
Measurable Residual Disease and Clonal Evolution in Acute Myeloid Leukemia from Diagnosis to Post-Transplant Follow-Up: The Role of Next-Generation Sequencing

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Acute Myeloid Leukemia
3. Next-Generation Sequencing Platforms as Used in Hematology
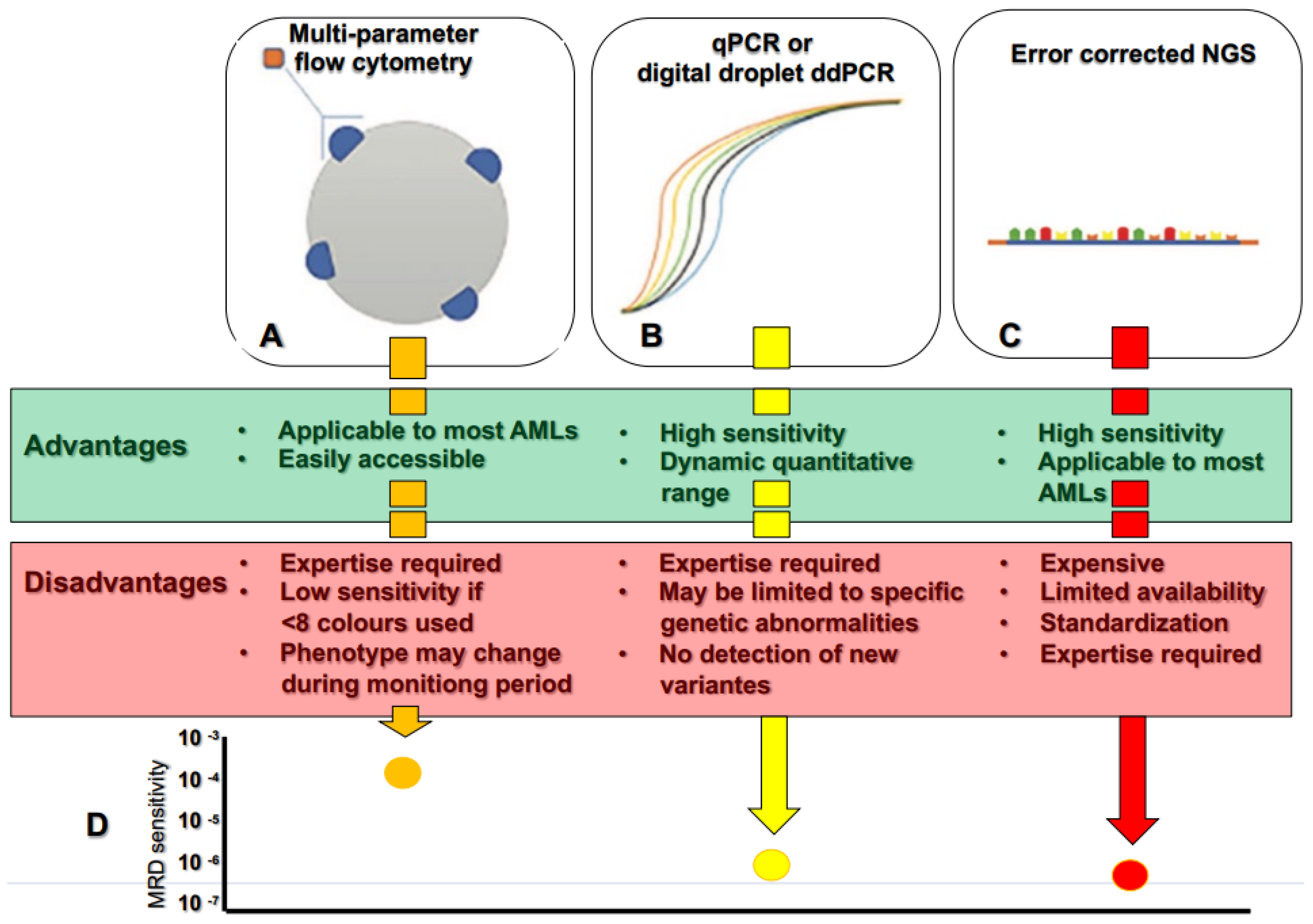
4. Measurable Residual Disease: Next-Generation Sequencing Vs. Conventional Techniques
5. Relevant Timepoints for Next-Generation Sequencing Measurable Residual Disease Monitoring
5.1. Chemotherapy Setting: Induction—Consolidation Phase
5.2. Allogeneic Hematopoietic Stem Cell Transplant Setting
5.2.1. Pre-Transplant Mutational Status
Pre-Transplant Mutational Status and Conditioning Regimen
5.2.2. Post-Transplant Mutational Status
6. Clonal Evolution Detected by Next-Generation Sequencing: From Diagnosis to Post-Transplant Relapse
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 20, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, P.; Zhang, Y.; Tang, R.; Joulin, V.; Boutroux, H.; Pronier, E.; Moatti, H.; Flandrin, P.; Marzac, C.; Bories, D.; et al. Genetic Hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat. Commun. 2016, 7, 12475. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelli, F.; Vignetti, M.; Suciu, S.; Stasi, R.; Petti, M.-C.; Meloni, G.; Muus, P.; Marmont, F.; Marie, J.-P.; LaBar, B.; et al. Daunorubicin Versus Mitoxantrone Versus Idarubicin as Induction and Consolidation Chemotherapy for Adults with Acute Myeloid Leukemia: The EORTC and GIMEMA Groups Study AML-10. J. Clin. Oncol. 2009, 27, 5397–5403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.B.; Othus, M.; Burnett, A.K.; Lowenberg, B.; Kantarjian, H.M.; Ossenkoppele, G.J.; Hills, R.K.; Ravandi, F.; Pabst, T.; Evans, A.; et al. Resistance prediction in AML: Analysis of 4601 patients from MRC/NCRI, HOVON/SAKK, SWOG and MD Anderson Cancer Center. Leukemia 2015, 29, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Toren, A.; Rechavi, G.; Nagler, A. Minimal Residual Disease Post-Bone Marrow Transplantation for Hemato-Oncological Diseases. Stem Cells 1996, 14, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Vashi, N.; Faham, M.; Carlton, V.; Kong, K.; Buño, I.; Zheng, J.; Moorhead, M.; Klinger, M.; Zhang, B.; et al. Immunoglobulin and T Cell Receptor Gene High-Throughput Sequencing Quantifies Minimal Residual Disease in Acute Lymphoblastic Leukemia and Predicts Post-Transplantation Relapse and Survival. Biol. Blood Marrow Transplant. 2014, 20, 1307–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncavage, E.J.; Tandon, B. The utility of next-generation sequencing in diagnosis and monitoring of acute myeloid leukemia and myelodysplastic syndromes. Int. J. Lab. Hematol. 2015, 37, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Freeman, S.D. Defining minimal residual disease in acute myeloid leukemia: Which platforms are ready for “prime time”? Blood 2014, 124, 3345–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salipante, S.J.; Fromm, J.R.; Shendure, J.; Wood, B.L.; Wu, D. Detection of minimal residual disease in NPM1-mutated acute myeloid leukemia by next-generation sequencing. Mod. Pathol. 2014, 27, 1438–1446. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Mrozek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef] [Green Version]
