
Concordant and Discordant Cerebrospinal Fluid and Plasma Cytokine and Chemokine Responses in Mild Cognitive Impairment and Early-Stage Alzheimer’s Disease

Abstract
:1. Introduction
2. Subjects and Methodological Approaches
2.1. Human Subjects
2.2. Direct Binding Enzyme-Linked Immunosorbent Assay (ELISA)
2.3. Multiplex ELISA
2.4. Materials and Reagents
2.5. Statistics
3. Results
3.1. Group Participant Features
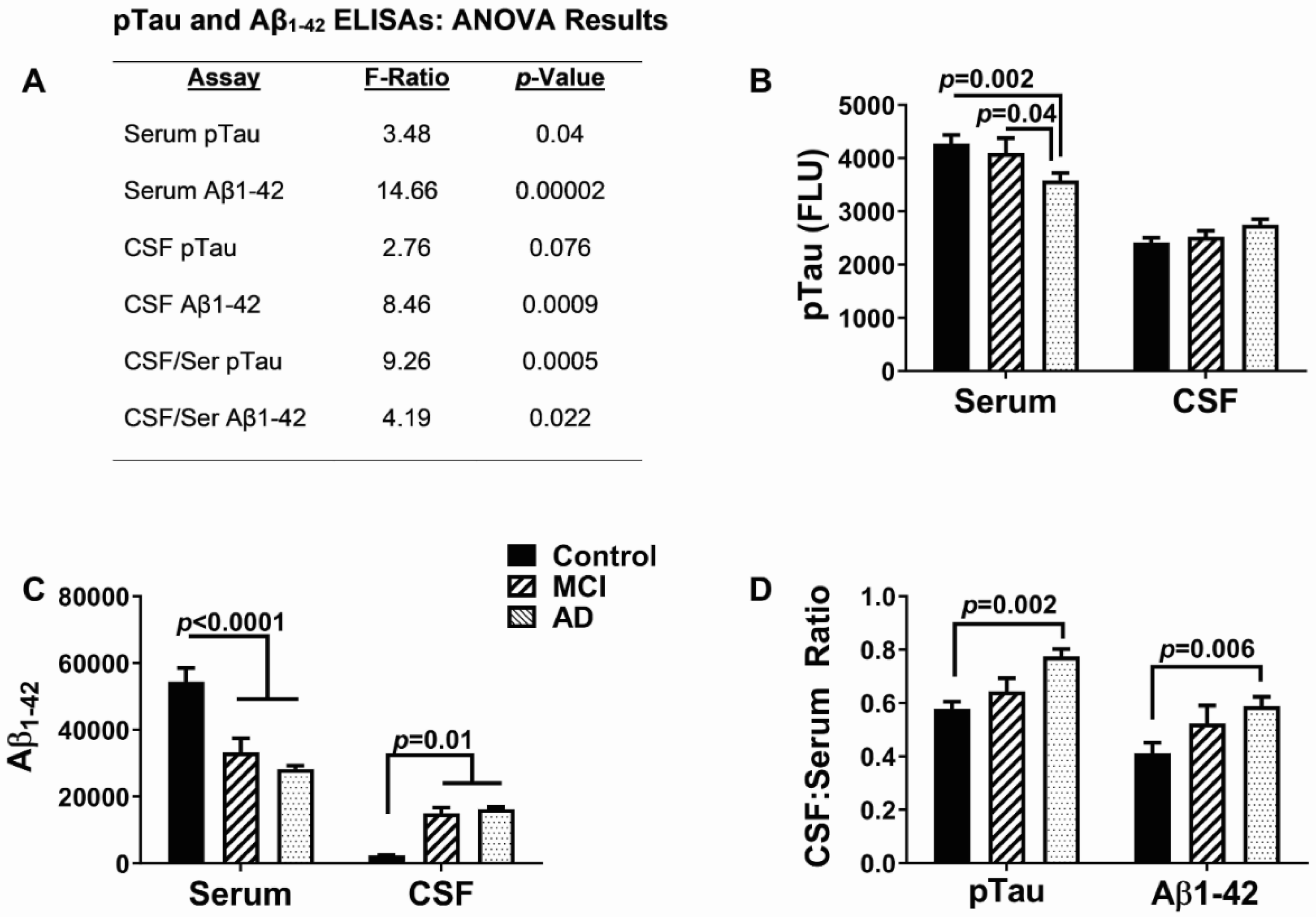
3.2. AD Biomarkers
3.3. Cytokine and Chemokine Responses
3.4. Serum-MCI/AD Effects on Cytokine/Chemokine/Trophic Factor Expression (Figure 2, Figure 3 and Figure 4)
3.5. CSF-MCI/AD Effects on Cytokine/Chemokine/Trophic Factor Expression (Figure 5, Figure 6 and Figure 7)
3.6. Concordant and Discordant CSF/Serum Responses in MCI/AD
4. Discussion
4.1. Overview
4.2. Subgroup Characteristics
4.3. Overall Cytokine/Chemokine Alterations in MCI/AD
4.4. Serum Cytokines/Chemokines—Implications of Specific Alterations
4.5. CSF Cytokines/Chemokines—Implications of Specific Alterations
4.6. Concordant Versus Discordant CSF-Serum Responses
4.7. Strengths and Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gomez, M.G.; Langois, C.M.; Langbaum, J.B.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol. 2012, 11, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Cselenyi, Z.; Jonhagen, M.E.; Forsberg, A.; Halldin, C.; Julin, P.; Schou, M.; Johnstrom, P.; Varnas, K.; Svensson, S.; Farde, L. Clinical validation of 18F-AZD4694, an amyloid-beta-specific PET radioligand. J. Nucl. Med. 2012, 53, 415–424. [Google Scholar] [CrossRef]
- Babic, M.; Svob Strac, D.; Muck-Seler, D.; Pivac, N.; Stanic, G.; Hof, P.R.; Simic, G. Update on the core and developing cerebrospinal fluid biomarkers for Alzheimer disease. Croat. Med. J. 2014, 55, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Cure, S.; Abrams, K. Systematic literature review and meta-analysis of diagnostic test accuracy in Alzheimer’s disease and other dementia using autopsy as standard of truth. J. Alzheimers Dis. 2014, 42, 169. [Google Scholar] [CrossRef]
- de Souza, L.C.; Sarazin, M.; Teixeira-Junior, A.L.; Caramelli, P.; Santos, A.E.; Dubois, B. Biological markers of Alzheimer’s disease. Arq. Neuropsiquiatr. 2014, 72, 227–231. [Google Scholar] [CrossRef]
- Sargent, L.; Brown, R. Assessing the Current State of Cognitive Frailty: Measurement Properties. J. Nutr. Health. Aging 2017, 21, 152–160. [Google Scholar] [CrossRef]
- Dandrea, M.R.; Reiser, P.A.; Gumula, N.A.; Hertzog, B.M.; Andrade-Gordon, P. Application of triple immunohistochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimer’s disease. Biotech. Histochem. 2001, 76, 97–106. [Google Scholar] [CrossRef]
- Mehlhorn, G.; Hollborn, M.; Schliebs, R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int. J. Dev. Neurosci. 2000, 18, 423–431. [Google Scholar] [CrossRef]
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Cerebrospinal fluid biomarkers for Alzheimer’s disease: Current limitations and recent developments. Curr. Opin. Psychiatry 2015, 28, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Singhal, G.; Jaehne, E.J.; Corrigan, F.; Toben, C.; Baune, B.T. Inflammasomes in neuroinflammation and changes in brain function: A focused review. Front. Neurosci. 2014, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38MAPK pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, R.J.; Palmer, R.F.; Royall, D.R. Serum interleukin (IL)-15 as a biomarker of Alzheimer’s disease. PLoS ONE 2015, 10, e0117282. [Google Scholar] [CrossRef]
