
Multi-Omic Profiling, Structural Characterization, and Potent Inhibitor Screening of Evasion-Related Proteins of a Parasitic Nematode, Haemonchus contortus, Surviving Vaccine Treatment


Abstract
:1. Introduction
2. Results and Discussion
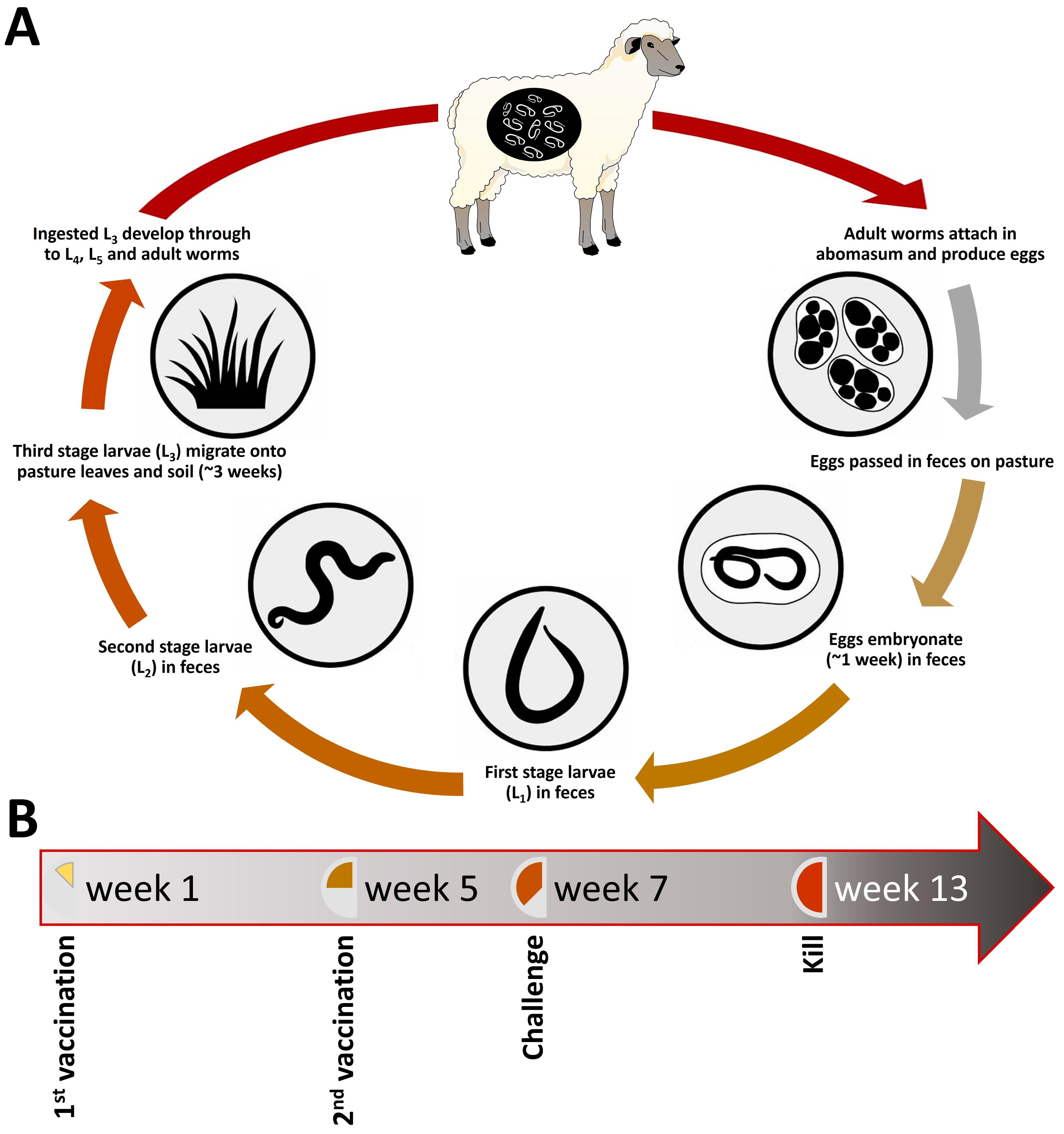
2.1. Vaccine Efficacy
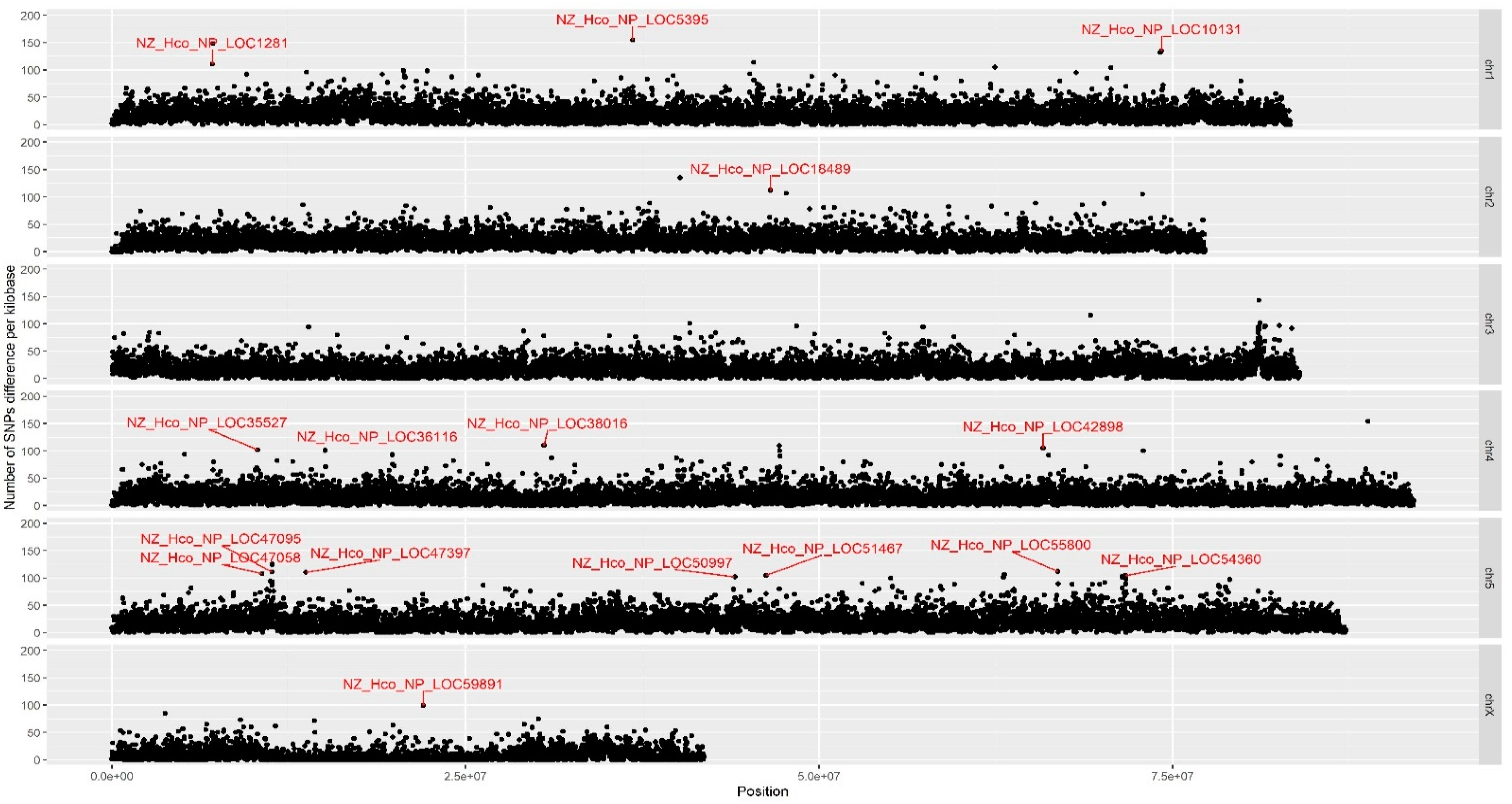
2.2. Genome-Wide SNP Analysis Reveals Putative Candidate Vaccine Evasion Genes
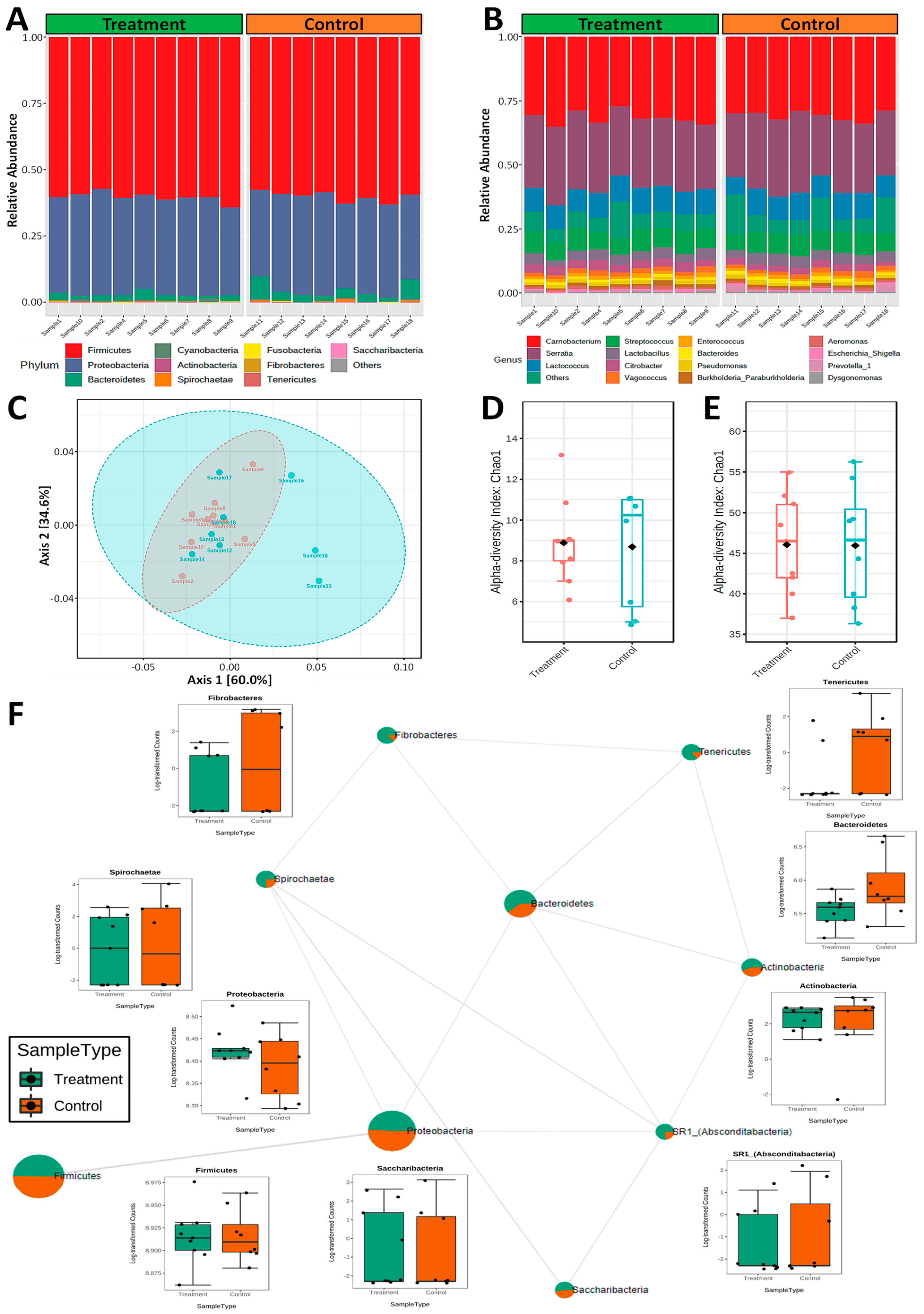
2.3. Nematode Microbiome Population Structure Associated with Vaccine Evasion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Chromosome | Position (bp) | SNP per KB | Fisher’s p-Value | Gene Description |
|---|---|---|---|---|---|
| LOC1281 | chr 1 | 7,110,000 | 148 | 1.80 × 10−41 | Zinc finger in N-recognin |
| LOC5395 | chr 1 | 36,800,000 | 155 | 1.62 × 10−45 | Putative integrase core domain protein |
| LOC10131 | chr 1 | 74,220,000 | 135 | 2.71 × 10−34 | Transcription initiation factor IIA, helical domain protein |
| LOC18489 | chr 2 | 46,570,000 | 112 | 8.60 × 10−23 | Unknown |
| LOC35527 | chr 4 | 10,300,000 | 102 | 2.45 × 10−18 | Hypothetical protein |
| LOC36116 | chr 4 | 15,080,000 | 101 | 6.53 × 10−18 | Unknown |
| LOC38016 | chr 4 | 30,550,000 | 110 | 7.14 × 10−22 | Bestrophin-1 domain protein |
| LOC42898 | chr 4 | 65,840,000 | 105 | 1.23 × 10−19 | GNS1/SUR4 family protein |
| LOC47058 | chr 5 | 10,610,000 | 108 | 5.75 × 10−21 | G_PROTEIN_RECEP_F1_2 domain-containing protein |
| LOC47095 | chr 5 | 11,310,000 | 111 | 2.49 × 10−22 | Long non-coding RNA lnc-LOC47095 |
| LOC47397 | chr 5 | 13,700,000 | 110 | 7.14 × 10−22 | Unknown |
| LOC50997 | chr 5 | 44,040,000 | 102 | 2.45 × 10−18 | Hypothetical protein |
| LOC51467 | chr 5 | 46,260,000 | 105 | 1.23 × 10−19 | Ligand-binding domain of nuclear hormone receptor |
| LOC55800 | chr 5 | 66,890,000 | 112 | 8.60 × 10−23 | Peptidase M1 domain-containing protein |
| LOC54360 | chr 5 | 71,650,000 | 104 | 3.37 × 10−19 | Protein tyrosine kinase |
| LOC59891 | chr X | 22,000,000 | 99 | 5.56 × 10−17 | Hypothetical protein |
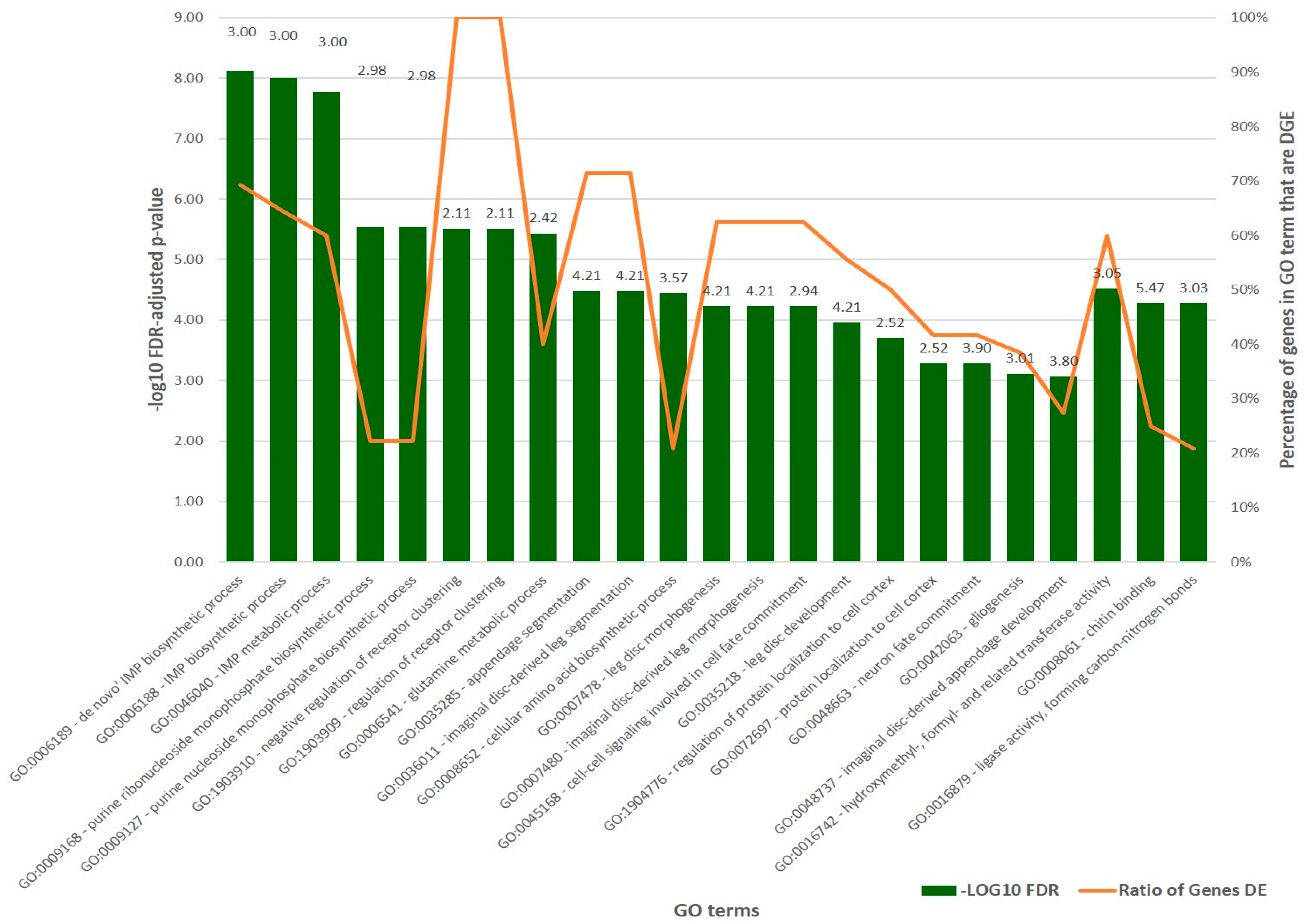
2.4. Transcriptional Changes and Differential Gene Expression
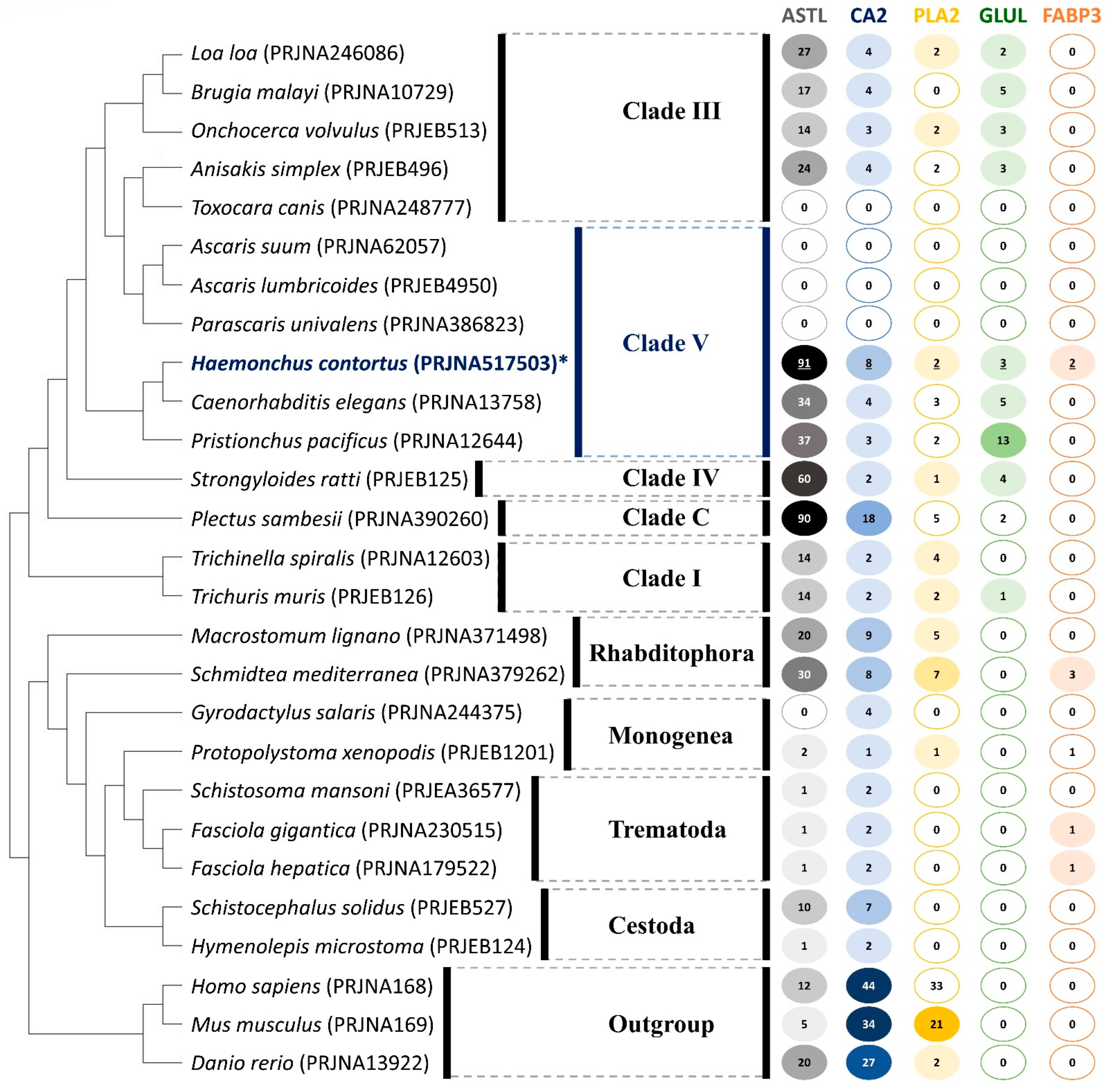
2.5. Orthology and Evolution of Significant DEGs
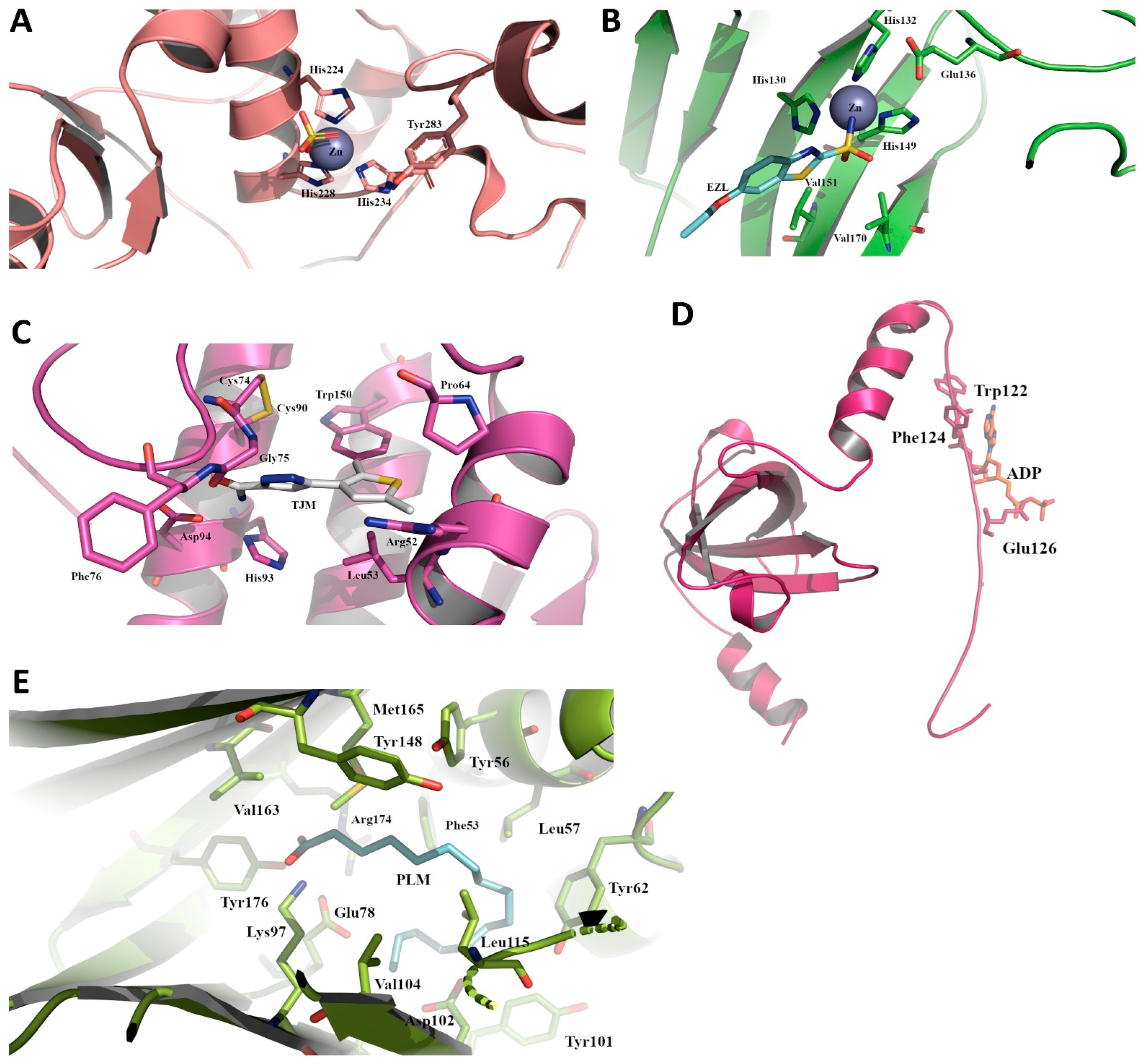
2.6. Structures of Selected Immune Evasion-Related Proteins and Potential Drug-Targeting
3. Materials and Methods
3.1. Animal Experiments and Collection of Parasite Material
3.2. Extraction of Nucleic Acids and Library Preparation
3.3. DNA and RNA-Seq Raw Reads Pre-Processing
3.4. Whole-Genome Sequencing and Identification of SNP Variants
