
Evaluation of the Anti-Leishmania mexicana and -Trypanosoma brucei Activity and Mode of Action of 4,4′-(Arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)

 , and
, and
Abstract
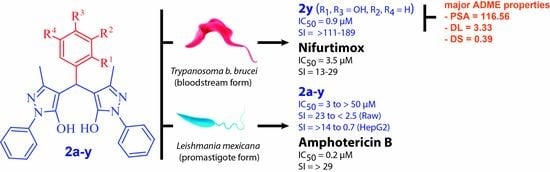
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General
2.1.2. General Synthesis of 4,4′-(Arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ols) 2a-y
2.2. Biological Evaluation
2.2.1. Determination of Drug Likeness and ADME Properties
2.2.2. Cytotoxicity Screening Assays against Leishmania mexicana Promastigotes
2.2.3. Cytotoxicity Screening Assays against Trypanosoma brucei brucei Bloodstream Form
2.2.4. Cytotoxicity Screening Assays against Murine Macrophages and HepG2 Cell Lines
2.2.5. In-Cell Redox Assays with Fluorescent Protein Thiol-Based Redox Biosensor
2.2.6. In Vitro Assays with Fluorescent Protein Thiol-Based Redox Biosensor
3. Results and Discussion
3.1. Compounds Synthesis
3.2. Biological Activity (Potency and Selectivity) against Leishmania mexicana Promastigotes
3.3. Biological Activity (Potency and Selectivity) against Bloodstream Trypanosoma brucei brucei
3.4. Evaluation of Intracellular Redox State of Low-Molecular-Weight Thiols in Infective T. b. brucei Treated with Hit Compounds
3.5. Drug Profiling of Bioactive Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Research priorities for Chagas disease, Human African Trypanosomiasis and Leishmaniasis: Technical report of the TDR Disease Reference Group on Chagas Disease, Human African Trypanosomiasis and Leishmaniasis. In WHO Technical Report Series; WHO Press: Geneve, Switzerland, 2012; Volume 975, pp. 26–29. [Google Scholar]
- World Health Organization. World Health Organization Control of the Leishmaniasis: Report of a meeting of the WHO Expert Committee on the control of Leishmaniases, Geneva, 22–26 March 2010. In WHO Technical Report Series; WHO Press: Geneve, Switzerland, 2010; Volume 949, pp. 54–74. [Google Scholar]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Schultzberg, M.; Ambatsis, M.; Samuelsson, E.-B.; Kristensson, K.; van Meirvenne, N. Spread of Trypanosoma brucei to the nervous system: Early attack on circumventricular organs and sensory ganglia. J. Neurosci. Res. 1988, 21, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Kansiime, F.; Adibaku, S.; Wamboga, C.; Idi, F.; Kato, C.D.; Yamuah, L.; Vaillant, M.; Kioy, D.; Olliaro, P.; Matovu, E. A multicentre, randomised, non-inferiority clinical trial comparing a nifurtimox-eflornithine combination to standard eflornithine monotherapy for late stage Trypanosoma brucei gambiense human African trypanosomiasis in Uganda. Parasit. Vectors 2018, 11, 105. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.-P.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Jamwal, A.; Javed, A.; Bhardwaj, V. A review on pyrazole derivatives of pharmacological potential. J. Pharm. Biosci. 2013, 3, 114–123. [Google Scholar]