- Slovak, M.L.; Kopecky, K.J.; Cassileth, P.A.; Harrington, D.H.; Theil, K.S.; Mohamed, A.; Paietta, E.; Willman, C.L.; Head, D.R.; Rowe, J.M.; et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology Group study. Blood 2000, 96, 4075–4083. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solè, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, P.; Pleyer, L. Next Generation Sequencing in AML-on the way to becoming a new standard for treatment initiation and/or modulation? Cancer 2019, 11, 252. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Shumilov, E.; Joncourt, R.; Porret, N.; Tchinda, J.; Legros, M.; Scarpelli, I.; Hewer, E.; Novak, U.; Schoumans, J.; et al. Detection of rare reciprocal RUNX1 rearrangements by next-generation sequencing in acute myeloid leukemia. Genes Chromosom. Cancer 2019, 59, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Nadarajah, N.; Haferlach, T.; Dicker, F.; Kern, W.; Meggendorfer, M.; Haferlach, C. Detection of recurrent and of novel fusion transcripts in myeloid malignancies by targeted RNA sequencing. Leukemia 2018, 32, 1229–1238. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Roloff, G.W.; Lai, C.; Hourigan, C.S.; Dillon, L.W. Technical Advances in the Measurement of Residual Disease in Acute Myeloid Leukemia. J. Clin. Med. 2017, 6, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowgey, E.L.; Mahajan, N.; Wong, W.H.; Gopalakrishnapillai, A.; Barwe, S.P.; Kolb, E.A.; Druley, T.E. Error-corrected sequencing strategies enable comprehensive detection of leukemic mutations relevant for diagnosis and minimal residual disease monitoring. BMC Med. Genom. 2020, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Young, A.L.; Wong, T.N.; Hughes, A.E.O.; Heath, S.E.; Ley, T.J.; Link, D.C.; Druley, T.E. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia 2015, 29, 1608–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.A.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levis, M.J.; Perl, A.E.; Altman, J.K.; Gocke, C.D.; Bahceci, E.; Hill, J.; Liu, C.; Xie, Z.; Carson, A.R.; McClain, V.; et al. A next-generation sequencing–based assay for minimal residual disease assessment in AML patients with FLT3-ITD mutations. Blood Adv. 2018, 2, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of Somatic Mutations at Remission and the Risk of Relapse in Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Ofran, Y.; Wierzbowska, A.; Ravandi, F.; Hourigan, C.S.; Ngai, L.M.; Venditti, A.; Buccisano, F.; Ossenkoppele, G.J.; Roboz, G.J. Measurable residual disease as a biomarker in acute myeloid leukemia: Theoretical and practical considerations. Leukemia 2021, 35, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hokland, P.; Ommen, H.B. Towards individualized follow-up in adult acute myeloid leukemia in remission. Blood 2011, 117, 2577–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, R.; Potter, N.; Freeman, S.; Russell, N. How we use molecular minimal residual disease (MRD) testing in acute myeloid leukaemia (AML). Br. J. Haematol. 2020, 193, 231–244. [Google Scholar] [CrossRef]
- Shimoni, A.; Nagler, A. Clinical implications of minimal residual disease monitoring for stem cell transplantation after reduced intensity and nonmyeloablative conditioning. Acta Haematol. 2004, 112, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNET MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef] [PubMed]