- Erhardt, E.B.; Adair, J.C.; Knoefel, J.E.; Caprihan, A.; Prestopnik, J.; Thompson, J.; Hobson, S.; Siegel, D.; Rosenberg, G.A. Inflammatory Biomarkers Aid in Diagnosis of Dementia. Front. Aging Neurosci. 2021, 13, 717344. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Yu, C.; Hsuchou, H.; Zhang, Y.; Kastin, A.J. Neuroinflammation facilitates LIF entry into brain: Role of TNF. Am. J. Physiol. Cell Physiol. 2008, 294, C1436–C1442. [Google Scholar] [CrossRef]
- Aisen, P.S. The potential of anti-inflammatory drugs for the treatment of Alzheimer’s disease. Lancet Neurol. 2002, 1, 279–284. [Google Scholar] [CrossRef]
- Meyer, P.F.; Labonte, A.; Rosa-Neto, P.; Poirier, J.; Breitner, J.C.S.; Group, P.-A.R. No apparent effect of naproxen on CSF markers of innate immune activation. Ann. Clin. Transl. Neurol. 2019, 6, 1127–1133. [Google Scholar] [CrossRef]
- Jelic, V.; Kivipelto, M.; Winblad, B. Clinical trials in mild cognitive impairment: Lessons for the future. J. Neurol. Neurosurg Psychiatry 2006, 77, 429–438. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- Winblad, B.; Palmer, K.; Kivipelto, M.; Jelic, V.; Fratiglioni, L.; Wahlund, L.O.; Nordberg, A.; Backman, L.; Albert, M.; Almkvist, O.; et al. Mild cognitive impairment--beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 2004, 256, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Neusner, A.; Longato, L.; Lawton, M.; Wands, J.R.; de la Monte, S.M. Nitrosamine exposure causes insulin resistance diseases: Relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 827–844. [Google Scholar] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, M.E.; Ikezu, T. Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 92–101. [Google Scholar] [CrossRef]
- Yoshimura, S.; Teramoto, T.; Whalen, M.J.; Irizarry, M.C.; Takagi, Y.; Qiu, J.; Harada, J.; Waeber, C.; Breakefield, X.O.; Moskowitz, M.A. FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J. Clin. Investig. 2003, 112, 1202–1210. [Google Scholar] [CrossRef]
- Cummings, B.J.; Su, J.H.; Cotman, C.W. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp. Neurol. 1993, 124, 315–325. [Google Scholar] [CrossRef]
- Nakamichi, M.; Akishima-Fukasawa, Y.; Fujisawa, C.; Mikami, T.; Onishi, K.; Akasaka, Y. Basic Fibroblast Growth Factor Induces Angiogenic Properties of Fibrocytes to Stimulate Vascular Formation during Wound Healing. Am. J. Pathol. 2016, 186, 3203–3216. [Google Scholar] [CrossRef]
- Clark, C.; Richiardi, J.; Marechal, B.; Bowman, G.L.; Dayon, L.; Popp, J. Systemic and central nervous system neuroinflammatory signatures of neuropsychiatric symptoms and related cognitive decline in older people. J. Neuroinflammation 2022, 19, 127. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.K.; Giles, D.A.; Segal, B.M.; Irani, D.N. An emerging role for eotaxins in neurodegenerative disease. Clin. Immunol. 2018, 189, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Tsai, R.K.; Chang, C.H.; Wang, H.Z. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp. Eye Res. 2008, 87, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Han, S.H.; Mook-Jung, I. Peripheral inflammatory biomarkers in Alzheimer’s disease: A brief review. BMB Rep. 2020, 53, 10–19. [Google Scholar] [CrossRef]
- Schmitz, M.; Hermann, P.; Oikonomou, P.; Stoeck, K.; Ebert, E.; Poliakova, T.; Schmidt, C.; Llorens, F.; Zafar, S.; Zerr, I. Cytokine profiles and the role of cellular prion protein in patients with vascular dementia and vascular encephalopathy. Neurobiol. Aging 2015, 36, 2597–2606. [Google Scholar] [CrossRef]