- Dewangan, D.; Kumar, T.; Alexander, A.; Nagori, K.; Tripathi, D.K. Tripathi Pyrazole: Their chemistry and pharmacological potentials: A review. Curr. Pharma Res. 2011, 1, 369–377. [Google Scholar]
- Pavlov, P.T.; Goleneva, A.F.; Lesnov, A.E.; Prokhorova, T.S. Biological activity of some pyrazolone derivatives. Pharm. Chem. J. 1998, 32, 370–372. [Google Scholar] [CrossRef]
- Mariappan, G.; Saha, B.; Sutharson, L.; Ankit; Garg, S.; Pandey, L.; Kumar, D. The diverse pharmacological importance of pyrazolone derivatives: A review. J. Pharm. Res. 2010, 3, 2856–2859. [Google Scholar]
- Naim, M.J.; Alam, O.; Nawaz, F.; Alam, J.; Alam, P. Current status of pyrazole and its biological activities. J. Pharm. Bioallied Sci. 2016, 8, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Jitsuiki, D.; Chayama, K.; Yoshizumi, M. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 85–93. [Google Scholar] [CrossRef]
- Cadena-Cruz, J.E.; Guamán-Ortiz, L.M.; Romero-Benavides, J.C.; Bailon-Moscoso, N.; Murillo-Sotomayor, K.E.; Ortiz-Guamán, N.V.; Heredia-Moya, J.; Eduardo, J.; Cruz, C.; Ortiz, L.M.G.; et al. Synthesis of 4,4′-(arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ols) and evaluation of their antioxidant and anticancer activities. BMC Chem. 2021, 15, 38. [Google Scholar] [CrossRef]
- Mahajan, P.S.; Nikam, M.D.; Khedkar, V.; Jha, P.; Badadhe, P.V.; Gill, C.H. An organocatalyzed efficient one-pot synthesis, biological evaluation, and molecular docking studies of 4,4′-(Arylmethylene)bis-(3-methyl-1-phenyl-1H -pyrazol-5-ols). J. Heterocycl. Chem. 2017, 54, 1109–1120. [Google Scholar] [CrossRef]
- Sujatha, K.; Shanthi, G.; Selvam, N.P.; Manoharan, S.; Perumal, P.T.; Rajendran, M. Synthesis and antiviral activity of 4,4′-(arylmethylene)bis(1H-pyrazol-5-ols) against peste des petits ruminant virus (PPRV). Bioorg. Med. Chem. Lett. 2009, 19, 4501–4503. [Google Scholar] [CrossRef]
- Diwan, F.; Shaikh, M.; Farooqui, M. Lemon juice catalyzed efficient one-pot synthesis, antioxidant and antimicrobial evaluation of bispyrazolyl methanes. Chem. Biol. Interface 2018, 8, 255–268. [Google Scholar]
- Bhavanarushi, S.; Kanakaiah, V.; Bharath, G.; Gangagnirao, A.; Vatsala Rani, J. Synthesis and antibacterial activity of 4,4′-(aryl or alkyl methylene)-bis(1H-pyrazol-5-ol) derivatives. Med. Chem. Res. 2014, 23, 158–167. [Google Scholar] [CrossRef]
- Kamble, S.S.; Shankarling, G.S. A unique blend of water, DES and ultrasound for one-pot Knorr Pyrazole synthesis and Knoevenagel-Michael addition reaction. ChemistrySelect 2018, 3, 2032–2036. [Google Scholar] [CrossRef]
