
Antitumor Potential of Antiepileptic Drugs in Human Glioblastoma: Pharmacological Targets and Clinical Benefits
 , , , and
, , , and
Abstract
:1. Introduction
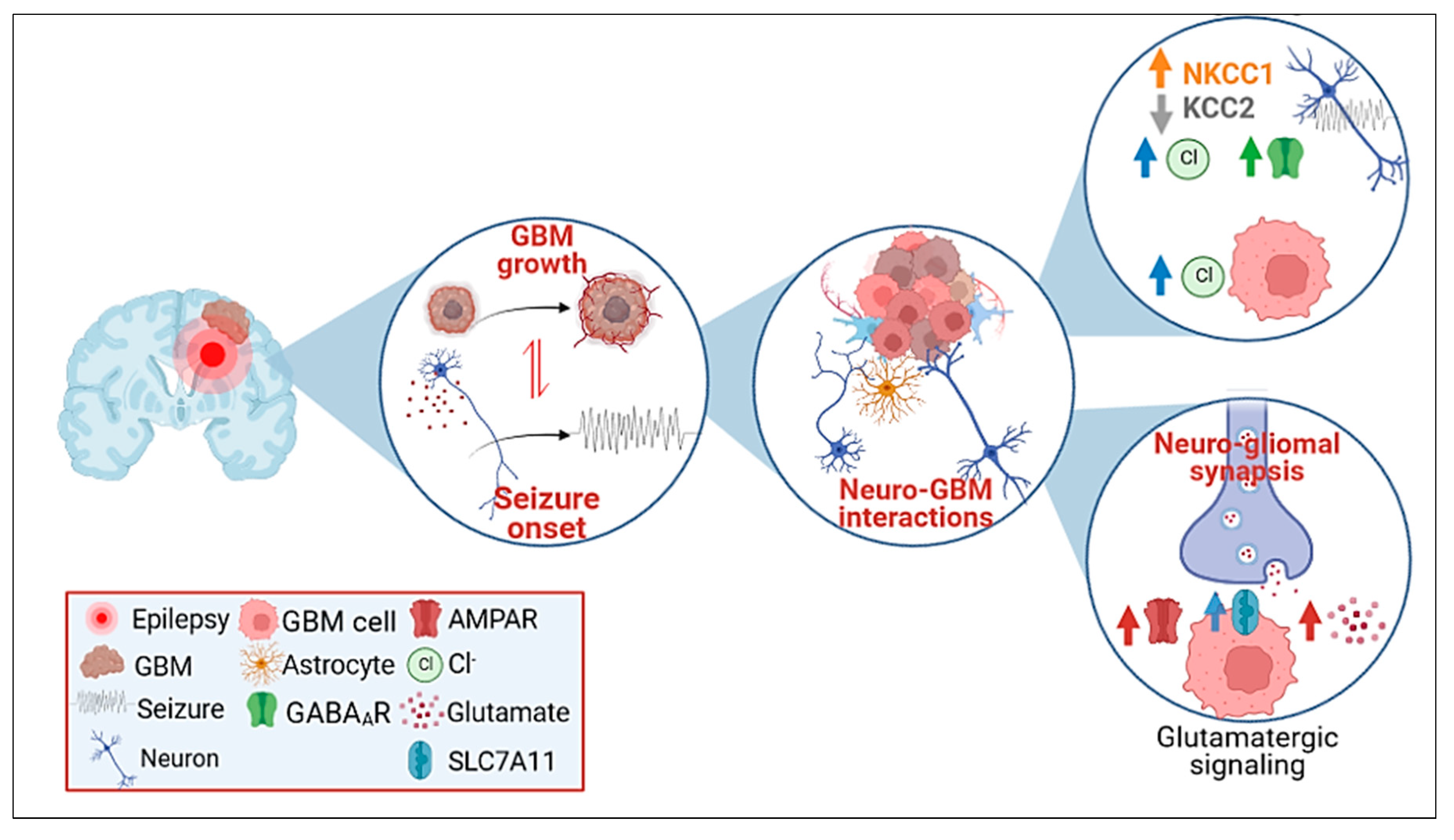
2. Glioblastoma-Related Epilepsy (GRE)
3. Antiepileptic Drugs in GRE: Clinical Management
4. Molecules at the Crossroads of GBM Development and Epilepsy
4.1. Intrinsic Molecular Features of GBM Progression and Their Role in GRE
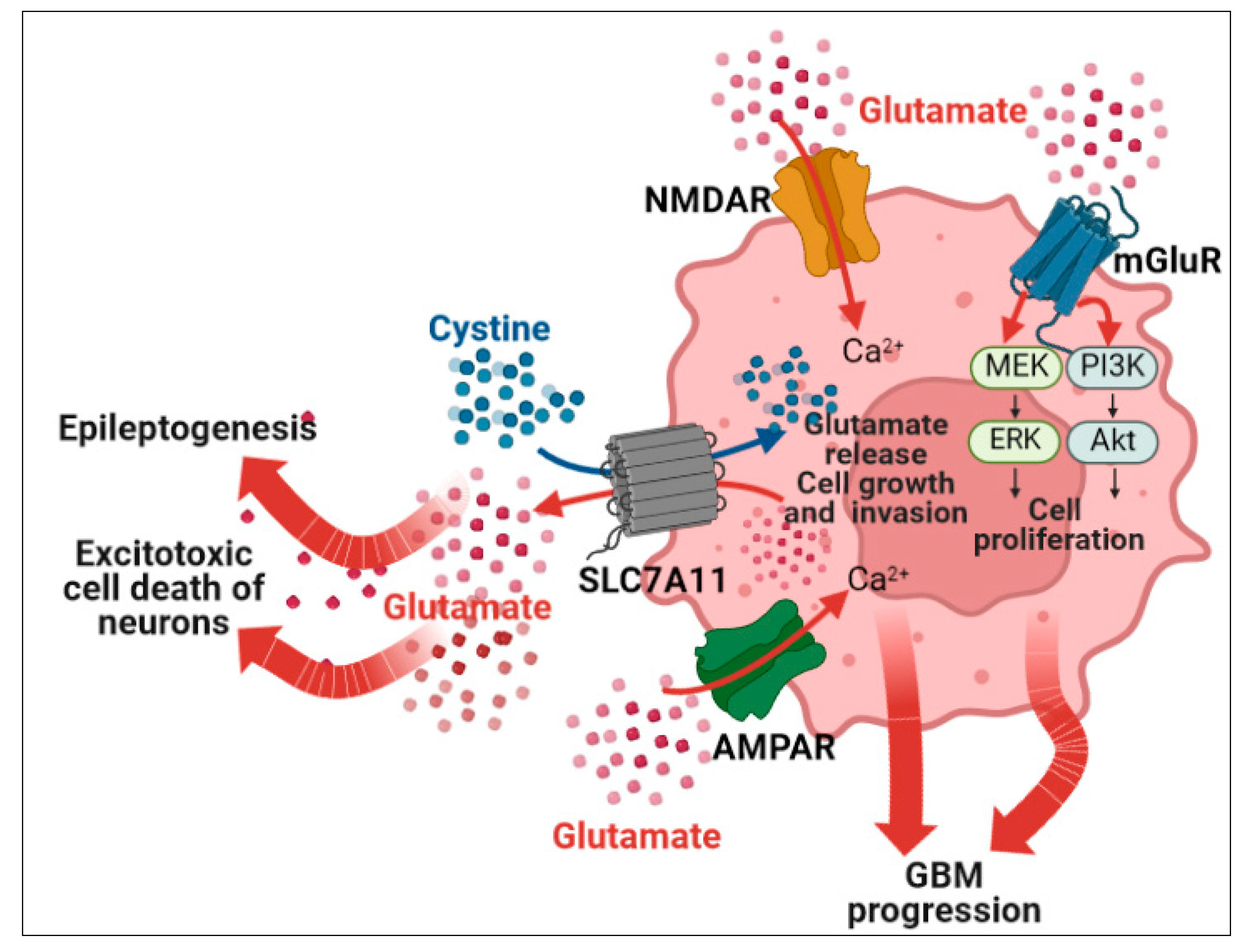
4.2. Neurotransmitter Synaptic Inputs to GBM Progression and GRE
4.3. GABAergic Signaling
4.4. Glutamatergic Signaling
4.5. Endogenous Cannabinoids
5. Repurposing Antiepileptic Drugs for the Treatment of Glioblastoma: Pharmacologic Targets
5.1. Antitumor Efficacy of AEDs Used in GRE: Preclinical and Clinical Perspectives
5.1.1. Levetiracetam
5.1.2. Valproic Acid
5.1.3. Perampanel
5.1.4. Cannabidiol
5.2. Potential Repositioning of Other AEDs Used in GRE
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S.; Duncan, D.L. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015 Introduction. Neuro. Oncol. 2018, 20, 1–86. [Google Scholar] [CrossRef] [Green Version]
- Fabbro-Peray, P.; Zouaoui, S.; Darlix, A.; Fabbro, M.; Pallud, J.; Rigau, V.; Mathieu-Daude, H.; Bessaoud, F.; Fabienne Bauchet, L.; Riondel, A.; et al. Association of Patterns of Care, Prognostic Factors, and Use of Radiotherapy-Temozolomide Therapy with Survival in Patients with Newly Diagnosed Glioblastoma: A French National Population-Based Study. J. Neurooncol. 2019, 142, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejada Solís, S.; Plans Ahicart, G.; Iglesias Lozano, I.; De Quintana Schmidt, C.; Fernández Coello, A.; Hostalot Panisello, C.; Ley Urzaiz, L.; García Romero, J.C.; Díez Valle, R.; González Sánchez, J.; et al. Glioblastoma Treatment Guidelines: Consensus by the Spanish Society of Neurosurgery Tumor Section. Neurocirugía 2020, 31, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Ostrom, Q.T.; Cioffi, G.; Neff, C.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. Primary Brain and Other Central Nervous System Tumors in the United States (2014–2018): A Summary of the CBTRUS Statistical Report for Clinicians. Neurooncol. Pract. 2022, 9, 165–182. [Google Scholar] [CrossRef]
- Carrano, A.; Jose Juarez, J.; Incontri, D.; Ibarra, A.; Cazares, H.G.; Minchiotti, G.; Fico, A.; Castresana, J.S. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef]
- Leece, R.; Xu, J.; Ostrom, Q.T.; Chen, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Global Incidence of Malignant Brain and Other Central Nervous System Tumors by Histology, 2003–2007. Neuro. Oncol. 2017, 19, 1553–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune Microenvironment of Gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Mattei, V.; Santilli, F.; Martellucci, S.; Monache, S.D.; Fabrizi, J.; Colapietro, A.; Angelucci, A.; Festuccia, C. The Importance of Tumor Stem Cells in Glioblastoma Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 3863. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of Glioblastoma Multiforme: From Molecular Mechanisms to Therapeutic Approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Antonio Chiocca, E.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro. Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatzki, D.; Kickingereder, P.; Hentschel, B.; Felsberg, J.; Herrlinger, U.; Schackert, G.; Tonn, J.C.; Westphal, M.; Sabel, M.; Schlegel, U.; et al. Limited Role for Extended Maintenance Temozolomide for Newly Diagnosed Glioblastoma. Neurology 2017, 88, 1422–1430. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Lamborn, K.R.; Yung, W.K.A.; Chang, S.M.; Wen, P.Y.; Cloughesy, T.F.; DeAngelis, L.M.; Robins, H.I.; Lieberman, F.S.; Fine, H.A.; Fink, K.L.; et al. Progression-Free Survival: An Important End Point in Evaluating Therapy for Recurrent High-Grade Gliomas. Neuro. Oncol. 2008, 10, 162–170. [Google Scholar] [CrossRef]
- Vargas López, A.J. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro. Oncol. 2021, 23, 502–503. [Google Scholar] [CrossRef]
- Nabors, L.B.; Portnow, J.; Ahluwalia, M.; Baehring, J.; Brem, H.; Brem, S.; Butowski, N.; Campian, J.L.; Clark, S.W.; Fabiano, A.J.; et al. Central Nervous System Cancers, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 1537–1570. [Google Scholar] [CrossRef]
- Weller, M.; Tabatabai, G.; Kästner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Roth, P.; Rushing, E.J.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M.; et al. Bevacizumab May Improve Quality of Life, but Not Overall Survival in Glioblastoma: An Epidemiological Study. Ann. Oncol. 2018, 29, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; Van Den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular Targeted Therapy of Glioblastoma. Cancer Treat Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Andersen, B.M.; Reardon, D.A. Immunotherapy Approaches for Adult Glioma: Knowledge Gained from Recent Clinical Trials. Curr. Opin. Neurol. 2022, 35, 803–813. [Google Scholar] [CrossRef]
- Fisher, R.S.; Van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Liang, S.; Fan, X.; Zhao, M.; Shan, X.; Li, W.; Ding, P.; You, G.; Hong, Z.; Yang, X.; Luan, G.; et al. Clinical Practice Guidelines for the Diagnosis and Treatment of Adult Diffuse Glioma-Related Epilepsy. Cancer Med 2019, 8, 4527–4535. [Google Scholar] [CrossRef] [Green Version]
- Herman, S.T. Epilepsy after Brain Insult: Targeting Epileptogenesis. Neurology 2002, 59, S21–S26. [Google Scholar] [CrossRef]