- Schabitz, W.R.; Kruger, C.; Pitzer, C.; Weber, D.; Laage, R.; Gassler, N.; Aronowski, J.; Mier, W.; Kirsch, F.; Dittgen, T.; et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J. Cereb. Blood Flow. Metab. 2008, 28, 29–43. [Google Scholar] [CrossRef]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Yokoyama, I.; Mosley, R.L.; Gendelman, H.E. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J. Neuroimmunol. 2018, 319, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kosloski, L.M.; Kosmacek, E.A.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J. Neuroimmunol. 2013, 265, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, A.; Usui, T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediat. Inflamm. 2015, 2015, 568543. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.A.; Collier, L.A.; Chapman, C.B.; Benkovic, S.A.; Willing, A.E.; Pennypacker, K.R. Pro-inflammatory interferon gamma signaling is directly associated with stroke induced neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 679–689. [Google Scholar] [CrossRef]
- Walter, J.; Dihne, M. Species-dependent differences of embryonic stem cell-derived neural stem cells after Interferon gamma treatment. Front. Cell Neurosci. 2012, 6, 52. [Google Scholar] [CrossRef]
- Su, C.; Zhao, K.; Xia, H.; Xu, Y. Peripheral inflammatory biomarkers in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Psychogeriatrics 2019, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Belkhelfa, M.; Rafa, H.; Medjeber, O.; Arroul-Lammali, A.; Behairi, N.; Abada-Bendib, M.; Makrelouf, M.; Belarbi, S.; Masmoudi, A.N.; Tazir, M.; et al. IFN-gamma and TNF-alpha are involved during Alzheimer disease progression and correlate with nitric oxide production: A study in Algerian patients. J. Interferon Cytokine Res. 2014, 34, 839–847. [Google Scholar] [CrossRef]
- King, E.; O’Brien, J.T.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.P.; Thomas, A.J. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J. Neurol. Neurosurg. Psychiatry 2018, 89, 339–345. [Google Scholar] [CrossRef]
- Jana, M.; Mondal, S.; Jana, A.; Pahan, K. Interleukin-12 (IL-12), but not IL-23, induces the expression of IL-7 in microglia and macrophages: Implications for multiple sclerosis. Immunology 2014, 141, 549–563. [Google Scholar] [CrossRef]
- Jana, M.; Pahan, K. IL-12 p40 homodimer, but not IL-12 p70, induces the expression of IL-16 in microglia and macrophages. Mol. Immunol. 2009, 46, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Mancino, R.; Bergami, A.; Mori, F.; Castelli, M.; De Chiara, V.; Studer, V.; Mataluni, G.; Sancesario, G.; Parisi, V.; et al. Potential role of IL-13 in neuroprotection and cortical excitability regulation in multiple sclerosis. Mult. Scler. 2011, 17, 1301–1312. [Google Scholar] [CrossRef]
- Mori, S.; Maher, P.; Conti, B. Neuroimmunology of the Interleukins 13 and 4. Brain. Sci. 2016, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Dayon, L.; Kirkland, R.; Wojcik, J.; Peyratout, G.; Severin, I.C.; Henry, H.; Oikonomidi, A.; Migliavacca, E.; Bacher, M.; et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement. 2018, 14, 1640–1650. [Google Scholar] [CrossRef]
