
Formal Meta-Analysis of Hypoxic Gene Expression Profiles Reveals a Universal Gene Signature

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-seq Data Download and Processing
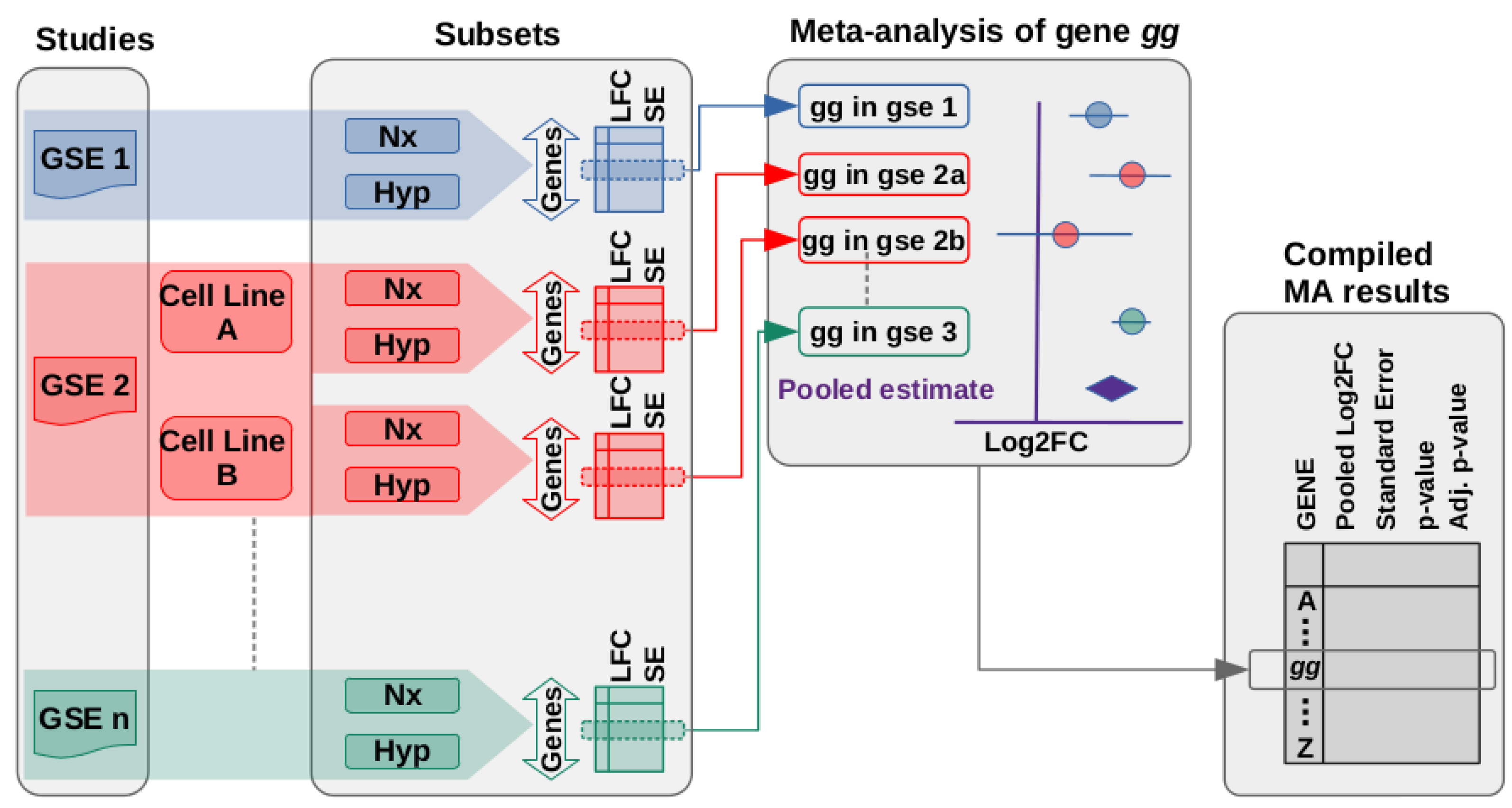
2.2. Meta-Analysis
2.3. Functional Enrichment Analysis
3. Results
3.1. Hypoxia-Induced Transcriptional Profiles Show Limited Overlap
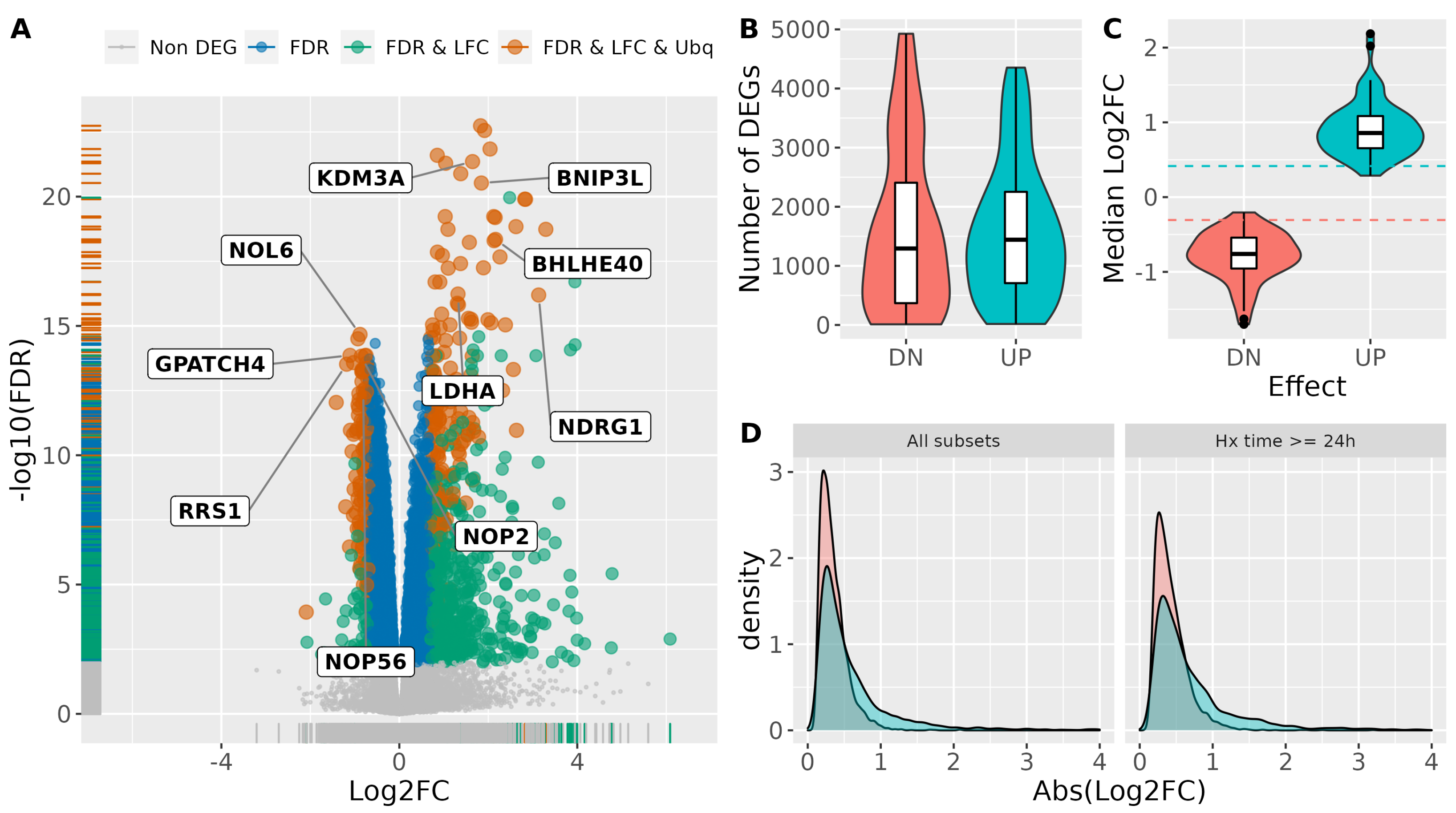
3.2. Identification of Robust Transcriptional Responses to Hypoxia
3.3. Identification of a Universal Core of Hypoxia-Inducible Genes
3.4. Consistency of Meta-Analysis Results
3.5. Comparison of Meta-Analyses Results with a Reference Hypoxia Signature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG(s) | Differentially expressed gene(s) |
References
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Kasper, L.H.; Boussouar, F.; Boyd, K.; Xu, W.; Biesen, M.; Rehg, J.; Baudino, T.A.; Cleveland, J.L.; Brindle, P.K. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005, 24, 3846–3858. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; del Peso, L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010, 38, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef]
- Hong, F.; Breitling, R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics 2008, 24, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Makinde, F.L.; Tchamga, M.S.S.; Jafali, J.; Fatumo, S.; Chimusa, E.R.; Mulder, N.; Mazandu, G.K. Reviewing and assessing existing meta-analysis models and tools. Brief. Bioinform. 2021, 22, bbab324. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-Tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef]
- Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Balduzzi, S.; Rücker, G.; Schwarzer, G. How to perform a meta-analysis with R: A practical tutorial. Evid.-Based Ment. Health 2019, 22, 153–160. [Google Scholar] [CrossRef]