- Englot, D.J.; Chang, E.F.; Vecht, C.J. Epilepsy and Brain Tumors. Handb. Clin. Neurol. 2016, 134, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.Y.; Chen, C.C.; Crawford, J.R.; Wang, S.G. Tumor-Related Epilepsy: Epidemiology, Pathogenesis and Management. J. Neurooncol. 2018, 139, 13–21. [Google Scholar] [CrossRef]
- Pallud, J.; Capelle, L.; Huberfeld, G. Tumoral Epileptogenicity: How Does It Happen? Epilepsia 2013, 54 (Suppl. 9), 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Huang, T.L.; Chen, X.; Yu, S.X.; Li, W.; Chen, T.; Li, Y.; Kuang, Y.Q.; Shu, H.F. Glioma-Derived TSP2 Promotes Excitatory Synapse Formation and Results in Hyperexcitability in the Peritumoral Cortex of Glioma. J. Neuropathol. Exp. Neurol. 2021, 80, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Jansen, G.H.; Leenstra, S.; Yankaya, B.; Troost, D. Expression of Connexin 43 and Connexin 32 Gap-Junction Proteins in Epilepsy-Associated Brain Tumors and in the Perilesional Epileptic Cortex. Acta Neuropathol. 2001, 101, 449–459. [Google Scholar] [CrossRef]
- Dong, H.; Zhou, X.W.; Wang, X.; Yang, Y.; Luo, J.W.; Liu, Y.H.; Mao, Q. Complex Role of Connexin 43 in Astrocytic Tumors and Possible Promotion of Glioma-associated Epileptic Discharge (Review). Mol. Med. Rep. 2017, 16, 7890–7900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, K.; Iijima, K.; Kawabata-Iwakawa, R.; Fujihara, K.; Kakizaki, T.; Yanagawa, Y.; Yoshimoto, Y.; Miyata, S. Glioma Facilitates the Epileptic and Tumor-Suppressive Gene Expressions in the Surrounding Region. Sci. Rep. 2022, 12, 6805. [Google Scholar] [CrossRef]
- Li, L.; Fang, S.; Li, G.; Zhang, K.; Huang, R.; Wang, Y.; Zhang, C.; Li, Y.; Zhang, W.; Zhang, Z.; et al. Glioma-Related Epilepsy in Patients with Diffuse High-Grade Glioma after the 2016 WHO Update: Seizure Characteristics, Risk Factors, and Clinical Outcomes. J. Neurosurg. 2021, 136, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Koekkoek, J.A.F.; Kerkhof, M.; Dirven, L.; Heimans, J.J.; Reijneveld, J.C.; Taphoorn, M.J.B. Seizure Outcome after Radiotherapy and Chemotherapy in Low-Grade Glioma Patients: A Systematic Review. Neuro. Oncol. 2015, 17, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Climans, S.A.; Brandes, A.A.; Cairncross, J.G.; Ding, K.; Fay, M.; Laperriere, N.; Menten, J.; Nishikawa, R.; O’Callaghan, C.J.; Perry, J.R.; et al. Temozolomide and Seizure Outcomes in a Randomized Clinical Trial of Elderly Glioblastoma Patients. J. Neurooncol. 2020, 149, 65–71. [Google Scholar] [CrossRef]
- Van Der Meer, P.B.; Taphoorn, M.J.B.; Koekkoek, J.A.F. C URRENT OPINION Management of Epilepsy in Brain Tumor Patients. Curr. Opin. Oncol. 2022, 34, 685–690. [Google Scholar] [CrossRef]
- Maschio, M.; Aguglia, U.; Avanzini, G.; Banfi, P.; Buttinelli, C.; Capovilla, G.; Casazza, M.M.L.; Colicchio, G.; Coppola, A.; Costa, C.; et al. Management of Epilepsy in Brain Tumors. Neurol. Sci. 2019, 40, 2217–2234. [Google Scholar] [CrossRef]
- Walbert, T.; Harrison, R.A.; Schiff, D.; Avila, E.K.; Chen, M.; Kandula, P.; Lee, J.W.; le Rhun, E.; Stevens, G.H.J.; Vogelbaum, M.A.; et al. SNO and EANO Practice Guideline Update: Anticonvulsant Prophylaxis in Patients with Newly Diagnosed Brain Tumors. Neuro. Oncol. 2021, 23, 1835–1844. [Google Scholar] [CrossRef]
- Stocksdale, B.; Nagpal, S.; Hixson, J.D.; Johnson, D.R.; Rai, P.; Shivaprasad, A.; Tremont-Lukats, I.W. Neuro-Oncology Practice Clinical Debate: Long-Term Antiepileptic Drug Prophylaxis in Patients with Glioma. Neurooncol. Pract. 2020, 7, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Medicines Complete. Martindale: The Complete Drug Reference. 2020. Available online: https://about.medicinescomplete.com/publication/martindale-the-complete-drug-reference/ (accessed on 10 January 2023).
- Bénit, C.P.; Vecht, C.J. Seizures and Cancer: Drug Interactions of Anticonvulsants with Chemotherapeutic Agents, Tyrosine Kinase Inhibitors and Glucocorticoids. Neurooncol. Pract. 2016, 3, 245–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourg, V.; Lebrun, C.; Chichmanian, R.M.; Thomas, P.; Frenay, M. Nitroso-Urea-Cisplatin-Based Chemotherapy Associated with Valproate: Increase of Haematologic Toxicity. Ann. Oncol. 2001, 12, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.S.; Grant, R.; Gilbert, M.R.; Lee, J.W.; Norden, A.D. Epilepsy in Glioma Patients: Mechanisms, Management, and Impact of Anticonvulsant Therapy. Neuro. Oncol. 2016, 18, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruin, M.E.; Van Der Meer, P.B.; Dirven, L.; Taphoorn, M.J.B.; Koekkoek, J.A.F. Efficacy of Antiepileptic Drugs in Glioma Patients with Epilepsy: A Systematic Review. Neurooncol. Pract. 2021, 8, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meer, P.B.; Dirven, L.; Fiocco, M.; Vos, M.J.; Kouwenhoven, M.C.M.; Van Den Bent, M.J.; Taphoorn, M.J.B.; Koekkoek, J.A.F. First-Line Antiepileptic Drug Treatment in Glioma Patients with Epilepsy: Levetiracetam vs Valproic Acid. Epilepsia 2021, 62, 1119–1129. [Google Scholar] [CrossRef]
- Maschio, M.; Zarabla, A.; Maialetti, A.; Giannarelli, D.; Koudriavtseva, T.; Villani, V.; Zannino, S. Perampanel in Brain Tumor-Related Epilepsy: Observational Pilot Study. Brain Behav. 2020, 10, e01612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, A.; Zarabla, A.; Maialetti, A.; Villani, V.; Koudriavtseva, T.; Russo, E.; Nozzolillo, A.; Sueri, C.; Belcastro, V.; Balestrini, S.; et al. Perampanel Confirms to Be Effective and Well-Tolerated as an Add-On Treatment in Patients With Brain Tumor-Related Epilepsy (PERADET Study). Front. Neurol. 2020, 11, 592. [Google Scholar] [CrossRef]