- Gautam, A.S.; Pulivarthi, C.B.; Singh, R.K. Proinflammatory IL-17 levels in serum/cerebrospinal fluid of patients with neurodegenerative diseases: A meta-analysis study. Naunyn. Schmiedebergs Arch. Pharmacol. 2023, 396, 577–588. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Swardfager, W.; Winer, D.A.; Herrmann, N.; Winer, S.; Lanctot, K.L. Interleukin-17 in post-stroke neurodegeneration. Neurosci. Biobehav. Rev. 2013, 37, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Studer, V.; Macchiarulo, G.; Volpe, E.; Barbieri, F.; Ruocco, G.; Buttari, F.; Finardi, A.; Mancino, R.; et al. Interleukin-1beta causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol. Neurodegener. 2014, 9, 56. [Google Scholar] [CrossRef]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and inflammatory neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Meola, D.; Huang, Z.; Ha, G.K.; Petitto, J.M. Loss of Neuronal Phenotype and Neurodegeneration: Effects of T Lymphocytes and Brain Interleukin-2. J. Alzheimers Dis. Park. 2013, 3 (Suppl. 10), 003. [Google Scholar] [CrossRef] [PubMed]
- King, E.; O’Brien, J.T.; Donaghy, P.; Morris, C.; Barnett, N.; Olsen, K.; Martin-Ruiz, C.; Taylor, J.P.; Thomas, A.J. Peripheral inflammation in mild cognitive impairment with possible and probable Lewy body disease and Alzheimer’s disease. Int. Psychogeriatr. 2019, 31, 551–560. [Google Scholar] [CrossRef]
- Ayari, S.; Abellard, A.; Carayol, M.; Guedj, E.; Gavarry, O. A systematic review of exercise modalities that reduce pro-inflammatory cytokines in humans and animals’ models with mild cognitive impairment or dementia. Exp. Gerontol. 2023, 175, 112141. [Google Scholar] [CrossRef]
- Motta, C.; Finardi, A.; Toniolo, S.; Di Lorenzo, F.; Scaricamazza, E.; Loizzo, S.; Mercuri, N.B.; Furlan, R.; Koch, G.; Martorana, A. Protective Role of Cerebrospinal Fluid Inflammatory Cytokines in Patients with Amnestic Mild Cognitive Impairment and Early Alzheimer’s Disease Carrying Apolipoprotein E4 Genotype. J. Alzheimers Dis. 2020, 76, 681–689. [Google Scholar] [CrossRef]
- Elomaa, A.P.; Niskanen, L.; Herzig, K.H.; Viinamaki, H.; Hintikka, J.; Koivumaa-Honkanen, H.; Honkalampi, K.; Valkonen-Korhonen, M.; Harvima, I.T.; Lehto, S.M. Elevated levels of serum IL-5 are associated with an increased likelihood of major depressive disorder. BMC Psychiatry 2012, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Liva, S.M.; de Vellis, J. IL-5 induces proliferation and activation of microglia via an unknown receptor. Neurochem. Res. 2001, 26, 629–637. [Google Scholar] [CrossRef]
- Hazen, J.; Vistnes, M.; Barca, M.L.; Eldholm, R.S.; Persson, K.; Braekhus, A.; Saltvedt, I.; Selbaek, G.; Engedal, K.; Knapskog, A.B. The Association Between Circulating Inflammatory Markers and the Progression of Alzheimer Disease in Norwegian Memory Clinic Patients With Mild Cognitive Impairment or Dementia. Alzheimer. Dis. Assoc. Disord. 2020, 34, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-alpha, TGF-beta and IFN-gamma. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Spittau, B.; Zhou, X.; Ming, M.; Krieglstein, K. IL6 protects MN9D cells and midbrain dopaminergic neurons from MPP+-induced neurodegeneration. Neuromolecular. Med. 2012, 14, 317–327. [Google Scholar] [CrossRef]
- Anderson, K.M.; Olson, K.E.; Estes, K.A.; Flanagan, K.; Gendelman, H.E.; Mosley, R.L. Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl. Neurodegener. 2014, 3, 25. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Sagare, A.; Pachicano, M.; Sweeney, M.D.; Toga, A.; Zlokovic, B.; Chui, H.; Joe, E.; Schneider, L.; Morris, J.C.; et al. Early neuroinflammation is associated with lower amyloid and tau levels in cognitively normal older adults. Brain. Behav. Immun. 2021, 94, 299–307. [Google Scholar] [CrossRef]