- Hasserjian, R.P.; Steensma, D.P.; Graubert, T.A.; Ebert, B.L. Clonal hematopoiesis and measurable residual disease assessment in acute myeloid leukemia. Blood 2020, 135, 1729–1738. [Google Scholar] [CrossRef]
- Terwijn, M.; van Putten, W.L.J.; Kelder, A.; van der Velden, V.H.J.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.E.; Valk, P.J.M.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: Data from the HOVON/SAKK AML 42A study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Freeman, S.D.; Hills, R.K.; Virgo, P.; Khan, N.; Couzens, S.; Dillon, R.; Gilkes, A.; Upton, L.; Nielsen, O.J.; Cavenagh, J.D.; et al. Measurable residual disease at induction redefines partial response in acute myeloid leukemia and stratifies outcomes in patients at standard risk without NPM1 mutations. J. Clin. Oncol. 2018, 36, 1486–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, A.; Piciocchi, A.; Candoni, A.; Melillo, L.; Calafiore, V.; Cairoli, R.; de Fabritiis, P.; Storti, G.; Salutari, P.; Lanza, F.; et al. GIMEMA AML1310 trial of risk-adapted, MRD-directed therapy for young adults with newly diagnosed acute myeloid leukemia. Blood 2019, 134, 935–945. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Ghannam, J.; Dillon, L.W.; Hourigan, C.S. Next–generation sequencing for measurable residual disease detection in acute myeloid leukaemia. Br. J. Haematol. 2019, 188, 77–85. [Google Scholar] [CrossRef]
- Heuser, M.; Heida, B.; Buttner, K.; Wienecke, C.P.; Teich, K.; Funke, C.; Brandes, M.; Klement, P.; Liebich, A.; Wichmann, M.; et al. Posttransplantation MRD monitoring in patients with AML by next-generation sequencing using DTA and non-DTA mutations. Blood Adv. 2021, 5, 2294–2304. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs CHOP mutations in AML with NPM1 alteration. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes Chromosom. Cancer 2012, 51, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Abel, H.J.; Lockwood, C.; Payton, J.E.; Szankasi, P.; Kelley, T.W.; Kulkarni, S.; Pfeifer, J.D.; Duncavage, E.J. Detection of FLT3 Internal Tandem Duplication in Targeted, Short-Read-Length, Next-Generation Sequencing Data. J. Mol. Diagn. 2013, 15, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 2015, 314, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Onecha, E.; Linares, M.; Rapado, I.; Ruiz-Heredia, Y.; Martinez-Sanchez, P.; Cedena, T.; Pratcorona, M.; DE Oteyza, J.P.; Herrera, P.; Barragan, E.; et al. A novel deep targeted sequencing method for minimal residual disease monitoring in acute myeloid leukemia. Haematologica 2019, 104, 288–296. [Google Scholar] [CrossRef]
- Delsing Malmberg, E.; Rehammar, A.; Pereira, M.B.; Abrahammasson, J.; Samuelsson, T.; Stahlman, S.; Asp, J.; Tierens, A.; Palmqvist, L.; Kristiansson, E.; et al. Accurate and sensitive analysis of minimal residual disease in acute myeloid leukemia using deep sequencing of single nucleotide variations. J. Mol. Diagn. 2019, 21, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Shumilov, E.; Flach, J.; Joncourt, R.; Porret, N.; Wiedemann, G.; Angelillo-Scherrer, A.; Trumper, L.; Fiedler, M.; Jeker, B.; Amstutz, U.; et al. Critical evaluation of current molecular MRD strategies including NGS for the management of AML patients with multiple mutations. Hematol. Oncol. 