desmazes, S.; Lillicrap, D. Alloantibodies to therapeutic factor VIII in hemophilia A: The role of von Willebrand factor in regulating factor VIII immunogenicity. Haematologica 2015, 100, 149–156. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerra, P.E.; Parker, E.T.; Baldwin, W.H.; Healey, J.F.; Patel, S.R.; McCoy, J.W.; Cox, C.; Stowell, S.R.; Meeks, S.L. Engineering a Therapeutic Protein to Enhance the Study of Anti-Drug Immunity. Biomedicines 2022, 10, 1724. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071724
Zerra PE, Parker ET, Baldwin WH, Healey JF, Patel SR, McCoy JW, Cox C, Stowell SR, Meeks SL. Engineering a Therapeutic Protein to Enhance the Study of Anti-Drug Immunity. Biomedicines. 2022; 10(7):1724. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071724
Chicago/Turabian StyleZerra, Patricia E., Ernest T. Parker, Wallace Hunter Baldwin, John F. Healey, Seema R. Patel, James W. McCoy, Courtney Cox, Sean R. Stowell, and Shannon L. Meeks. 2022. "Engineering a Therapeutic Protein to Enhance the Study of Anti-Drug Immunity" Biomedicines 10, no. 7: 1724. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10071724