- Chen, D.; Tang, T.X.; Deng, H.; Yang, X.P.; Tang, Z.H. Interleukin-7 Biology and Its Effects on Immune Cells: Mediator of Generation, Differentiation, Survival, and Homeostasis. Front. Immunol. 2021, 12, 747324. [Google Scholar] [CrossRef] [PubMed]
- Belarif, L.; Mary, C.; Jacquemont, L.; Mai, H.L.; Danger, R.; Hervouet, J.; Minault, D.; Thepenier, V.; Nerriere-Daguin, V.; Nguyen, E.; et al. IL-7 receptor blockade blunts antigen-specific memory T cell responses and chronic inflammation in primates. Nat. Commun. 2018, 9, 4483. [Google Scholar] [CrossRef]
- Delaby, C.; Gabelle, A.; Blum, D.; Schraen-Maschke, S.; Moulinier, A.; Boulanghien, J.; Severac, D.; Buee, L.; Reme, T.; Lehmann, S. Central Nervous System and Peripheral Inflammatory Processes in Alzheimer’s Disease: Biomarker Profiling Approach. Front. Neurol. 2015, 6, 181. [Google Scholar] [CrossRef]
- Aksnes, M.; Aass, H.C.D.; Tiiman, A.; Edwin, T.H.; Terenius, L.; Bogdanovic, N.; Vukojevic, V.; Knapskog, A.B. Associations of cerebrospinal fluid amyloidogenic nanoplaques with cytokines in Alzheimer’s disease. Transl. Neurodegener. 2021, 10, 18. [Google Scholar] [CrossRef]
- Sajjad, M.U.; Blennow, K.; Knapskog, A.B.; Idland, A.V.; Chaudhry, F.A.; Wyller, T.B.; Zetterberg, H.; Watne, L.O. Cerebrospinal Fluid Levels of Interleukin-8 in Delirium, Dementia, and Cognitively Healthy Patients. J. Alzheimers Dis. 2020, 73, 1363–1372. [Google Scholar] [CrossRef]
- Contreras, J.A.; Aslanyan, V.; Albrecht, D.S.; Mack, W.J.; Alzheimer’s Disease Neuroimaging, I.; Pa, J. Higher baseline levels of CSF inflammation increase risk of incident mild cognitive impairment and Alzheimer’s disease dementia. Alzheimers Dement. (Amst.) 2022, 14, e12346. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain. Behav. Immun. 2011, 25, 539–547. [Google Scholar] [CrossRef]
- Kuhn, M.K.; Fleeman, R.M.; Beidler, L.M.; Snyder, A.M.; Chan, D.C.; Proctor, E.A. Alzheimer’s disease-specific cytokine secretion suppresses neuronal mitochondrial metabolism. BioRxiv 2023. [Google Scholar] [CrossRef]
- Yang, J.; Richmond, A. The angiostatic activity of interferon-inducible protein-10/CXCL10 in human melanoma depends on binding to CXCR3 but not to glycosaminoglycan. Mol. Ther. 2004, 9, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Scarpini, E. Inflammation and oxidative damage in Alzheimer’s disease: Friend or foe? Front. Biosci. (Schol. Ed.) 2011, 3, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Schoonenboom, N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.K. Inflammatory Biomarkers in AD: Implications for Diagnosis. Curr. Alzheimer Res. 2020, 17, 962–971. [Google Scholar] [CrossRef]
- Sogorb-Esteve, A.; Swift, I.J.; Woollacott, I.O.C.; Warren, J.D.; Zetterberg, H.; Rohrer, J.D. Differential chemokine alteration in the variants of primary progressive aphasia-a role for neuroinflammation. J. Neuroinflammation 2021, 18, 224. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Bethel-Brown, C.; Yao, H.; Hu, G.; Buch, S. Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: Implications for HIV-associated neuroinflammation. J. Neuroinflammation 2012, 9, 262. [Google Scholar] [CrossRef]
- Askovic, S.; Favara, C.; McAtee, F.J.; Portis, J.L. Increased expression of MIP-1 alpha and MIP-1 beta mRNAs in the brain correlates spatially and temporally with the spongiform neurodegeneration induced by a murine oncornavirus. J. Virol. 2001, 75, 2665–2674. [Google Scholar] [CrossRef]
- Wu, Y.P.; Proia, R.L. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8425–8430. [Google Scholar] [CrossRef]