- Sharma, G.; Chowdhury, S.; Sinha, S.; Majumder, H.K.; Kumar, S.V. Antileishmanial activity evaluation of bis-lawsone analogs and DNA topoisomerase-I inhibition studies. J. Enzyme Inhib. Med. Chem. 2014, 29, 185–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, F.; Samreen; Ullah, H.; Fakhri, M.I.; Salar, U.; Perveen, S.; Khan, K.M.; Choudhary, M.I. Anti-leishmanial activities of synthetic biscoumarins. J. Chem. Soc. Pakistan 2017, 39, 79–82. [Google Scholar]
- Bharate, S.B.; Bharate, J.B.; Khan, S.I.; Tekwani, B.L.; Jacob, M.R.; Mudududdla, R.; Yadav, R.R.; Singh, B.B.; Sharma, P.R.; Maity, S.; et al. Discovery of 3,3′-diindolylmethanes as potent antileishmanial agents. Eur. J. Med. Chem. 2013, 63, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreiro-Costa, O.; Morales-Noboa, G.; Rojas-Silva, P.; Lara-Barba, E.; Santamaría-Aguirre, J.; Bailón-Moscoso, N.; Romero-Benavides, J.C.; Herrera, A.; Cueva, C.; Ron-Garrido, L.; et al. Synthesis and evaluation of biological activities of bis(spiropyrazolone)cyclopropanes: A potential application against leishmaniasis. Molecules 2021, 26, 4960. [Google Scholar] [CrossRef]
- Hamama, W.S. Pyrazolones as versatile precursors for the synthesis of fused and binary heterocycles. Synth. Commun. 2001, 31, 1335–1345. [Google Scholar] [CrossRef]
- Li, X.-L.; Wang, Y.-M.; Tian, B.; Matsuura, T.; Meng, J.-B. The solid-state Michael addition of 3-methyl-1-phenyl-5-pyrazolone. J. Heterocycl. Chem. 1998, 35, 129–134. [Google Scholar] [CrossRef]
- Moosavi-Zare, A.R.; Zolfigol, M.A.; Noroozizadeh, E.; Khaledian, O.; Shaghasemi, B.S. Cyclocondensation-Knoevenagel–Michael domino reaction of phenyl hydrazine, acetoacetate derivatives and aryl aldehydes over acetic acid functionalized ionic liquid. Res. Chem. Intermed. 2016, 42, 4759–4772. [Google Scholar] [CrossRef]
- Hasaninejed, A.; Kazerooni, M.R.; Zare, A. Room-temperature, catalyst-free, one-pot pseudo-five-component synthesis of 4,4-(arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)s under ultrasonic irradiation. ACS Sustain. Chem. Eng. 2013, 1, 679–684. [Google Scholar] [CrossRef]
- Gouda, M.A. Chemistry of 4,4′-(arylmethylene)-bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)s. J. Heterocycl. Chem. 2015, 53, 356–376. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.K.; Rajput, G.; Srivastava, K.; Singh, R.K.; Mishra, R.; Drew, M.G.B.; Singh, N. Anti-leishmanial activity of Ni(ii), Pd(ii) and Pt(ii) β-oxodithioester complexes. New, J. Chem. 2015, 39, 6358–6366. [Google Scholar] [CrossRef]
- Rodríguez-Gutiérrez, S.V.; Barreiro-Costa, O.; León, C.D.A.; Heredia-Moya, J. Synthesis and leishmanicidal activity of molecular hybrids 1,2,3-triazole-chalcones. Chem. Proc. 2021, 3, 55. [Google Scholar] [CrossRef]