- Maschio, M.; Maialetti, A.; Mocellini, C.; Domina, E.; Pauletto, G.; Costa, C.; Mascia, A.; Romoli, M.; Giannarelli, D. Effect of Brivaracetam on Efficacy and Tolerability in Patients With Brain Tumor-Related Epilepsy: A Retrospective Multicenter Study. Front. Neurol. 2020, 11, 813. [Google Scholar] [CrossRef]
- Golub, V.; Reddy, D.S. Cannabidiol Therapy for Refractory Epilepsy and Seizure Disorders. Adv. Exp. Med. Biol. 2021, 1264, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.D.R.; Pacheco, J.C.; Rossi, G.N.; De-Paulo, B.O.; Zuardi, A.W.; Guimarães, F.S.; Hallak, J.E.C.; Crippa, J.A.; Dos Santos, R.G. Adverse Effects of Oral Cannabidiol: An Updated Systematic Review of Randomized Controlled Trials (2020–2022). Pharmaceutics 2022, 14, 2598. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Judkins, J.; Thomas, C.; Wu, M.; Khoury, L.; Benjamin, C.G.; Pacione, D.; Golfinos, J.G.; Kumthekar, P.; Ghamsari, F.; et al. Mutant IDH1 and Seizures in Patients with Glioma. Neurology 2017, 88, 1805–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opijnen, M.P.; Tesileanu, C.M.S.; Dirven, L.; Van Der Meer, P.B.; Wijnenga, M.M.J.; Vincent, A.J.P.E.; Broekman, M.L.D.; Dubbink, H.J.; Kros, J.M.; van Duinen, S.G.; et al. IDH1/2 Wildtype Gliomas Grade 2 and 3 with Molecular Glioblastoma-like Profile Have a Distinct Course of Epilepsy Compared to IDH1/2 Wildtype Glioblastomas. Neuro. Oncol. 2022; Epub ahead of print. [Google Scholar] [CrossRef]
- Mortazavi, A.; Fayed, I.; Bachani, M.; Dowdy, T.; Jahanipour, J.; Khan, A.; Owotade, J.; Walbridge, S.; Inati, S.K.; Steiner, J.; et al. IDH-Mutated Gliomas Promote Epileptogenesis through d-2-Hydroxyglutarate-Dependent MTOR Hyperactivation. Neuro. Oncol. 2022, 24, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.; Sperry, J.; Braas, D.; Yan, W.; Le, T.M.; Mottahedeh, J.; Ludwig, K.; Eskin, A.; Qin, Y.; Levy, R.; et al. Metabolic Characterization of Isocitrate Dehydrogenase (IDH) Mutant and IDH Wildtype Gliomaspheres Uncovers Cell Type-Specific Vulnerabilities. Cancer Metab. 2018, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.-E.; Germain, M.-A.; Motorina, A.; Guiot, M.-C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The Oncometabolite 2-Hydroxyglutarate Activates the MTOR Signalling Pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crino, P.B. The MTOR Signalling Cascade: Paving New Roads to Cure Neurological Disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef]
- Venkatesan, S.; Hoogstraat, M.; Caljouw, E.; Pierson, T.; Spoor, J.K.H.; Zeneyedpour, L.; Dubbink, H.J.; Dekker, L.J.; Van Der Kaaij, M.; Kloezeman, J.; et al. TP53 Mutated Glioblastoma Stem-like Cell Cultures Are Sensitive to Dual MTORC1/2 Inhibition While Resistance in TP53 Wild Type Cultures Can Be Overcome by Combined Inhibition of MTORC1/2 and Bcl-2. Oncotarget 2016, 7, 58435–58444. [Google Scholar] [CrossRef]
- Huberfeld, G.; Vecht, C.J. Seizures and Gliomas--towards a Single Therapeutic Approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef]
- Hatcher, A.; Yu, K.; Meyer, J.; Aiba, I.; Deneen, B.; Noebels, J.L. Pathogenesis of Peritumoral Hyperexcitability in an Immunocompetent CRISPR-Based Glioblastoma Model. J. Clin. Investig. 2020, 130, 2286–2300. [Google Scholar] [CrossRef] [Green Version]
- Winter, R.; Pembrey, M. Interpretation of the Heterogeneity in the Linkage Relationships of DNA Markers around the Fragile X Locus. Hum. Genet. 1987, 77, 297–298. [Google Scholar] [CrossRef]
- Hu, S.; Kao, H.-Y.; Yang, T.; Wang, Y. Early and Bi-Hemispheric Seizure Onset in a Rat Glioblastoma Multiforme Model. Neurosci. Lett. 2022, 766, 136351. [Google Scholar] [CrossRef]
- Toledo, M.; Sarria-Estrada, S.; Quintana, M.; Maldonado, X.; Martinez-Ricarte, F.; Rodon, J.; Auger, C.; Aizpurua, M.; Salas-Puig, J.; Santamarina, E.; et al. Epileptic Features and Survival in Glioblastomas Presenting with Seizures. Epilepsy Res. 2017, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Lee, J.H. Mechanistic Target of Rapamycin Pathway in Epileptic Disorders. J. Korean Neurosurg. Soc. 2019, 62, 272–287. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Bordey, A. Convergent and Divergent Mechanisms of Epileptogenesis in MTORopathies. Front. Neuroanat. 2021, 15, 664695. [Google Scholar] [CrossRef]
- Sorrentino, U.; Bellonzi, S.; Mozzato, C.; Brasson, V.; Toldo, I.; Parrozzani, R.; Clementi, M.; Cassina, M.; Trevisson, E. Epilepsy in NF1: Epidemiologic, Genetic, and Clinical Features. A Monocentric Retrospective Study in a Cohort of 784 Patients. Cancers 2021, 13, 6336. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 Tumor Suppressor Critically Regulates TSC2 and MTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Lin, C.C.J.; Hatcher, A.; Lozzi, B.; Kong, K.; Huang-Hobbs, E.; Cheng, Y.T.; Beechar, V.B.; Zhu, W.; Zhang, Y.; et al. PIK3CA Variants Selectively Initiate Brain Hyperactivity during Gliomagenesis. Nature 2020, 578, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucchiara, F.; Pasqualetti, F.; Giorgi, F.S.; Danesi, R.; Bocci, G. Epileptogenesis and Oncogenesis: An Antineoplastic Role for Antiepileptic Drugs in Brain Tumours? Pharmacol. Res. 2020, 156, 104786. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Rinehart, J.; Lifton, R.P. Phosphoregulation of the Na-K-2Cl and K-Cl Cotransporters by the WNK Kinases. Biochim. Biophys. Acta 2010, 1802, 1150–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, H.K.; Roos, D.; Blümcke, I.; Pietsch, T.; Wiestler, O.D. Perilesional Neurochemical Changes in Focal Epilepsies. Acta Neuropathol. 