2019, 37, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Gooley, T.A.; Chien, J.W.; Pergam, S.A.; Hingorani, S.; Sorror, M.L.; Boeckh, M.; Martin, P.J.; Sandmaier, B.M.; Marr, K.A.; Appelbaum, F.R.; et al. Reduced mortality after allogeneic hematopoietic-cell transplantation. N. Engl. J. Med. 2010, 363, 2091–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, G.B.; Sandmaier, B.M.; Mielcarek, M.; Sorror, M.; Pergam, S.A.; Cheng, G.-S.; Hingorani, S.; Boeckh, M.; Flowers, M.D.; Lee, S.J.; et al. Survival, nonrelapse mortality, and relapse-related mortality after allogeneic hematopoietic cell transplantation: Comparing 2003–2007 versus 2013–2017 cohorts. Ann. Intern. Med. 2020, 172, 229–239. [Google Scholar] [CrossRef]
- Hourigan, C.S.; Karp, J.E. Minimal residual disease in acute myeloid leukemia. Nat. Rev. Clin. Oncol. 2013, 10, 460–471. [Google Scholar] [CrossRef]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: Is it time to move toward a minimal residual disease-based definition of complete remission? Blood 2015, 126, 2571. [Google Scholar] [CrossRef]
- Buckley, S.A.; Wood, B.L.; Othus, M.; Hourigan, C.S.; Ustun, C.; Linden, M.A.; DeFor, T.E.; Malagola, M.; Anthias, C.; Valkova, V.; et al. Minimal residual disease prior to allogeneic hematopoietic cell transplantation in acute myeloid leukemia: A meta-analysis. Haematologica 2017, 102, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hourigan, C.S.; Gale, R.P.; Gormley, N.J.; Ossenkoppele, G.J.; Walter, R.B. Measurable residual disease testing in acute myeloid leukaemia. Leukemia 2017, 31, 1482–1490. [Google Scholar] [CrossRef]
- Hourigan, C.S.; Goswami, M.; Battiwalla, M.; Barrett, A.J.; Sheela, S.; Karp, J.E.; Lai, C. When the Minimal Becomes Measurable. J. Clin. Oncol. 2016, 34, 2557–2558. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Grimwade, D.; Jovanovic, J.V.; Hills, R.K.; Nugent, E.A.; Patel, Y.; Flora, R.; Diverio, D.; Jones, K.; Aslett, H.; Batson, E.; et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J. Clin. Oncol. 2009, 27, 3650–3658. [Google Scholar] [CrossRef]
- Getta, B.M.; Devlin, S.M.; Levine, R.L.; Arcila, M.E.; Mohanty, A.S.; Zehir, A.; Tallman, M.S.; Giralt, S.A.; Roshal, M. Multicolor Flow Cytometry and Multigene Next-Generation Sequencing Are Complementary and Highly Predictive for Relapse in Acute Myeloid Leukemia after Allogeneic Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Luskin, M.R.; Carroll, M.; Lieberman, D.; Morrissette, J.J.; Zhao, J.; Crisalli, L.; Roth, D.B.; Luger, S.M.; Porter, D.L.; Reshef, R. Clinical Utility of Next-Generation Sequencing for Oncogenic Mutations in Patients with Acute Myeloid Leukemia Undergoing Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1961–1967. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Köhnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksienzyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2018, 32, 1598–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.-S.; Kim, T.; Jung, S.-H.; Ahn, S.-Y.; Jung, S.-Y.; Song, G.-Y.; Kim, M.; Yang, D.-H.; Lee, J.-J.; Choi, S.; et al. Allogeneic transplant can abrogate the risk of relapse in the patients of first remission acute myeloid leukemia with detectable measurable residual disease by next-generation sequencing. Bone Marrow Transplant. 2021, 56, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Hourigan, C.S.; Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; et al. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease. J. Clin. Oncol. 2020, 12, 1273. [Google Scholar] [CrossRef]