- Benítez, D.; Dibello, E.; Bonilla, M.; Comini, M.A. A simple, robust, and affordable bioluminescent assay for drug discovery against infective African trypanosomes. Drug Dev. Res. 2022, 83, 253–263. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Castera-Ducros, C.; Azas, N.; Verhaeghe, P.; Hutter, S.; Garrigue, P.; Dumètre, A.; Mbatchi, L.; Laget, M.; Remusat, V.; Sifredi, F.; et al. Targeting the human malaria parasite Plasmodium falciparum: In vitro identification of a new antiplasmodial hit in 4-phenoxy-2-trichloromethylquinazoline series. Eur. J. Med. Chem. 2011, 46, 4184–4191. [Google Scholar] [CrossRef]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef]
- Franco, J.; Sardi, F.; Szilágyi, L.; Kövér, K.E.; Fehér, K.; Comini, M.A. Diglycosyl diselenides alter redox homeostasis and glucose consumption of infective African trypanosomes. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 303–313. [Google Scholar] [CrossRef]
- Franco, J.; Medeiros, A.; Benítez, D.; Perelmuter, K.; Serra, G.; Comini, M.A.; Scarone, L. In vitro activity and mode of action of distamycin analogues against African trypanosomes. Eur. J. Med. Chem. 2017, 126, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Ebersoll, S.; Bogacz, M.; Günter, L.M.; Dick, T.P.; Krauth-Siegel, R.L. A tryparedoxin-coupled biosensor reveals a mitochondrial trypanothione metabolism in trypanosomes. Elife 2020, 9, e53227. [Google Scholar] [CrossRef] [PubMed]
- Ortíz, C.; Moraca, F.; Laverriere, M.; Jordan, A.; Hamilton, N.; Comini, M.A. Glucose 6-phosphate dehydrogenase from trypanosomes: Selectivity for steroids and chemical validation in bloodstream Trypanosoma brucei. Molecules 2021, 26, 358. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.; Löfdahl, P.-Å.; Härd, T.; Berglund, H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006, 121, 291–298. [Google Scholar] [CrossRef]
- Fersing, C.; Boudot, C.; Castera-Ducros, C.; Pinault, E.; Hutter, S.; Paoli-Lombardo, R.; Primas, N.; Pedron, J.; Seguy, L.; Bourgeade-Delmas, S.; et al. 8-Alkynyl-3-nitroimidazopyridines display potent antitrypanosomal activity against both T. b. brucei and cruzi. Eur. J. Med. Chem. 2020, 202, 112558. [Google Scholar] [CrossRef]
- Don, R.; Ioset, J.-R. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 2014, 141, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Shahamirian, M.; Szatylowicz, H.; Krygowski, T.M. How OH and O– groups affect electronic structure of meta-substituted and para-substituted phenols and phenolates. Struct. Chem. 2017, 28, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, I.; Tamura, M.; Nakajima, R.; Nakamura, M. Physiological aspects of free-radical reactions. Environ. Health Perspect. 1985, 64, 331–342. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Radical scavenging by thiols and the fate of thiyl radicals. In Oxidative Stress and Redox Regulation; Jakob, U., Reichmann, D., Eds.; Springer: Dordrecht, The Netherlands, 2013; Volume 9789400757, pp. 43–58. ISBN 9789400757875. [Google Scholar]