3.5. 16S rRNA Gene Library Preparation, MiSeq Sequencing and Microbiome Profiling
3.6. Transcriptome Sequencing, Assembly, Functional Annotation, and Differential Expression Analysis
3.7. Gene Family Evolution
3.8. Protein Modelling and Structural Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Besier, R.; Kahn, L.; Sargison, N.; Van Wyk, J. Diagnosis, treatment and management of Haemonchus contortus in small ruminants. In Advances in Parasitology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 93, pp. 181–238. [Google Scholar]
- Kaplan, R.M.; Vidyashankar, A.N. An inconvenient truth: Global worming and anthelmintic resistance. Vet. Parasitol. 2012, 186, 70–78. [Google Scholar] [CrossRef]
- Palevich, N.; Maclean, P.H.; Candy, P.M.; Taylor, W.; Mladineo, I.; Cao, M. Untargeted multimodal metabolomics investigation of the Haemonchus contortus exsheathment secretome. Cells 2022, 11, 2525. [Google Scholar] [CrossRef] [PubMed]
- Geary, T.G.; Sakanari, J.A.; Caffrey, C.R. Anthelmintic drug discovery: Into the future. J. Parasitol. 2015, 101, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Bassetto, C.; Amarante, A. Vaccination of sheep and cattle against haemonchosis. J. Helminthol. 2015, 89, 517. [Google Scholar] [CrossRef] [PubMed]
- Umair, S.; Bouchet, C.; Palevich, N.; Simpson, H. Teladorsagia circumcincta 1,6-bisphosphate aldolase: Molecular and biochemical characterisation, structure analysis and recognition by immune hosts. Parasitologia 2021, 1, 1–11. [Google Scholar] [CrossRef]
- Umair, S.; Bouchet, C.; Palevich, N.; Simpson, H.V. Characterisation and structural analysis of glyoxylate cycle enzymes of Teladorsagia circumcincta. Mol. Biochem. Parasitol. 2020, 240, 111335. [Google Scholar] [CrossRef]
- Umair, S.; Bouchet, C.L.G.; Deng, Q.; Palevich, N.; Simpson, H.V. Characterisation of a Teladorsagia circumcincta glutathione transferase. Mol. Biochem. Parasitol. 2020, 239, 111316. [Google Scholar] [CrossRef] [PubMed]
- Umair, S.; Knight, J.S.; Bouchet, C.; Palevich, N.; Cleland, S.B.; Grant, W.; Simpson, H.V. Characterisation of Macrophage Inhibitory Factor-2 (MIF-2) in Haemonchus contortus and Teladorsagia circumcincta. Parasitologia 2022, 2, 338–349. [Google Scholar] [CrossRef]
- Sallé, G.; Laing, R.; Cotton, J.A.; Maitland, K.; Martinelli, A.; Holroyd, N.; Tracey, A.; Berriman, M.; Smith, W.D.; Newlands, G.F. Transcriptomic profiling of nematode parasites surviving vaccine exposure. Int. J. Parasitol. 2018, 48, 395–402. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Read, A.F. Why does drug resistance readily evolve but vaccine resistance does not? Proc. R. Soc. B Biol. Sci. 2017, 284, 20162562. [Google Scholar] [CrossRef]