- Diez-Campelo, M.; Perez-Simon, J.A.; Perez, J.; Alcoceba, M.; Richtmon, J.; Vidriales, B.; San Miguel, J. Minimal residual disease monitoring after allogeneic transplantation may help to individualize post-transplant therapeutic strategies in acute myeloid malignancies. Am. J. Hematol. 2008, 84, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Fujita, H.; Fujimaki, K.; Hosoyama, T.; Watanabe, R.; Tachibana, T.; Fujita, A.; Matsumoto, K.; Tanaka, M.; Koharazawa, H.; et al. Clinical significance of minimal residual disease detected by multidimensional flow cytometry: Serial monitoring after allogeneic stem cell transplantation for acute leukemia. Leuk. Res. 2012, 36, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Anthias, C.; Dignan, F.L.; Morilla, R.; Morilla, A.; Ethell, M.E.; Potter, M.N.; Shaw, B.E. Pre-transplant MRD predicts outcome following reduced-intensity and myeloablative allogeneic hemopoietic SCT in AML. Bone Marrow Transplant. 2014, 49, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, C.J.; Kennedy, J.A.; Nikiforow, S.; Kuo, F.C.; Alyea, E.P.; Ho, V.; Ritz, J.; Soiffer, R.; Antin, J.H.; Lindsley, R.C. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood 2017, 130, 91–94. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Moon, J.H.; Ahn, J.-S.; Kim, Y.-K.; Lee, S.-S.; Ahn, S.-Y.; Jung, S.-H.; Yang, D.-H.; Lee, J.-J.; Choi, S.H.; et al. Next-generation sequencing–based posttransplant monitoring of acute myeloid leukemia identifies patients at high risk of relapse. Blood 2018, 132, 1604–1613. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, B.K.; Rybicki, L.; Hirsch, C.; Przychodzen, B.; Nazha, A.; Gerds, A.T.; Hanna, R.; Kalaycio, M.; Sekeres, M.A.; Sobecks, R.; et al. Mutation clonal burden and allogeneic hematopoietic cell transplantation outcomes in acute myeloid leukemia and myelodysplastic syndromes. Bone Marrow Transplant. 2019, 54, 1281–1286. [Google Scholar] [CrossRef]
- Clarke, J.R.S.; Douglas, L.R.; Duriez, P.J.; Balourdas, D.-I.; Joerger, A.C.; Khadiullina, R.; Bulatov, E.; Baud, M.G.J. Discovery of Nanomolar-Affinity Pharmacological Chaperones Stabilizing the Oncogenic p53 Mutant Y220C. ACS Pharmacol. Transl. Sci. 2022, 5, 1169–1180. [Google Scholar] [CrossRef]
- Wang, S.; Hu, S.; Luo, X.; Bao, X.; Li, J.; Liu, M.; Lv, M.; Zhao, C.; Zeng, M.; Chen, X.; et al. Prevalence and prognostic significance of DNMT3A- and TET2- clonal haematopoiesis-driver mutations in patients presenting with ST-segment elevation myocardial infarction. EBioMedicine 2022, 78, 103964. [Google Scholar] [CrossRef]
- Jawad, M.; Afkhami, M.; Ding, Y.; Zhang, X.; Li, P.; Young, K.; Xu, M.L.; Cui, W.; Zhao, Y.; Halene, S.; et al. DNMT3A R882 Mutations Confer Unique Clinicopathologic Features in MDS Including a High Risk of AML Transformation. Front. Oncol. 2022, 12, 849376. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brune, B.; Wagner, S.; Serve, H.; et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood–cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Kim, Y.; Kang, D.; Kim, H.S.; Lee, J.-M.; Kim, M.; Cho, B.-S. Prognostic value of measurable residual disease monitoring by next-generation sequencing before and after allogeneic hematopoietic cell transplantation in acute myeloid leukemia. Blood Cancer J. 2021, 11, 109. [Google Scholar] [CrossRef]