1996, 91, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic Excitation Contributes to Epileptic Activities around Human Glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.L.; Robel, S.; Cuddapah, V.A.; Robert, S.; Buckingham, S.C.; Kahle, K.T.; Sontheimer, H. GABAergic Disinhibition and Impaired KCC2 Cotransporter Activity Underlie Tumor-Associated Epilepsy. Glia 2015, 63, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huberfeld, G.; Wittner, L.; Clemenceau, S.; Baulac, M.; Kaila, K.; Miles, R.; Rivera, C. Perturbed Chloride Homeostasis and GABAergic Signaling in Human Temporal Lobe Epilepsy. J. Neurosci. 2007, 27, 9866–9873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, G.; O’Toole, K.K.; Maguire, J. Compromised GABAergic Inhibition Contributes to Tumor-Associated Epilepsy. Epilepsy Res. 2016, 126, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Habela, C.W.; Ernest, N.J.; Swindall, A.F.; Sontheimer, H. Chloride Accumulation Drives Volume Dynamics Underlying Cell Proliferation and Migration. J. Neurophysiol. 2009, 101, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A1 Expression Is Associated with Seizures, Predicts Poor Survival in Patients with Malignant Glioma. Sci. Transl. Med. 2015, 7, 289ra86. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, M.F.; Heimisdóttir, S.B.; Sørensen, M.D.; Mellegaard, C.S.; Wohlleben, H.; Kristensen, B.W.; Beier, C.P. High Expression of Cystine–Glutamate Antiporter XCT (SLC7A11) Is an Independent Biomarker for Epileptic Seizures at Diagnosis in Glioma. J. Neurooncol. 2018, 138, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Buccoliero, A.M.; Caporalini, C.; Scagnet, M.; Mussa, F.; Giordano, F.; Sardi, I.; Migliastro, I.; Moscardi, S.; Conti, V.; Barba, C.; et al. Angiocentric Glioma-Associated Seizures: The Possible Role of EATT2, Pyruvate Carboxylase and Glutamine Synthetase. Seizure 2021, 86, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 Promotes Cell Proliferation through Amino Acid Catabolism in Gliomas Carrying Wild-Type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate Release by Primary Brain Tumors Induces Epileptic Activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Rzeski, W.; Ikonomidou, C.; Turski, L. Glutamate Antagonists Limit Tumor Growth. Biochem. Pharmacol. 2002, 64, 1195–1200. [Google Scholar] [CrossRef]
- Schenk, U.; Menna, E.; Kim, T.; Passafaro, M.; Chang, S.; de Camilli, P.; Matteoli, M. A Novel Pathway for Presynaptic Mitogen-Activated Kinase Activation via AMPA Receptors. J. Neurosci. 2005, 25, 1654–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okado, H.; Miwa, A.; et al. Ca2+-Permeable AMPA Receptors Regulate Growth of Human Glioblastoma via Akt Activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.; Vissel, B. The Essential Role of AMPA Receptor GluR2 Subunit RNA Editing in the Normal and Diseased Brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of Glutamate Receptor GluR-B MRNA in Malignant Gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Cesarini, V.; Silvestris, D.A.; Galeano, F.; Tassinari, V.; Martini, M.; Locatelli, F.; Gallo, A. ADAR2 Protein Is Associated with Overall Survival in GBM Patients and Its Decrease Triggers the Anchorage-Independent Cell Growth Signature. Biomolecules 2022, 12, 1142. [Google Scholar] [CrossRef]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A Multigene Predictor of Outcome in Glioblastoma. Neuro. Oncol. 2010, 12, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and Synaptic Integration of Glioma into Neural Circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic Synaptic Input to Glioma Cells Drives Brain Tumour Progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, F.; Grandi, V.; Banerjee, A.; Trant, J.F. Cannabinoids and Cannabinoid Receptors: The Story so Far. iScience 2020, 23, 101301. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Modulation of Transmitter Release via Presynaptic Cannabinoid Receptors. Trends Pharm. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Ortega-Gutiérrez, S.; van der Stelt, M.; et al. CB1 Cannabinoid Receptors and On-Demand Defense against Excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, V.; Karanian, D.A.; Vadivel, S.K.; Locklear, J.R.; Wood, J.A.T.; Nasr, M.; Quizon, P.M.P.; Graves, E.E.; Shukla, V.; Makriyannis, A.; et al. Equipotent Inhibition of Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase—Dual Targets of the Endocannabinoid System to Protect against Seizure Pathology. Neurotherapeutics 2012, 9, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Monory, K.; Massa, F.; Egertová, M.; Eder, M.; Blaudzun, H.; Westenbroek, R.; Kelsch, W.; Jacob, W.; Marsch, R.; Ekker, M.; et al. The Endocannabinoid System Controls Key Epileptogenic Circuits in the Hippocampus. Neuron 2006, 51, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Ludányi, A.; Eross, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 Cannabinoid Receptor and Related Molecular Elements of the Endocannabinoid System in Epileptic Human Hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef] [Green Version]