- Brueggemann, A.B.; Pai, R.; Crook, D.W.; Beall, B. Vaccine escape recombinants emerge after pneumococcal vaccination in the United States. PLoS Pathog. 2007, 3, e168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palevich, N.; Maclean, P.H.; Baten, A.; Scott, R.W.; Leathwick, D.M. The genome sequence of the anthelmintic-susceptible new zealand Haemonchus contortus. Genome Biol. Evol. 2019, 11, 1965–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palevich, N.; Maclean, P.; Baten, A.; Scott, R.; Leathwick, D.M. The complete mitochondrial genome of the New Zealand parasitic roundworm Haemonchus contortus (Trichostrongyloidea: Haemonchidae) field strain NZ_Hco_NP. Mitochondrial DNA Part B 2019, 4, 2208–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnathamby, G.; Henderson, G.; Umair, S.; Janssen, P.; Bland, R.; Simpson, H. The bacterial community associated with the sheep gastrointestinal nematode parasite Haemonchus contortus. PLoS ONE 2018, 13, e0192164. [Google Scholar]
- Jourdan, P.M.; Lamberton, P.H.; Fenwick, A.; Addiss, D.G. Soil-transmitted helminth infections. Lancet 2018, 391, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, J.; van der Voort, M.; Kenyon, F.; Skuce, P.; Vercruysse, J. Chasing helminths and their economic impact on farmed ruminants. Trends Parasitol. 2014, 30, 361–367. [Google Scholar] [CrossRef]
- Gibbons, L.M. Revision of the genus Haemonchus Cobb, 1898 (Nematoda: Trichostrongylidae). Syst. Parasitol. 1979, 1, 3–24. [Google Scholar] [CrossRef]
- Lichtenfels, J.R.; Pilitt, P.; Hoberg, E.P. New morphological characters for identifying individual specimens of Haemonchus spp. (Nematoda: Trichostrongyloidea) and a key to species in ruminants of North America. J. Parasitol. 1994, 80, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Bisset, S.; Knight, J.; Bouchet, C. A multiplex PCR-based method to identify strongylid parasite larvae recovered from ovine faecal cultures and/or pasture samples. Vet. Parasitol. 2014, 200, 117–127. [Google Scholar] [CrossRef]
- Amarante, M.; Santos, M.; Bassetto, C.; Amarante, A. PCR primers for straightforward differentiation of Haemonchus contortus, Haemonchus placei and their hybrids. J. Helminthol. 2017, 91, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Palevich, N.; Britton, C.; Kamenetzky, L.; Mitreva, M.; de Moraes Mourão, M.; Bennuru, S.; Quack, T.; Scholte, L.L.S.; Tyagi, R. Tackling hypotheticals in helminth genomes. Trends Parasitol. 2018, 34, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Maclean, P.H.; Choi, Y.