- Kapoor, A.; Nation, D.A.; Alzheimer’s Disease Neuroimaging, I. Platelet-derived growth factor-BB and white matter hyperintensity burden in APOE4 carriers. Cereb. Circ. Cogn. Behav. 2022, 3, 100131. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Ohlsson, M.; Minthon, L.; Hansson, O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS ONE 2012, 7, e29868. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Abeta accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood. Flow Metab. 2018, 38, 103–115. [Google Scholar] [CrossRef]
- Deuel, T.F.; Senior, R.M.; Huang, J.S.; Griffin, G.L. Chemotaxis of monocytes and neutrophils to platelet-derived growth factor. J. Clin. Investig. 1982, 69, 1046–1049. [Google Scholar] [CrossRef]
- Mohapel, P.; Frielingsdorf, H.; Haggblad, J.; Zachrisson, O.; Brundin, P. Platelet-derived growth factor (PDGF-BB) and brain-derived neurotrophic factor (BDNF) induce striatal neurogenesis in adult rats with 6-hydroxydopamine lesions. Neuroscience 2005, 132, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Tokami, H.; Ago, T.; Sugimori, H.; Kuroda, J.; Awano, H.; Suzuki, K.; Kiyohara, Y.; Kamouchi, M.; Kitazono, T.; Investigators, R. RANTES has a potential to play a neuroprotective role in an autocrine/paracrine manner after ischemic stroke. Brain. Res. 2013, 1517, 122–132. [Google Scholar] [CrossRef]
- Lee, H.P.; Jun, Y.C.; Choi, J.K.; Kim, J.I.; Carp, R.I.; Kim, Y.S. The expression of RANTES and chemokine receptors in the brains of scrapie-infected mice. J. Neuroimmunol. 2005, 158, 26–33. [Google Scholar] [CrossRef]
- Vacinova, G.; Vejrazkova, D.; Rusina, R.; Holmerova, I.; Vankova, H.; Jarolimova, E.; Vcelak, J.; Bendlova, B.; Vankova, M. Regulated upon activation, normal T cell expressed and secreted (RANTES) levels in the peripheral blood of patients with Alzheimer’s disease. Neural. Regen. Res. 2021, 16, 796–800. [Google Scholar] [CrossRef]
- Sasayama, D.; Hattori, K.; Yokota, Y.; Matsumura, R.; Teraishi, T.; Yoshida, S.; Kunugi, H. Increased apolipoprotein E and decreased TNF-alpha in the cerebrospinal fluid of nondemented APOE-epsilon4 carriers. Neuropsychopharmacol. Rep. 2020, 40, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Chen, S.; Xuran, L.; Dong, J.; Chen, W.; Tao, S.; Yang, W.; Zhang, Y. The role of vascular endothelial growth factor in ischemic stroke. Pharmazie 2021, 76, 127–131. [Google Scholar] [CrossRef]
- Ribatti, D.; Guidolin, D. Morphogenesis of vascular and neuronal networks and the relationships between their remodeling processes. Brain. Res. Bull. 2022, 186, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hohman, T.J.; Bell, S.P.; Jefferson, A.L.; Alzheimer’s Disease Neuroimaging, I. The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: Exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol. 2015, 72, 520–529. [Google Scholar] [CrossRef]
- Rattner, A.; Wang, Y.; Nathans, J. Signaling Pathways in Neurovascular Development. Annu. Rev. Neurosci. 2022, 45, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.R.; Shah, B.; Ruiz de Almodovar, C. Cellular and Molecular Mechanisms of Spinal Cord Vascularization. Front. Physiol. 2020, 11, 599897. [Google Scholar] [CrossRef]
- Dabiri, S.; Ramirez Ruiz, M.I.; Jean-Louis, G.; Ntekim, O.E.; Obisesan, T.O.; Campbell, A.L.; Mwendwa, D.T.; Alzheimer’s Disease Neuroimaging, I. The Mediating Role of Inflammation in the Relationship Between alpha-Synuclein and Cognitive Functioning. J. Gerontol. A Biol. Sci. Med. Sci. 2023, 78, 206–212. [Google Scholar] [CrossRef]