- Romigi, A.; Bari, M.; Placidi, F.; Grazia Marciani, M.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F.; et al. Cerebrospinal Fluid Levels of the Endocannabinoid Anandamide Are Reduced in Patients with Untreated Newly Diagnosed Temporal Lobe Epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Attinà, M.; Cartoni, A.; Bari, M.; Finazzi-Agrò, A. Gas Chromatography-Mass Spectrometry Analysis of Endogenous Cannabinoids in Healthy and Tumoral Human Brain and Human Cells in Culture. J. Neurochem. 2001, 76, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of Endocannabinoid System in Human Gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.; Moesgaard, B.; Schmid, P.C.; Schmid, H.H.O.; Broholm, H.; Kosteljanetz, M.; Hansen, H.S. Endocannabinoid Metabolism in Human Glioblastomas and Meningiomas Compared to Human Non-Tumour Brain Tissue. J. Neurochem. 2005, 93, 299–309. [Google Scholar] [CrossRef]
- De Jesús, M.L.; Hostalot, C.; Garibi, J.M.; Sallés, J.; Meana, J.J.; Callado, L.F. Opposite Changes in Cannabinoid CB1 and CB2 Receptor Expression in Human Gliomas. Neurochem. Int. 2010, 56, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Cannabinoid Signaling in Glioma Cells. Adv. Exp. Med. Biol. 2020, 1202, 223–241. [Google Scholar] [CrossRef]
- Esposito, G.; Ligresti, A.; Izzo, A.A.; Bisogno, T.; Ruvo, M.; di Rosa, M.; di Marzo, V.; Iuvone, T. The Endocannabinoid System Protects Rat Glioma Cells against HIV-1 Tat Protein-Induced Cytotoxicity: Mechanism and Regulation. J. Biol. Chem. 2002, 277, 50348–50354. [Google Scholar] [CrossRef] [Green Version]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulamluis, R.; Alvarez, L.; Guzmán, M.; Galve-Roperh, I. Cannabinoids Induce Glioma Stem-like Cell Differentiation and Inhibit Gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Grinspoon, R.; Fleming, B.; Skirvin, L.A.; Wade, C.; Wolper, E.; Bruno, P.L.; Thiele, E.A. The Long-Term Efficacy of Cannabidiol in the Treatment of Refractory Epilepsy. Epilepsia 2021, 62, 1594–1603. [Google Scholar] [CrossRef]
- Ntafoulis, I.; Koolen, S.L.W.; Leenstra, S.; Lamfers, M.L.M. Drug Repurposing, a Fast-Track Approach to Develop Effective Treatments for Glioblastoma. Cancers 2022, 14, 3705. [Google Scholar] [CrossRef]
- Barbieri, F.; Verduci, I.; Carlini, V.; Zona, G.; Pagano, A.; Mazzanti, M.; Florio, T. Repurposed Biguanide Drugs in Glioblastoma Exert Antiproliferative Effects via the Inhibition of Intracellular Chloride Channel 1 Activity. Front. Oncol. 2019, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würth, R.; Barbieri, F.; Florio, T. New Molecules and Old Drugs as Emerging Approaches to Selectively Target Human Glioblastoma Cancer Stem Cells. Biomed. Res. Int. 2014, 126586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The Synaptic Vesicle Protein SV2A Is the Binding Site for the Antiepileptic Drug Levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigo, J.M.; Hans, G.; Nguyen, L.; Rocher, V.; Belachew, S.; Malgrange, B.; Leprince, P.; Moonen, G.; Selak, I.; Matagne, A.; et al. The Anti-Epileptic Drug Levetiracetam Reverses the Inhibition by Negative Allosteric Modulators of Neuronal GABA- and Glycine-Gated Currents. Br. J. Pharmacol. 2002, 136, 659–672. [Google Scholar] [CrossRef]
- Marutani, A.; Nakamura, M.; Nishimura, F.; Nakazawa, T.; Matsuda, R.; Hironaka, Y.; Nakagawa, I.; Tamura, K.; Takeshima, Y.; Motoyama, Y.; et al. Tumor-Inhibition Effect of Levetiracetam in Combination with Temozolomide in Glioblastoma Cells. Neurochem. J. 2017, 11, 43–49. [Google Scholar] [CrossRef]
- Bobustuc, G.C.; Baker, C.H.; Limaye, A.; Jenkins, W.D.; Pearl, G.; Avgeropoulos, N.G.; Konduri, S.D. Levetiracetam Enhances P53-Mediated MGMT Inhibition and Sensitizes Glioblastoma Cells to Temozolomide. Neuro. Oncol. 2010, 12, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scicchitano, B.M.; Sorrentino, S.; Proietti, G.; Lama, G.; Dobrowolny, G.; Catizone, A.; Binda, E.; Larocca, L.M.; Sica, G. Levetiracetam Enhances the Temozolomide Effect on Glioblastoma Stem Cell Proliferation and Apoptosis. Cancer Cell Int. 2018, 18, 136. [Google Scholar] [CrossRef]
- Ni, X.R.; Guo, C.C.; Yu, Y.J.; Yu, Z.H.; Cai, H.P.; Wu, W.C.; Ma, J.X.; Chen, F.R.; Wang, J.; Chen, Z.P. Combination of Levetiracetam and IFN-α Increased Temozolomide Efficacy in MGMT-Positive Glioma. Cancer Chemother. Pharmacol. 2020, 86, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, T.; Joo, J.D.; Han, J.H.; Kim, Y.J.; Kim, I.A.; Yun, C.H.; Kim, C.Y. Survival Benefit of Levetiracetam in Patients Treated with Concomitant Chemoradiotherapy and Adjuvant Chemotherapy with Temozolomide for Glioblastoma Multiforme. Cancer 2015, 121, 2926–2932. [Google Scholar] [CrossRef]
- Roh, T.H.; Moon, J.H.; Park, H.H.; Kim, E.H.; Hong, C.K.; Kim, S.H.; Kang, S.G.; Chang, J.H. Association between Survival and Levetiracetam Use in Glioblastoma Patients Treated with Temozolomide Chemoradiotherapy. Sci. Rep. 2020, 10, 10783. [Google Scholar] [CrossRef]
- Cardona, A.F.; Rojas, L.; Wills, B.; Bernal, L.; Ruiz-Patiño, A.; Arrieta, O.; Hakim, E.J.; Hakim, F.; Mejía, J.A.; Useche, N.; et al. Efficacy and Safety of Levetiracetam vs. Other Antiepileptic Drugs in Hispanic Patients with Glioblastoma. J. Neurooncol. 2018, 136, 363–371. [Google Scholar] [CrossRef]
- Happold, C.; Gorlia, T.; Chinot, O.; Gilbert, M.R.; Nabors, L.B.; Wick, W.; Pugh, S.L.; Hegi, M.; Cloughesy, T.; Roth, P.; et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2016, 34, 731–739. [Google Scholar] [CrossRef]
- Pallud, J.; Huberfeld, G.; Dezamis, E.; Peeters, S.; Moiraghi, A.; Gavaret, M.; Guinard, E.; Dhermain, F.; Varlet, P.; Oppenheim, C.; et al. Effect of Levetiracetam Use Duration on Overall Survival of Isocitrate Dehydrogenase Wild-Type Glioblastoma in Adults: An Observational Study. Neurology 2022, 98, E125–E140. [Google Scholar] [CrossRef]