-J.; Mitreva, M. Characterization of the complete mitochondrial genomes of two sibling species of parasitic roundworms, Haemonchus contortus and Teladorsagia circumcincta. Front. Genet. 2020, 11, 573395. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Maclean, P.H.; Mitreva, M.; Scott, R.; Leathwick, D. The complete mitochondrial genome of the New Zealand parasitic roundworm Teladorsagia circumcincta (Trichostrongyloidea: Haemonchidae) field strain NZ_Teci_NP. Mitochondrial DNA Part B 2019, 4, 2869–2871. [Google Scholar] [CrossRef] [Green Version]
- El-Ashram, S.; Suo, X. Exploring the microbial community (microflora) associated with ovine Haemonchus contortus (macroflora) field strains. Sci. Rep. 2017, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midha, A.; Schlosser, J.; Hartmann, S. Reciprocal interactions between nematodes and their microbial environments. Front. Cell. Infect. Microbiol. 2017, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Mafuna, T.; Soma, P.; Tsotetsi-Khambule, A.; Hefer, C.; Muchadeyi, F.; Thekisoe, O.; Pierneef, R. Bacterial profiling of Haemonchus contortus gut microbiome infecting Dohne Merino sheep in South Africa. Sci. Rep. 2021, 11, 5905. [Google Scholar] [CrossRef]
- Hogan, G.; Walker, S.; Turnbull, F.; Curiao, T.; Morrison, A.A.; Flores, Y.; Andrews, L.; Claesson, M.J.; Tangney, M.; Bartley, D.J. Microbiome analysis as a platform R&D tool for parasitic nematode disease management. ISME J. 2019, 13, 2664–2680. [Google Scholar]
- Oh, C.-S.; Toke, D.A.; Mandala, S.; Martin, C.E. ELO2 and ELO3, Homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J. Biol. Chem. 1997, 272, 17376–17384. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arranz, M.; Maldonado, A.M.; Mazon, M.J.; Portillo, F. Transcriptional control of yeast plasma membrane H (+)-ATPase by glucose. Cloning and characterization of a new gene involved in this regulation. J. Biol. Chem. 1994, 269, 18076–18082. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, D.; Yuan, Y.; Wang, D. Intestinal long non-coding RNAs in response to simulated microgravity stress in Caenorhabditis elegans. Sci. Rep. 2021, 11, 1997. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, T.; Korhonen, P.K.; Stroehlein, A.J.; Young, N.D.; Gasser, R.B. Dauer signalling pathway model for Haemonchus contortus. Parasites Vectors 2019, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Duque-Correa, M.A.; Maizels, R.M.; Grencis, R.K.; Berriman, M. Organoids—New models for host-helminth interactions. Trends Parasitol. 2020, 36, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, I.H.G. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163. [Google Scholar]