- Bhaskar, A.; Chawla, M.; Mehta, M.; Parikh, P.; Chandra, P.; Bhave, D.; Kumar, D.; Carroll, K.S.; Singh, A. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 2014, 10, e1003902. [Google Scholar] [CrossRef] [Green Version]
- Loi, V.V.; Harms, M.; Müller, M.; Huyen, N.T.T.; Hamilton, C.J.; Hochgräfe, F.; Pané-Farré, J.; Antelmann, H. Real-time imaging of the bacillithiol redox potential in the human pathogen Staphylococcus aureus using a genetically encoded bacilliredoxin-fused redox biosensor. Antioxid. Redox Signal. 2017, 26, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Manta, B.; Möller, M.N.; Bonilla, M.; Deambrosi, M.; Grunberg, K.; Bellanda, M.; Comini, M.A.; Ferrer-Sueta, G. Kinetic studies reveal a key role of a redox-active glutaredoxin in the evolution of the thiol-redox metabolism of trypanosomatid parasites. J. Biol. Chem. 2019, 294, 3235–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manta, B.; Comini, M.; Medeiros, A.; Hugo, M.; Trujillo, M.; Radi, R. Trypanothione: A unique bis-glutathionyl derivative in trypanosomatids. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3199–3216. [Google Scholar] [CrossRef] [PubMed]
- Polkam, N.; Ramaswamy, V.R.; Rayam, P.; Allaka, T.R.; Anantaraju, H.S.; Dharmarajan, S.; Perumal, Y.; Gandamalla, D.; Yellu, N.R.; Balasubramanian, S.; et al. Synthesis, molecular properties prediction and anticancer, antioxidant evaluation of new edaravone derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 2562–2568. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds a | Mp (°C) | Yield (%) |
|---|---|---|
| 2a | 219.5–220.6 | 98 |
| 2b | 159.5–161.1 | 97 |
| 2c | 210.0–211.0 | 95 |
| 2d | 150.7–152.0 | 95 b |
| 2e | 176.0–177.0 | 92 |
| 2f | 213.2–214.7 | 95 |
| 2g | 188.8–189.6 | 84 d |
| 2h | 205–207 (d) | 93 |
| 2i | 205.7–207.1 | 94 c |
| 2j | 200.0–202.0 | 91 d |
| 2k | 218.0–219.0 | 97 |
| 2l | 165.0–167.0 | 98 d |
| 2m | 208.0–209.5 | 97 c |
| 2n | 182.7–184.0 | 93 c |
| 2o | 217.2–218.9 | 97 d |
| 2p | 183.8–185.8 | 87 |
| 2q | 217.2–218.7 | Quant. |
| 2r | 203.0–205.0 | 96 |
| 2s | 174.5–176.0 | Quant. |
| 2t | 178.0–179.0 | Quant. |
| 2u | 209.1–211.3 | 60 |
| 2v | 245.0–248.0 | 92 |
| 2w | 207.0–209.0 | Quant. |
| 2x | 207.0–209.0 | Quant. |
| 2y | 205.0 | 87 c |
| Compound | L. mexicana Promastigote | CC50 (µM) | |||
|---|---|---|---|---|---|
| IC50 (µM) a | SI vs. Macrophage b | SI vs. Hepatocytes c | RAW Macrophages | HepG2 Hepatocytes | |
| 2a | 12 ± 3 * | 4.3 | >4.8 | 51 ± 6 | >100 |
| 2b | 32 ± 7 **** | 6.4 | >3.1 | 206 ± 28 | >100 |
| 2c | 17 ± 8 **** | 3.1 | 1.5 | 53 ± 6 | 26 ± 16 |
| 2d | 10 ± 3 * | 7.5 | >10 | 75 ± 15 | >100 |
| 2e | > 50 **** | ND | ND | ND | ND |
| 2f | 22 ± 9 **** | 4.2 | >4.5 | 93 ± 7 | >100 |
| 2g | > 50 **** | ND | ND | ND | ND |
| 2h | 21 ± 4 **** | 5.1 | >4.8 | 108 ± 22 | >100 |