- Hwang, K.; Kim, J.; Kang, S.G.; Jung, T.Y.; Kim, J.H.; Kim, S.H.; Kang, S.H.; Hong, Y.K.; Kim, T.M.; Kim, Y.J.; et al. Levetiracetam as a Sensitizer of Concurrent Chemoradiotherapy in Newly Diagnosed Glioblastoma: An Open-Label Phase 2 Study. Cancer Med. 2022, 11, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Huang, N.; Tao, Y.; Wen, R.; Zhao, G.; Zhang, X.; Xie, Z.; Cheng, Y.; Mao, J.; Liu, G. The Efficacy of Temozolomide Combined with Levetiracetam for Glioblastoma (GBM) after Surgery: A Study Protocol for a Double-Blinded and Randomized Controlled Trial. Trials 2022, 23, 234. [Google Scholar] [CrossRef] [PubMed]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Candelaria, M.; Perez-Plascencia, C.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Herrera, L.A. Valproic Acid as Epigenetic Cancer Drug: Preclinical, Clinical and Transcriptional Effects on Solid Tumors. Cancer Treat. Rev. 2008, 34, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [Green Version]
- Barciszewska, A.M.; Belter, A.; Gawrońska, I.; Giel-Pietraszuk, M.; Naskręt-Barciszewska, M.Z. Cross-Reactivity between Histone Demethylase Inhibitor Valproic Acid and DNA Methylation in Glioblastoma Cell Lines. Front. Oncol. 2022, 12, 1033035. [Google Scholar] [CrossRef]
- Castro, L.M.R.; Gallant, M.; Niles, L.P. Novel Targets for Valproic Acid: Up-Regulation of Melatonin Receptors and Neurotrophic Factors in C6 Glioma Cells. J. Neurochem. 2005, 95, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Yuan, X.; Hu, Z.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic Acid Promotes Human Glioma U87 Cells Apoptosis and Inhibits Glycogen Synthase Kinase-3β Through ERK/Akt Signaling. Cell. Physiol. Biochem. 2016, 39, 2173–2185. [Google Scholar] [CrossRef]
- Han, W.; Yu, F.; Cao, J.; Dong, B.; Guan, W.; Shi, J. Valproic Acid Enhanced Apoptosis by Promoting Autophagy Via Akt/MTOR Signaling in Glioma. Cell Transpl. 2020, 29. [Google Scholar] [CrossRef]
- Riva, G.; Cilibrasi, C.; Bazzoni, R.; Cadamuro, M.; Negroni, C.; Butta, V.; Strazzabosco, M.; Dalprà, L.; Lavitrano, M.; Bentivegna, A. Valproic Acid Inhibits Proliferation and Reduces Invasiveness in Glioma Stem Cells Through Wnt/β Catenin Signalling Activation. Genes 2018, 9, 522. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Shao, C.J.; Chen, F.R.; Ng, H.K.; Chen, Z.P. Autophagy Induced by Valproic Acid Is Associated with Oxidative Stress in Glioma Cell Lines. Neuro. Oncol. 2010, 12, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tsai, Y.H.; Tseng, S.H. Valproic Acid Affected the Survival and Invasiveness of Human Glioma Cells through Diverse Mechanisms. J. Neurooncol. 2012, 109, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Lai, H.Y.; Chiu, A.; Chan, S.H.; Hsiao, L.P.; Lee, S.T. The Effects of Antiepileptic Drugs on the Growth of Glioblastoma Cell Lines. J. Neurooncol. 2016, 127, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, F.; Weßlau, K.; Porath, K.; Hörnschemeyer, J.; Bergner, C.; Krause, B.J.; Mullins, C.S.; Linnebacher, M.; Köhling, R.; Kirschstein, T. AMPA Receptor Antagonist Perampanel Affects Glioblastoma Cell Growth and Glutamate Release in Vitro. PLoS ONE 2019, 14, e0211644. [Google Scholar] [CrossRef] [Green Version]
- Ciusani, E.; Balzarotti, M.; Calatozzolo, C.; De Grazia, U.; Boiardi, A.; Salmaggi, A.; Croci, D. Valproic Acid Increases the in Vitro Effects of Nitrosureas on Human Glioma Cell Lines. Oncol. Res. 2007, 16, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xia, Y.; Bu, X.; Yang, D.; Yuan, Y.; Guo, X.; Zhang, G.; Wang, Z.; Jiao, J. Effects of Valproic Acid on the Susceptibility of Human Glioma Stem Cells for TMZ and ACNU. Oncol. Lett. 2018, 15, 9877–9883. [Google Scholar] [CrossRef]
- Ryu, C.H.; Yoon, W.S.; Park, K.Y.; Kim, S.M.; Lim, J.Y.; Woo, J.S.; Jeong, C.H.; Hou, Y.; Jeun, S.S. Valproic Acid Downregulates the Expression of MGMT and Sensitizes Temozolomide-Resistant Glioma Cells. J. Biomed. Biotechnol. 2012, 987495. [Google Scholar] [CrossRef] [Green Version]
- Van Nifterik, K.A.; Van Den Berg, J.; Slotman, B.J.; Lafleur, M.V.M.; Sminia, P.; Stalpers, L.J.A. Valproic Acid Sensitizes Human Glioma Cells for Temozolomide and γ-Radiation. J. Neurooncol. 2012, 107, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Li, J.R.; Wu, C.C.; Ou, Y.C.; Chen, W.Y.; Kuan, Y.H.; Wang, W.Y.; Chen, C.J. Valproic Acid Sensitizes Human Glioma Cells to Gefitinib-Induced Autophagy. IUBMB Life 2015, 67, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Das, C.M.; Aguilera, D.; Vasquez, H.; Prasad, P.; Zhang, M.; Wolff, J.E.; Gopalakrishnan, V. Valproic Acid Induces P21 and Topoisomerase-II (Alpha/Beta) Expression and Synergistically Enhances Etoposide Cytotoxicity in Human Glioblastoma Cell Lines. J. Neurooncol. 2007, 85, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Y.; Wang, H.; Niu, J.; Hou, H.; Jiang, Y. Histone Deacetylase Inhibitor, Valproic Acid, Radiosensitizes the C6 Glioma Cell Line in Vitro. Oncol. Lett. 2014, 7, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.J.; Yang, Y.H.; Lee, I.Y.; Chen, P.C.; Yang, J.T.; Wang, T.C.; Lin, M.H.C.; Yang, W.H.; Cheng, C.Y.; Chen, K.T.; et al. Effect of Valproic Acid on Overall Survival in Patients with High-Grade Gliomas Undergoing Temozolomide: A Nationwide Population-Based Cohort Study in Taiwan. Medicine 2020, 99, e21147. [Google Scholar] [CrossRef]
- Redjal, N.; Reinshagen, C.; Le, A.; Walcott, B.P.; McDonnell, E.; Dietrich, J.; Nahed, B.V. Valproic Acid, Compared to Other Antiepileptic Drugs, Is Associated with Improved Overall and Progression-Free Survival in Glioblastoma but Worse Outcome in Grade II/III Gliomas Treated with Temozolomide. J. Neurooncol. 2016, 127, 505–514. [Google Scholar] [CrossRef]