- Ruskamo, S.; Nieminen, T.; Kristiansen, C.K.; Vatne, G.H.; Baumann, A.; Hallin, E.I.; Raasakka, A.; Joensuu, P.; Bergmann, U.; Vattulainen, I. Molecular mechanisms of Charcot-Marie-Tooth neuropathy linked to mutations in human myelin protein P2. Sci. Rep. 2017, 7, 6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Knerr, L.; Åkerud, T.; Hallberg, K.; Öster, L.; Rohman, M.; Österlund, K.; Beisel, H.-G.; Olsson, T.; Brengdhal, J. Discovery of a novel pyrazole series of group X secreted phospholipase A2 inhibitor (sPLA2X) via fragment based virtual screening. Bioorganic Med. Chem. Lett. 2014, 24, 5251–5255. [Google Scholar] [CrossRef]
- Okada, A.; Sano, K.; Nagata, K.; Yasumasu, S.; Ohtsuka, J.; Yamamura, A.; Kubota, K.; Iuchi, I.; Tanokura, M. Crystal structure of zebrafish hatching enzyme 1 from the zebrafish Danio rerio. J. Mol. Biol. 2010, 402, 865–878. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, N.A.; Ellis, P. Drug development: From concept to marketing! Nephron Clin. Pract. 2009, 113, c125–c131. [Google Scholar] [CrossRef]
- Umair, S.; Pernthaner, A.; Deng, Q.; Gibson, B.; Hook, S.; Heath, D. Preliminary evaluation of a thermosensitive chitosan hydrogel for Echinococcus granulosus vaccine delivery. Vet. Parasitol. 2017, 236, 117–120. [Google Scholar] [CrossRef]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef] [Green Version]
- Palevich, N.; Kelly, W.J.; Leahy, S.C.; Altermann, E.; Rakonjac, J.; Attwood, G.T. The complete genome sequence of the rumen bacterium Butyrivibrio hungatei MB2003. Stand. Genom. Sci. 2017, 12, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palevich, N.; Kelly, W.J.; Leahy, S.C.; Denman, S.; Altermann, E.; Rakonjac, J.; Attwood, G.T. Comparative genomics of rumen Butyrivibrio spp. uncovers a continuum of polysaccharide-degrading capabilities. Appl. Environ. Microbiol. 2019, 86, e01993-19. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Maclean, P.H.; Kelly, W.J.; Leahy, S.C.; Rakonjac, J.; Attwood, G.T. Complete genome sequence of the polysaccharide-degrading rumen bacterium Pseudobutyrivibrio xylanivorans MA3014 reveals an incomplete glycolytic pathway. Genome Biol. Evol. 2020, 12, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Palevich, F.P.; Maclean, P.H.; Altermann, E.; Gardner, A.; Burgess, S.; Mills, J.; Brightwell, G. Comparative genomics of Clostridium species associated with vacuum-packed meat spoilage. Food Microbiol. 2021, 95, 103687. [Google Scholar] [CrossRef]
- Burgess, S.A.; Palevich, F.P.; Gardner, A.; Mills, J.; Brightwell, G.; Palevich, N. Occurrence of genes encoding spore germination in Clostridium species that cause meat spoilage. Microb. Genom. 2022, 8, 000767. [Google Scholar] [CrossRef]