- Walter, J.; Honsek, S.D.; Illes, S.; Wellen, J.M.; Hartung, H.P.; Rose, C.R.; Dihne, M. A new role for interferon gamma in neural stem/precursor cell dysregulation. Mol. Neurodegener. 2011, 6, 18. [Google Scholar] [CrossRef]
- Brenneman, D.E.; Hauser, J.; Spong, C.Y.; Phillips, T.M. Chemokines released from astroglia by vasoactive intestinal peptide. Mechanism of neuroprotection from HIV envelope protein toxicity. Ann. N. Y. Acad. Sci. 2000, 921, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Brenneman, D.E.; Hauser, J.M.; Spong, C.; Phillips, T.M. Chemokine release is associated with the protective action of PACAP-38 against HIV envelope protein neurotoxicity. Neuropeptides 2002, 36, 271–280. [Google Scholar] [CrossRef]
- Cabezas, R.; Avila, M.F.; Gonzalez, J.; El-Bacha, R.S.; Barreto, G.E. PDGF-BB protects mitochondria from rotenone in T98G cells. Neurotox Res. 2015, 27, 355–367. [Google Scholar] [CrossRef]
- Cabezas, R.; Vega-Vela, N.E.; Gonzalez-Sanmiguel, J.; Gonzalez, J.; Esquinas, P.; Echeverria, V.; Barreto, G.E. PDGF-BB Preserves Mitochondrial Morphology, Attenuates ROS Production, and Upregulates Neuroglobin in an Astrocytic Model Under Rotenone Insult. Mol. Neurobiol. 2018, 55, 3085–3095. [Google Scholar] [CrossRef]
- Krupinski, J.; Issa, R.; Bujny, T.; Slevin, M.; Kumar, P.; Kumar, S.; Kaluza, J. A putative role for platelet-derived growth factor in angiogenesis and neuroprotection after ischemic stroke in humans. Stroke 1997, 28, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar] [CrossRef]
- de la Monte, S.M. Metabolic derangements mediate cognitive impairment and Alzheimer’s disease: Role of peripheral insulin-resistance diseases. Panminerva. Medica. 2012, 54, 171–178. [Google Scholar] [PubMed]
- de la Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp. Mol. Med. 2015, 47, e149. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Sridhar, G.R.; Lakshmi, G.; Nagamani, G. Emerging links between type 2 diabetes and Alzheimer’s disease. World J. Diabetes 2015, 6, 744–751. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Ott, B.R.; Jones, R.; Daiello, L.A.; de la Monte, S.M.; Stopa, E.G.; Johanson, C.E.; Denby, C.; Grammas, P. Blood-Cerebrospinal Fluid Barrier Gradients in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Inflammatory Cytokines and Chemokines. Front. Aging Neurosci. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Tong, M.; Daiello, L.A.; Ott, B.R. Early-Stage Alzheimer’s Disease Is Associated with Simultaneous Systemic and Central Nervous System Dysregulation of Insulin-Linked Metabolic Pathways. J. Alzheimers Dis. 2019, 68, 657–668. [Google Scholar] [CrossRef]
- Lewczuk, P.; Mroczko, B.; Fagan, A.; Kornhuber, J. Biomarkers of Alzheimer’s disease and mild cognitive impairment: A current perspective. Adv. Med. Sci. 2015, 60, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Takwoingi, Y.; Flicker, L.; Mason, S.E.; McShane, R. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 6, CD008782. [Google Scholar] [CrossRef]
- Pillai, J.A.; Maxwell, S.; Bena, J.; Bekris, L.M.; Rao, S.M.; Chance, M.; Lamb, B.T.; Leverenz, J.B.; Alzheimer’s Disease Neuroimaging, I. Key inflammatory pathway activations in the MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 1248–1262. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Amin, J.; Erskine, D.; Donaghy, P.C.; Surendranathan, A.; Swann, P.; Kunicki, A.P.; Boche, D.; Holmes, C.; McKeith, I.G.; O’Brien, J.T.; et al. Inflammation in dementia with Lewy bodies. Neurobiol. Dis. 2022, 168, 105698. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S. Glial cytokines in Alzheimer’s disease: Review and pathogenic implications. Hum. Pathol. 1995, 26, 816–823. [Google Scholar] [CrossRef]