- Kerkhof, M.; Dielemans, J.C.M.; Van Breemen, M.S.; Zwinkels, H.; Walchenbach, R.; Taphoorn, M.J.; Vecht, C.J. Effect of Valproic Acid on Seizure Control and on Survival in Patients with Glioblastoma Multiforme. Neuro. Oncol. 2013, 15, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; van den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged Survival with Valproic Acid Use in the EORTC/NCIC Temozolomide Trial for Glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kuwabara, Y.; Suehiro, S.; Yamashita, D.; Tanaka, M.; Tanaka, A.; Ohue, S.; Araki, H. Valproic Acid Reduces Hair Loss and Improves Survival in Patients Receiving Temozolomide-Based Radiation Therapy for High-Grade Glioma. Eur. J. Clin. Pharmacol. 2017, 73, 357–363. [Google Scholar] [CrossRef]
- Krauze, A.V.; Megan, M.; Theresa, C.Z.; Peter, M.; Shih, J.H.; Tofilon, P.J.; Rowe, L.; Gilbert, M.; Camphausen, K. The Addition of Valproic Acid to Concurrent Radiation Therapy and Temozolomide Improves Patient Outcome: A Correlative Analysis of RTOG 0525, SEER and a Phase II NCI Trial. Cancer Stud. Ther. 2020, 5, 1–15. [Google Scholar] [CrossRef]
- Yuan, Y.; Xiang, W.; Qing, M.; Yanhui, L.; Jiewen, L.; Yunhe, M. Survival Analysis for Valproic Acid Use in Adult Glioblastoma Multiforme: A Meta-Analysis of Individual Patient Data and a Systematic Review. Seizure 2014, 23, 830–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langan, Y.M.; Lucas, R.; Jewell, H.; Toublanc, N.; Schaefer, H.; Sander, J.W.A.S.; Patsalos, P.N. Talampanel, a New Antiepileptic Drug: Single- and Multiple-Dose Pharmacokinetics and Initial 1-Week Experience in Patients with Chronic Intractable Epilepsy. Epilepsia 2003, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Howes, J.F.; Bell, C. Talampanel. Neurotherapeutics 2007, 4, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, F.M.; Kreisl, T.N.; Kim, L.; Duic, J.P.; Butman, J.A.; Albert, P.S.; Fine, H.A. Phase 2 Trial of Talampanel, a Glutamate Receptor Inhibitor, for Adults with Recurrent Malignant Gliomas. Cancer 2010, 116, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel with Standard Radiation and Temozolomide in Patients with Newly Diagnosed Glioblastoma: A Multicenter Phase II Trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef] [Green Version]
- Yagi, C.; Tatsuoka, J.; Sano, E.; Hanashima, Y.; Ozawa, Y.; Yoshimura, S.; Yamamuro, S.; Sumi, K.; Hara, H.; Katayama, Y.; et al. Anti-tumor Effects of Anti-epileptic Drugs in Malignant Glioma Cells. Oncol. Rep. 2022, 48, 216. [Google Scholar] [CrossRef]
- Salmaggi, A.; Corno, C.; Maschio, M.; Donzelli, S.; D’urso, A.; Perego, P.; Ciusani, E. Synergistic Effect of Perampanel and Temozolomide in Human Glioma Cell Lines. J. Pers. Med. 2021, 11, 390. [Google Scholar] [CrossRef]
- Tatsuoka, J.; Sano, E.; Hanashima, Y.; Yagi, C.; Yamamuro, S.; Sumi, K.; Hara, H.; Takada, K.; Kanemaru, K.; Komine-Aizawa, S.; et al. Anti-Tumor Effects of Perampanel in Malignant Glioma Cells. Oncol. Lett. 2022, 24, 1–9. [Google Scholar] [CrossRef]
- Mayer, J.; Kirschstein, T.; Resch, T.; Porath, K.; Krause, B.J.; Köhling, R.; Lange, F. Perampanel Attenuates Epileptiform Phenotype in C6 Glioma. Neurosci. Lett. 2020, 715, 134629. [Google Scholar] [CrossRef]
- Lange, F.; Hartung, J.; Liebelt, C.; Boisserée, J.; Resch, T.; Porath, K.; Hörnschemeyer, J.; Reichart, G.; Sellmann, T.; Neubert, V.; et al. Perampanel Add-on to Standard Radiochemotherapy in Vivo Promotes Neuroprotection in a Rodent F98 Glioma Model. Front. Neurosci. 2020, 14, 598266. [Google Scholar] [CrossRef]
- Izumoto, S.; Miyauchi, M.; Tasaki, T.; Okuda, T.; Nakagawa, N.; Nakano, N.; Kato, A.; Fujita, M. Seizures and Tumor Progression in Glioma Patients with Uncontrollable Epilepsy Treated with Perampanel. Anticancer Res. 2018, 38, 4361–4366. [Google Scholar] [CrossRef] [PubMed]
- Gherzi, M.; Milano, G.; Fucile, C.; Calevo, M.G.; Mancardi, M.M.; Nobili, L.; Astuni, P.; Marini, V.; Barco, S.; Cangemi, G.; et al. Safety and Pharmacokinetics of Medical Cannabis Preparation in a Monocentric Series of Young Patients with Drug Resistant Epilepsy. Complement. Ther. Med. 2020, 51, 102402. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.A.; Whalley, B.J. The Proposed Mechanisms of Action of CBD in Epilepsy. Epileptic Disord. 2020, 22, 10–15. [Google Scholar] [CrossRef]
- Vaccani, A.; Massi, P.; Colombo, A.; Rubino, T.; Parolaro, D. Cannabidiol Inhibits Human Glioma Cell Migration through a Cannabinoid Receptor-Independent Mechanism. Br. J. Pharmacol. 2005, 144, 1032–1036. [Google Scholar] [CrossRef] [Green Version]
- López-Valero, I.; Saiz-Ladera, C.; Torres, S.; Hernández-Tiedra, S.; García-Taboada, E.; Rodríguez-Fornés, F.; Barba, M.; Dávila, D.; Salvador-Tormo, N.; Guzmán, M.; et al. Targeting Glioma Initiating Cells with A Combined Therapy of Cannabinoids and Temozolomide. Biochem. Pharmacol. 2018, 157, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Volmar, M.N.M.; Cheng, J.; Alenezi, H.; Richter, S.; Haug, A.; Hassan, Z.; Goldberg, M.; Li, Y.; Hou, M.; Herold-Mende, C.; et al. Cannabidiol Converts NF-ΚB into a Tumor Suppressor in Glioblastoma with Defined Antioxidative Properties. Neuro. Oncol. 2021, 23, 1898. [Google Scholar] [CrossRef] [PubMed]