- Palevich, N.; Palevich, F.P.; Gardner, A.; Brightwell, G.; Mills, J. Genome collection of Shewanella spp. isolated from spoiled lamb. Front. Microbiol. 2022, 13, 976152. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Wickham, H. ggplot2. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Palevich, N.; Maclean, P.H. Sequencing and reconstructing helminth mitochondrial genomes directly from genomic next-generation sequencing data. In Parasite Genomics: Methods and Protocols; de Pablos, L.M., Sotillo, J., Eds.; Humana Press: New York, NY, USA, 2021. [Google Scholar]
- Palevich, N.; Maclean, P.H.; Carvalho, L.; Jauregui, R. 16S rRNA Gene Amplicon Profiling of the New Zealand parasitic blowfly Calliphora vicina. Microbiol. Resour. Announc. 2021, 10, e00289-21. [Google Scholar] [CrossRef] [PubMed]
- Palevich, N.; Maclean, P.H.; Carvalho, L.; Jauregui, R. Bacterial diversity profiling of the New Zealand parasitic blowfly Lucilia sericata based on 16S rRNA gene amplicon sequencing. Microbiol. Resour. Announc. 2021, 10, e00257-21. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2. 0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Bolt, B.J.; Shafie, M.; Kersey, P.; Berriman, M. WormBase ParaSite—A comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Trumbić, Ž.; Hrabar, J.; Palevich, N.; Carbone, V.; Mladineo, I. Molecular and evolutionary basis for survival, its failure, and virulence factors of the zoonotic nematode Anisakis pegreffii. Genomics 2021, 113, 2891–2905. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Abagyan, R.; Batalov, S.; Cardozo, T.; Totrov, M.; Webber, J.; Zhou, Y. Homology modeling with internal coordinate mechanics: Deformation zone mapping and improvements of models via conformational search. Proteins Struct. Funct. Bioinform. 1997, 29, 29–37. [Google Scholar] [CrossRef]
- Cardozo, T.; Totrov, M.; Abagyan, R. Homology modeling by the ICM method. Proteins Struct. Funct. Bioinform. 1995, 23, 403–414. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System, Version 2.0; Schrodinger, LLC: New York, NY, USA.




| Locus Tag | Chromosome | Gene Description | logCPM a | FC | logFC b | adj. p c | |
|---|---|---|---|---|---|---|---|
| Top Differentially Expressed (DE) | LOC4701 | chr 1 | Hexokinase domain-containing protein | 374.11 | 13.64 | 3.77 | 4.47 × 10−72 |
| LOC8487 | chr 1 | Carbonate dehydratase, eukaryotic-type | 273.67 | 221.36 | 7.79 | 4.06 × 10−49 | |
| LOC6491 | chr 1 | Transporter, major facilitator family protein | 299.39 | 4.34 | 2.12 | 4.71 × 10−34 | |