- Harrer, M.; Cuijpers, P.; Furukawa, T.A.; Ebert, D.D. Doing Meta-Analysis with R: A Hands-On Guide, 1st ed.; Chapman and Hall/CRC Press: Boca Raton, FL, USA; London, UK, 2021. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.; del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Tiana, M.; Acosta-Iborra, B.; Hernandez, R.; Galiana, C.; Fernandez-Moreno, M.A.; Jimenez, B.; del Peso, L. Metabolic labeling of RNA uncovers the contribution of transcription and decay rates on hypoxia-induced changes in RNA levels. RNA 2020, 26, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Warren, D.; Spencer, F.; Kim, I.F.; Biswal, S.; Frank, B.C.; Gabrielson, E.; Garcia, J.G.; Geoghegan, J.; Germino, G.; et al. Multiple-laboratory comparison of microarray platforms. Nat. Methods 2005, 2, 345–350. [Google Scholar] [CrossRef]
- Kuo, W.P.; Jenssen, T.K.; Butte, A.J.; Ohno-Machado, L.; Kohane, I.S. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics 2002, 18, 405–412. [Google Scholar] [CrossRef]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef]
- Batie, M.; Del Peso, L.; Rocha, S. Hypoxia and Chromatin: A Focus on Transcriptional Repression Mechanisms. Biomedicines 2018, 6, 47. [Google Scholar] [CrossRef]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Dunn, L.L.; Kong, S.M.; Tumanov, S.; Chen, W.; Cantley, J.; Ayer, A.; Maghzal, G.J.; Midwinter, R.G.; Chan, K.H.; Ng, M.K.; et al. Hmox1 (Heme Oxygenase-1) Protects against Ischemia-Mediated Injury via Stabilization of HIF-1α (Hypoxia-Inducible Factor-1α). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 317–330. [Google Scholar] [CrossRef]
- Fan, J.; Lv, H.; Li, J.; Che, Y.; Xu, B.; Tao, Z.; Jiang, W. Roles of Nrf2/HO-1 and HIF-1α/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 7695–7707. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, B.; Ren, Q. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1α/BNIP3 pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3657–3663. [Google Scholar] [CrossRef]