- Likar, R.; Koestenberger, M.; Stutschnig, M.; Nahler, G. Cannabidiol Μay Prolong Survival in Patients with Glioblastoma Multiforme. Cancer Diagn. Progn. 2021, 1, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C.; McBain, C.; Haylock, B.; et al. A Phase 1b Randomised, Placebo-Controlled Trial of Nabiximols Cannabinoid Oromucosal Spray with Temozolomide in Patients with Recurrent Glioblastoma. Br. J. Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef]
- Doello, K.; Mesas, C.; Quiñonero, F.; Rama, A.R.; Vélez, C.; Perazzoli, G.; Ortiz, R. Antitumor Effect of Traditional Drugs for Neurological Disorders: Preliminary Studies in Neural Tumor Cell Lines. Neurotox. Res. 2022, 40, 1645–1652. [Google Scholar] [CrossRef]
- Rizzo, A.; Donzelli, S.; Girgenti, V.; Sacconi, A.; Vasco, C.; Salmaggi, A.; Blandino, G.; Maschio, M.; Ciusani, E. In Vitro Antineoplastic Effects of Brivaracetam and Lacosamide on Human Glioma Cells. J. Exp. Clin. Cancer Res. 2017, 36, 76. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.R.; Ambavade, S.D.; Jagdale, P.G.; Adkar, P.P.; Waghmare, A.B.; Ambavade, P.D. Lacosamide Reduces HDAC Levels in the Brain and Improves Memory: Potential for Treatment of Alzheimer’s Disease. Pharm. Biochem. Behav. 2015, 134, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Villa, L.S.; Yeon, S.K.; Householder, K.T.; Park, K.D.; Sirianni, R.W.; Khanna, R. CRMP2 Phosphorylation Drives Glioblastoma Cell Proliferation. Mol. Neurobiol. 2018, 55, 4403–4416. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Rizza, P.; Nigro, A.; Ceraldi, R.; Ricci, E.; Perrotta, I.; Aquila, S.; Lanzino, M.; Andò, S.; Morelli, C.; et al. FoxO3a Mediates the Inhibitory Effects of the Antiepileptic Drug Lamotrigine on Breast Cancer Growth. Mol. Cancer Res. 2018, 16, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Kumakura, S.; Someya, A.; Iseki, M.; Inada, E.; Nagaoka, I. Anti-inflammatory Actions of Gabapentin and Pregabalin on the Substance P-induced Mitogen-activated Protein Kinase Activation in U373 MG Human Glioblastoma Astrocytoma Cells. Mol. Med. Rep. 2017, 16, 6109–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Stiripentol: A Review in Dravet Syndrome. Drugs 2019, 79, 1785–1796. [Google Scholar] [CrossRef]
- Yadav, A.; Alnakhli, A.; Vemana, H.P.; Bhutkar, S.; Muth, A.; Dukhande, V.V. Repurposing an Antiepileptic Drug for the Treatment of Glioblastoma. Pharm. Res. 2022, 39, 2871–2883. [Google Scholar] [CrossRef]
- Guyon, J.; Fernandez-Moncada, I.; Larrieu, C.M.; Bouchez, C.L.; Pagano Zottola, A.C.; Galvis, J.; Chouleur, T.; Burban, A.; Joseph, K.; Ravi, V.M.; et al. Lactate Dehydrogenases Promote Glioblastoma Growth and Invasion via a Metabolic Symbiosis. EMBO Mol. Med. 2022, 14, e15343. [Google Scholar] [CrossRef]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Epilepsy Treatment. Targeting LDH Enzymes with a Stiripentol Analog to Treat Epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An in Vivo Study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Min, K.L.; Chang, M.J. Effect of Anti-Epileptic Drugs on the Survival of Patients with Glioblastoma Multiforme: A Retrospective, Single-Center Study. PLoS ONE 2019, 14, e0225599. [Google Scholar] [CrossRef] [Green Version]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients with Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | Antiepileptic Targets |
Antitumor Targets | Preclinical Studies |
Clinical Studies |
#Clinical Trials * |
|---|---|---|---|---|---|
| Brivaracetam |
Na+ channels SV2A GABA | MGMT | [172] | [51] | - |
| Cannabidiol/cannabinoids |
TRPV1 GPR55 ENT-1 | NF-κb | [166,167,168] | [169,170] |
NCT03529448 NR NCT03607643 NR NCT03529448 NR NCT0181260 C NCT01812616 C |
| Carbamazepine | Na+ channels | u.k. | [158] | - | - |
| Lamotrigine |
Na+ channels Ca2+ channels | PTEN PI3K/Akt | [138] | - | - |
| Lacosamide | Na+ channels | HDAC CRMP2 | [172,174] | - | - |
| Levetiracetam | SV2A Ca2+ channels GABA | MGMT HDAC | [117,118,119,120,171] | [121,122,123,125,126,182] | NCT03048084 R NCT00629889 C NCT02815410 |
| Perampanel | AMPAR | AMPA/glutamate | [139,160,159] | [163] | NCT04650204 R |
| Pregabalin | α2δ subunit Ca2+ channels | p38 MAPK NF-κB | [176] | - | NCT00629889 C |
| Stiripentol | GABAAR | LDH | [178,179] | - | - |
| Topiramate | Na+ channels GABAAR AMPA/kainateR Ca2+ channels | u.k. | [138] | - | - |
| Valproic Acid | GABA Na+ channels NMDAR Ca2+ channels | HDAC BDNF ERK/Akt Akt/mTOR Wnt | [133,134,135,136,137,138,140,141,142,143,144,145,146,171] | [147,148,149,150,151,182,183] | NCT01817751 NR NCT03243461 R NCT03048084 R NCT00879437 C NCT00302159 C NCT02648633 C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, M.; Baiardi, G.; Pasquariello, S.; Sacco, F.; Dellacasagrande, I.; Corsaro, A.; Mattioli, F.; Barbieri, F. Antitumor Potential of Antiepileptic Drugs in Human Glioblastoma: Pharmacological Targets and Clinical Benefits. Biomedicines 2023, 11, 582. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020582
Stella M, Baiardi G, Pasquariello S, Sacco F, Dellacasagrande I, Corsaro A, Mattioli F, Barbieri F. Antitumor Potential of Antiepileptic Drugs in Human Glioblastoma: Pharmacological Targets and Clinical Benefits. Biomedicines. 2023; 11(2):582. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020582
Chicago/Turabian StyleStella, Manuela, Giammarco Baiardi, Stefano Pasquariello, Fabio Sacco, Irene Dellacasagrande, Alessandro Corsaro, Francesca Mattioli, and Federica Barbieri. 2023. "Antitumor Potential of Antiepileptic Drugs in Human Glioblastoma: Pharmacological Targets and Clinical Benefits" Biomedicines 11, no. 2: 582. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11020582