| LOC10216 | chr 1 | Patched family protein | 202.18 | 6.14 | 2.62 | 9.13 × 10−23 | |
| LOC13594 | chr 2 | Peptidase S1/S6 domain-containing protein | 379.72 | 24.66 | 4.62 | 6.14 × 10−71 | |
| LOC12468 | chr 2 | Zinc finger domain-containing protein | 938.69 | 9.67 | 3.27 | 1.68 × 10−56 | |
| LOC19286 | chr 2 | Multifunctional protein ADE2, SAICAR synthetase | 228.43 | 35.58 | 5.15 | 1.11 × 10−37 | |
| LOC13325 | chr 2 | Phospholipase A2 | 1553.99 | 14.60 | 3.87 | 1.02 × 10−09 | |
| LOC34257 | chr 3 | RNA recognition motif domain-containing protein | 650.65 | 6.43 | 2.68 | 6.01 × 10−44 | |
| LOC36485 | chr 4 | PERMeable eggshell protein PERM-2 | 1045.16 | 11.97 | 3.58 | 1.72 × 10−91 | |
| LOC34077 | chr 4 | Glutamine synthetase, beta-grasp domain protein | 460.76 | 12.81 | 3.68 | 6.55 × 10−70 | |
| LOC44234 | chr 4 | Chondroitin proteoglycan 4 domain-containing protein | 6439.30 | 176.79 | 7.47 | 1.29 × 10−55 | |
| LOC39532 | chr 4 | Glycosyl hydrolase, family 18 | 875.07 | 17.10 | 4.10 | 4.44 × 10−50 | |
| LOC45198 | chr 4 | Hsp20/alpha crystallin family protein | 3904.34 | 300.66 | 8.23 | 3.91 × 10−42 | |
| LOC39044 | chr 4 | Phosphoribosylglycinamide formyltransferase | 231.50 | 5.94 | 2.57 | 7.83 × 10−42 | |
| LOC35958 | chr 4 | Class II glutamine amidotransferase | 1167.18 | 5.45 | 2.45 | 5.14 × 10−35 | |
| LOC35968 | chr 4 | Glutamine-fructose-6-phosphate transaminase | 1318.78 | 4.11 | 2.04 | 1.42 × 10−32 | |
| LOC35463 | chr 4 | Peptidase A1 domain-containing protein | 447.74 | 193.29 | 7.59 | 2.55 × 10−18 | |
| LOC55104 | chr 5 | Chondroitin proteoglycan 3 | 495.52 | 107.16 | 6.74 | 3.99 × 10−106 | |
| LOC54152 | chr 5 | Peptidase M1 domain-containing protein | 558.27 | 16.17 | 4.01 | 1.17 × 10−57 | |
| LOC55927 | chr 5 | Fibrous sheath CABYR-binding protein | 2197.76 | 882.98 | 9.79 | 1.97 × 10−33 | |
| LOC54628 | chr 5 | Diacylglycerol acyltransferase | 240.81 | 38.96 | 5.28 | 3.94 × 10−25 | |
| LOC56712 | chr X | Low-density lipoprotein receptor domain class A | 722.49 | 81.50 | 6.35 | 5.39 × 10−120 | |
| LOC56407 | chr X | Innexin inx-3 | 479.37 | 13.91 | 3.80 | 1.67 × 10−74 | |
| LOC57394 | chr X | Fatty acid-binding protein 3 | 2172.79 | 4.01 | 2.00 | 8.00 × 10−57 | |
| LOC58311 | chr X | Protein-tyrosine phosphatase | 319.44 | 5.54 | 2.47 | 3.42 × 10−39 | |
| LOC56508 | chr X | Lactamase_B domain-containing protein | 404.68 | 4.85 | 2.28 | 1.61 × 10−30 | |
| LOC61374 | chr X | von Willebrand factor type D domain protein | 342.28 | 49.01 | 5.61 | 6.94 × 10−28 | |
| LOC58549 | chr X | Chitin binding Peritrophin-A domain protein | 8763.11 | 232.54 | 7.86 | 1.86 × 10−19 | |