- Chen, D.; Wu, Y.X.; Qiu, Y.B.; Wan, B.B.; Liu, G.; Chen, J.L.; Lu, M.D.; Pang, Q.F. Hyperoside suppresses hypoxia-induced A549 survival and proliferation through ferrous accumulation via AMPK/HO-1 axis. Phytomedicine 2020, 67, 153138. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Han, F.; Li, B.; Takeda, K. Hypoxia and heme oxygenases: Oxygen sensing and regulation of expression. Antioxid. Redox Signal. 2007, 9, 2209–2225. [Google Scholar] [CrossRef]
- Zhao, Y.Z.; Liu, X.L.; Shen, G.M.; Ma, Y.N.; Zhang, F.L.; Chen, M.T.; Zhao, H.L.; Yu, J.; Zhang, J.W. Hypoxia induces peroxisome proliferator-activated receptor γ expression via HIF-1-dependent mechanisms in HepG2 cell line. Arch. Biochem. Biophys. 2014, 543, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Itoigawa, Y.; Kishimoto, K.N.; Okuno, H.; Sano, H.; Kaneko, K.; Itoi, E. Hypoxia induces adipogenic differentitation of myoblastic cell lines. Biochem. Biophys. Res. Commun. 2010, 399, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1α-PPARγ Axis Underlies the Integration of Glycolytic and Lipid Anabolic Pathways in Pathologic Cardiac Hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiang, Q.; Lin, G.; Fu, X.; Zhou, K.; Jiang, P.; Zheng, S.; Wang, T. Estrogen improved metabolic syndrome through down-regulation of VEGF and HIF-1α to inhibit hypoxia of periaortic and intra-abdominal fat in ovariectomized female rats. Mol. Biol. Rep. 2012, 39, 8177–8185. [Google Scholar] [CrossRef] [PubMed]
- Ezzeddini, R.; Taghikhani, M.; Amir, S.F.; Somi, M.H.; Samadi, N.; Esfahani, A.; Rasaee, M.J. Downregulation of fatty acid oxidation by involvement of HIF-1α and PPARγ in human gastric adenocarcinoma and related clinical significance. J. Physiol. Biochem. 2021, 77, 249–260. [Google Scholar] [CrossRef]