- Loke, J.; Vyas, H.; Craddock, C. Optimizing transplant approaches and post-transplant strategies for patients with acute myeloid leukemia. Front Oncol. 2021, 11, 666091. [Google Scholar] [CrossRef] [PubMed]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Hellmann, I.; Metzeler, K.H.; Herold, T.; Bamopoulos, S.A.; Kerbs, P.; et al. Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: An exome sequencing study of 50 patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quek, L.; Ferguson, P.; Metzner, M.; Ahmed, I.; Kennedy, A.; Garnett, C.; Jeffries, S.; Walter, C.; Piechocki, K.; Timbs, A.; et al. Mutational analysis of disease relapse in patients allografted for acute myeloid leukemia. Blood Adv. 2016, 1, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Yoshida, K.; Kim, Y.K.; Tyndel, M.S.; Park, H.J.; Choi, S.H.; Ahn, J.-S.; Jung, S.-H.; Yang, D.-H.; Lee, J.-J.; et al. Clonal dynamics in a single AML case tracked for 9 years reveals the complexity of leukemia progression. Leukemia 2016, 30, 295–302. [Google Scholar] [CrossRef]
- Ommen, H.B. Monitoring minimal residual disease in acute myeloid leukaemia: A review of the current evolving strategies. Ther. Adv. Hematol. 2016, 7, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Durruthy-Durruthy, R.; Eastburn, D.J.; Pellegrino, M.; Shah, O.; Meyer, E.; Zehnder, J. Clonal evolution and changes in two AML patients detected with a novel single-cell DNA sequencing platform. Sci. Rep. 2019, 9, 11119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ediriwickrema, A.; Aleshin, A.; Reiter, J.G.; Corces, M.R.; Köhnke, T.; Stafford, M.; Liedtke, M.; Medeiros, B.C.; Majeti, R. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020, 4, 943–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sperotto, A.; Bochicchio, M.T.; Simonetti, G.; Buccisano, F.; Peccatori, J.; Piemontese, S.; Calistri, E.; Ciotti, G.; Pierdomenico, E.; De Marchi, R.; et al. Measurable Residual Disease and Clonal Evolution in Acute Myeloid Leukemia from Diagnosis to Post-Transplant Follow-Up: The Role of Next-Generation Sequencing. Biomedicines 2023, 11, 359. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020359
Sperotto A, Bochicchio MT, Simonetti G, Buccisano F, Peccatori J, Piemontese S, Calistri E, Ciotti G, Pierdomenico E, De Marchi R, et al. Measurable Residual Disease and Clonal Evolution in Acute Myeloid Leukemia from Diagnosis to Post-Transplant Follow-Up: The Role of Next-Generation Sequencing. Biomedicines. 2023; 11(2):359. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020359
Chicago/Turabian StyleSperotto, Alessandra, Maria Teresa Bochicchio, Giorgia Simonetti, Francesco Buccisano, Jacopo Peccatori, Simona Piemontese, Elisabetta Calistri, Giulia Ciotti, Elisabetta Pierdomenico, Roberta De Marchi, and et al. 2023. "Measurable Residual Disease and Clonal Evolution in Acute Myeloid Leukemia from Diagnosis to Post-Transplant Follow-Up: The Role of Next-Generation Sequencing" Biomedicines 11, no. 2: 359. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020359