| 2i | 10 ± 3 * | 23.2 | >10 | 232 ± 25 | >100 |
| 2j | 10 ± 4 ns | 11.3 | >10 | 113 ± 15 | >100 |
| 2k | 9 ± 1 ns | 13 | 1.7 | 117 ± 15 | 15 ± 6 |
| 2l | 18 ± 5 **** | 6 | >5.6 | 108 ± 33 | >100 |
| 2m | 23 ± 5 **** | 12.4 | >4.3 | 286 ± 51 | >100 |
| 2n | 7 ± 1 ns | 10. | >14.3 | 70 ± 7 | >100 |
| 2o | > 50 **** | ND | ND | ND | ND |
| 2p | 17 ± 1 **** | 2.5 | 0.8 | 43 ± 6 | 13.0 ± 2 |
| 2q | 13 ± 3 *** | 8.5 | 2.2 | 111 ± 2 | 29 ± 4 |
| 2r | 3.3 ± 0.2 ns | 16.4 | 6.1 | 54 ± 3 | 20 ± 4 |
| 2s | 3 ± 1 ns | 14.3 | 8.7 | 49 ± 15 | 26 ± 15 |
| 2t | 11 ± 1 * | 10.5 | 1.8 | 115 ± 21 | 20 ± 11 |
| 2u | 30 ± 10 **** | 3.2 | 0.7 | 96 ± 27 | 21 ± 10 |
| 2v | 6 ± 1 ns | 15.1 | 3.5 | 91 ± 18 | 21 ± 2 |
| 2w | 4 ± 1 ns | 15.2 | 6.5 | 61 ± 4 | 26 ± 3 |
| 2x | 7 ± 2 ns | 4.8 | 4.3 | 34 ± 5 | 30 ± 2 |
| 2y | > 50 **** | ND | ND | 170 ± 1 | >100 |
| Amphotericin B | 0.17 ± 0.05 | 29.4 | ND | >5 | ND |
| Saponin | ND | ND | ND | 0.16 ± 0.03 d | ND |
| Nifurtimox | ND | ND | ND | >100 | 45 ± 1 e |
| Compound | T. brucei bloodstream | |||
|---|---|---|---|---|
| % Growth (10 µM) | IC50 (µM) a | SI vs. Macrophages b | SI vs. Hepatocytes c | |
| 2a | 77 ± 5 | ND | ND | ND |
| 2b | 74 ± 7 | ND | ND | ND |
| 2c | 68 ± 5 | ND | ND | ND |
| 2d | 73 ± 4 | ND | ND | ND |
| 2e | >100 | ND | ND | ND |
| 2f | 57 ± 6 | 8 ± 0.27 *** | 11.6 | >12.5 |
| 2g | >100 | ND | ND | ND |
| 2h | 89 ± 3 | ND | ND | ND |
| 2i | >100 | ND | ND | ND |
| 2j | >100 | ND | ND | ND |
| 2k | 88 ± 4 | ND | ND | ND |
| 2l | >100 | ND | ND | ND |
| 2m | >100 | ND | ND | ND |
| 2n | 54 ± 4 | 10.2 ± 1.75 ** | 7 | >9.8 |
| 2o | >100 | ND | ND | ND |
| 2p | 79 ± 8 | ND | ND | ND |
| 2q | 33 ± 2 | 5.9 ± 0.6 ns | 19 | 4.9 |
| 2r | 49 ± 7 | 10 ± 3 *** | 5.4 | 2.0 |
| 2s | >100 | ND | ND | ND |
| 2t | >100 | ND | ND | ND |
| 2u | >100 | ND | ND | ND |
| 2v | >100 | ND | ND | ND |
| 2w | 46 ± 2 | 8.9 ± 0.7 ** | 6.8 | 2.9 |
| 2x | 26 ± 1 | 1.9 ± 0.9 ns | 18 | 16 |
| 2y | 29 ± 4 | 0.9 ± 0.2 *** | 189 | >111 |
| Nifurtimox | ND | 3.5 | 28.5 | 13 |
| Compounds | MW | HBA | HBD | nrotb | PSA | M | T | RE | I | cLogP | cLogS | DL | DS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2d | 481.51 | 9 | 2 | 6 | 121.92 | none | high | none | none | 2.58 | −4.98 | −1.81 | 0.19 |
| 2i | 482.54 | 8 | 3 | 6 | 105.56 | none | high | none | none | 3.09 | −4.25 | 3.30 | 0.36 |
| 2j | 482.54 | 8 | 3 | 6 | 105.56 | none | high | none | none | 3.09 | −4.25 | 3.30 | 0.36 |
| 2k | 481.51 | 9 | 2 | 6 | 121.92 | none | high | none | none | 2.58 | −4.98 | −1.81 | 0.19 |
| 2n | 468.51 | 8 | 4 | 5 | 116.56 | none | high | none | none | 2.81 | −3.93 | 3.33 | 0.39 |
| 2q | 494.55 | 8 | 2 | 7 | 102.40 | none | high | none | none | 3.41 | −4.67 | 0.99 | 0.28 |
| 2r | 504.51 | 6 | 2 | 6 | 76.10 | none | high | none | none | 4.35 | −5.30 | −3.86 | 0.13 |