- Lane, S.L.; Blair Dodson, R.; Doyle, A.S.; Park, H.; Rathi, H.; Matarrazo, C.J.; Moore, L.G.; Lorca, R.A.; Wolfson, G.H.; Julian, C.G. Pharmacological activation of peroxisome proliferator-activated receptor γ (PPAR-γ) protects against hypoxia-associated fetal growth restriction. FASEB J. 2019, 33, 8999–9007. [Google Scholar] [CrossRef]
- Ameshima, S.; Golpon, H.; Cool, C.D.; Chan, D.; Vandivier, R.W.; Gardai, S.J.; Wick, M.; Nemenoff, R.A.; Geraci, M.W.; Voelkel, N.F. Peroxisome proliferator-activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 2003, 92, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Studies | Samples | Subsets | Cell Lines |
|---|---|---|---|---|
| Initial | 46 | 472 | 81 | 38 |
| Filtered | 43 | 430 | 70 | 34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puente-Santamaria, L.; Sanchez-Gonzalez, L.; Pescador, N.; Martinez-Costa, O.; Ramos-Ruiz, R.; del Peso, L. Formal Meta-Analysis of Hypoxic Gene Expression Profiles Reveals a Universal Gene Signature. Biomedicines 2022, 10, 2229. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10092229
Puente-Santamaria L, Sanchez-Gonzalez L, Pescador N, Martinez-Costa O, Ramos-Ruiz R, del Peso L. Formal Meta-Analysis of Hypoxic Gene Expression Profiles Reveals a Universal Gene Signature. Biomedicines. 2022; 10(9):2229. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10092229
Chicago/Turabian StylePuente-Santamaria, Laura, Lucia Sanchez-Gonzalez, Nuria Pescador, Oscar Martinez-Costa, Ricardo Ramos-Ruiz, and Luis del Peso. 2022. "Formal Meta-Analysis of Hypoxic Gene Expression Profiles Reveals a Universal Gene Signature" Biomedicines 10, no. 9: 2229. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10092229