| LOC61472 | chr X | Astacin | 231.32 | 385.77 | 8.59 | 8.97 × 10−18 | |
| Top SNP Associated Genes | LOC1281 | chr 1 | Zinc finger in N-recognin | 2052.89 | 1.02 | 0.03 | 8.43 × 10−01 |
| LOC5395 | chr 1 | Integrase core domain protein | 220.55 | 1.22 | 0.28 | 4.36 × 10−01 | |
| LOC10131 | chr 1 | Transcription initiation factor IIA | 75.74 | 1.07 | 0.09 | 7.77 × 10−01 | |
| LOC18489 | chr 2 | Unknown | 130.46 | 1.08 | 0.11 | 8.61 × 10−01 | |
| LOC35527 | chr 4 | Frag1 DRAM Sfk1 domain-containing protein | 24.61 | 1.13 | 0.17 | 7.50 × 10−01 | |
| LOC36116 | chr 4 | Unknown | 14.07 | 1.12 | 0.17 | 7.82 × 10−01 | |
| LOC38016 | chr 4 | Bestrophin-1 domain protein | 29.02 | 1.10 | 0.14 | 8.09 × 10−01 | |
| LOC42898 | chr 4 | GNS1/SUR4 family protein | 1409.99 | 1.21 | 0.28 | 9.66 × 10−03 | |
| LOC47058 | chr 5 | G_PROTEIN_RECEP_F1_2 domain-containing protein | 35.29 | 1.21 | 0.28 | 3.49 × 10−01 | |
| LOC47095 | chr 5 | Long non-coding RNA lnc-LOC47095 | 182.47 | 1.10 | 0.13 | 7.82 × 10−01 | |
| LOC47397 | chr 5 | Unknown | 200.62 | 1.94 | 0.95 | 1.21 × 10−02 | |
| LOC50997 | chr 5 | Hypothetical protein | 318.21 | 1.14 | 0.19 | 1.74 × 10−01 | |
| LOC51467 | chr 5 | Ligand-binding domain of nuclear hormone receptor | 1293.87 | 1.03 | 0.05 | 8.24 × 10−01 | |
| LOC55800 | chr 5 | Peptidase M1 domain-containing protein | 18,187.44 | 1.01 | 0.02 | 9.63 × 10−01 | |
| LOC54360 | chr 5 | Protein tyrosine kinase | 198.19 | 1.08 | 0.11 | 7.34 × 10−01 | |
| LOC59891 | chr X | Hypothetical protein | 161.38 | 1.09 | 0.13 | 7.36 × 10−01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palevich, N.; Maclean, P.H.; Carbone, V.; Jauregui, R.; Umair, S. Multi-Omic Profiling, Structural Characterization, and Potent Inhibitor Screening of Evasion-Related Proteins of a Parasitic Nematode, Haemonchus contortus, Surviving Vaccine Treatment. Biomedicines 2023, 11, 411. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020411
Palevich N, Maclean PH, Carbone V, Jauregui R, Umair S. Multi-Omic Profiling, Structural Characterization, and Potent Inhibitor Screening of Evasion-Related Proteins of a Parasitic Nematode, Haemonchus contortus, Surviving Vaccine Treatment. Biomedicines. 2023; 11(2):411. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020411
Chicago/Turabian StylePalevich, Nikola, Paul H. Maclean, Vincenzo Carbone, Ruy Jauregui, and Saleh Umair. 2023. "Multi-Omic Profiling, Structural Characterization, and Potent Inhibitor Screening of Evasion-Related Proteins of a Parasitic Nematode, Haemonchus contortus, Surviving Vaccine Treatment" Biomedicines 11, no. 2: 411. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020411