| 2s | 520.51 | 7 | 2 | 7 | 85.33 | none | high | none | none | 4.60 | −5.55 | −4.75 | 0.12 |
| 2v | 504.51 | 6 | 2 | 6 | 76.10 | none | high | none | none | 4.35 | −5.30 | −3.86 | 0.13 |
| 2w | 504.51 | 6 | 2 | 6 | 76.10 | none | high | none | none | 4.35 | −5.30 | −3.86 | 0.13 |
| 2x | 572.51 | 6 | 2 | 7 | 76.10 | none | high | none | none | 5.20 | −6.08 | −3.86 | 0.09 |
| 2y | 468.51 | 8 | 4 | 5 | 116.56 | none | high | none | none | 2.81 | −3.93 | 3.33 | 0.39 |
| Amphotericin B | 924.09 | 18 | 12 | 3 | 319.61 | none | none | none | none | 0.32 | −5.08 | −0.14 | 0.27 |
| Saponin | 414.63 | 3 | 1 | 0 | 38.69 | none | none | low | none | 4.88 | −5.58 | 0.84 | 0.31 |
| Nifurtimox | 287.30 | 8 | 0 | 3 | 117.08 | high | high | high | none | −0.25 | −3.01 | 0.75 | 0.16 |
| Compound | GPCR a | ICM | KI | NRL | PI | EI |
|---|---|---|---|---|---|---|
| 2d | −0.33 | −0.23 | −0.51 | −0.39 | −0.44 | −0.25 |
| 2i | −0.23 | −0.23 | −0.42 | −0.32 | −0.40 | −0.17 |
| 2j | −0.23 | −0.23 | −0.42 | −0.32 | −0.40 | −0.17 |
| 2k | −0.32 | −0.22 | −0.51 | −0.38 | −0.44 | −0.24 |
| 2n | −0.21 | −0.16 | −0.42 | −0.27 | −0.36 | −0.15 |
| 2q | −0.29 | −0.28 | −0.48 | −0.33 | −0.39 | −0.21 |
| 2r | −0.16 | −0.14 | −0.36 | −0.20 | −0.30 | −0.15 |
| 2s | −0.17 | −0.17 | −0.41 | −0.15 | −0.25 | −0.17 |
| 2v | −0.19 | −0.14 | −0.31 | −0.14 | −0.31 | −0.15 |
| 2w | −0.16 | −0.15 | −0.36 | −0.19 | −0.30 | −0.15 |
| 2x | −0.17 | −0.37 | −0.42 | −0.28 | −0.27 | −0.25 |
| 2y | −0.26 | −0.20 | −0.38 | −0.24 | −0.38 | −0.14 |
| Amphotericin B | −3.06 | −3.51 | −3.54 | −3.45 | −2.45 | −2.95 |
| Nifurtimox | −0.93 | −1.40 | −0.73 | −1.61 | −0.81 | −0.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreiro-Costa, O.; Quiroga Lozano, C.; Muñoz, E.; Rojas-Silva, P.; Medeiros, A.; Comini, M.A.; Heredia-Moya, J. Evaluation of the Anti-Leishmania mexicana and -Trypanosoma brucei Activity and Mode of Action of 4,4′-(Arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol). Biomedicines 2022, 10, 1913. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081913
Barreiro-Costa O, Quiroga Lozano C, Muñoz E, Rojas-Silva P, Medeiros A, Comini MA, Heredia-Moya J. Evaluation of the Anti-Leishmania mexicana and -Trypanosoma brucei Activity and Mode of Action of 4,4′-(Arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol). Biomedicines. 2022; 10(8):1913. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081913
Chicago/Turabian StyleBarreiro-Costa, Olalla, Cristina Quiroga Lozano, Erika Muñoz, Patricio Rojas-Silva, Andrea Medeiros, Marcelo A. Comini, and Jorge Heredia-Moya. 2022. "Evaluation of the Anti-Leishmania mexicana and -Trypanosoma brucei Activity and Mode of Action of 4,4′-(Arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)" Biomedicines 10, no. 8: 1913. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10081913