- Banks, W.A. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Banks, W.A.; Plotkin, S.R.; Kastin, A.J. Permeability of the blood-brain barrier to soluble cytokine receptors. Neuroimmunomodulation 1995, 2, 161–165. [Google Scholar] [CrossRef]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry (Edgmont) 2009, 6, 18–22. [Google Scholar]
- Etiene, D.; Kraft, J.; Ganju, N.; Gomez-Isla, T.; Gemelli, B.; Hyman, B.T.; Hedley-Whyte, E.T.; Wands, J.R.; de la Monte, S.M. Cerebrovascular pathology contributes to the heterogeneity of Alzheimer’s Disease. J. Alzheimers Dis. 1998, 1, 119–134. [Google Scholar] [CrossRef]
- Rauchmann, B.S.; Sadlon, A.; Perneczky, R.; Alzheimer’s Disease Neuroimaging, I. Soluble TREM2 and Inflammatory Proteins in Alzheimer’s Disease Cerebrospinal Fluid. J. Alzheimers Dis. 2020, 73, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Richartz, E.; Stransky, E.; Batra, A.; Simon, P.; Lewczuk, P.; Buchkremer, G.; Bartels, M.; Schott, K. Decline of immune responsiveness: A pathogenetic factor in Alzheimer’s disease? J. Psychiatr. Res. 2005, 39, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Eagar, T.N.; Miller, S.D. Helper T-Cell Subsets and Control of the Inflammatory Response. In Clinical Immunology, 6th ed.; Robert, R.R., Fleisher, T.A., Shearer, W.T., Harry, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 151–161. [Google Scholar]
- de la Monte, S.M.; Grammas, P. Insulin Resistance and Oligodendrocyte/Microvascular Endothelial Cell Dysfunction as Mediators of White Matter Degeneration in Alzheimer’s Disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Elsevier: Brisbane, Australia, 2019. [Google Scholar]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 26. [Google Scholar] [CrossRef]
- Grammas, P.; Martinez, J.; Miller, B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert. Rev. Mol. Med. 2011, 13, e19. [Google Scholar] [CrossRef]
- Brun, A.; Englund, E. A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study. Ann. Neurol. 1986, 19, 253–262. [Google Scholar] [CrossRef]
- Scheibel, A.B.; Duong, T.H.; Jacobs, R. Alzheimer’s disease as a capillary dementia. Ann. Med. 1989, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, L.S.; Chui, H.C. Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review. Brain. Res. Bull. 1990, 24, 677–686. [Google Scholar] [CrossRef]
- Verny, M.; Duyckaerts, C.; Pierot, L.; Hauw, J.J. Leuko-araiosis. Dev. Neurosci. 1991, 13, 245–250. [Google Scholar] [CrossRef]
- Englund, E. Neuropathology of white matter changes in Alzheimer’s disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 1998, 9 (Suppl. 1), 6–12. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C.; Stefano, G.B. Evidence that Alzheimer’s disease is a microvascular disorder: The role of constitutive nitric oxide. Brain. Res. Brain. Res. Rev. 2000, 34, 119–136. [Google Scholar] [CrossRef]
- Jellinger, K.A. The pathology of ischemic-vascular dementia: An update. J. Neurol. Sci. 2002, 203–204, 153–157. [Google Scholar] [CrossRef]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflammation 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- de la Monte, S.M. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann. Neurol. 1989, 25, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Polidori, M.C.; Monastero, R.; Ercolani, S.; Camarda, C.; Cecchetti, R.; Mecocci, P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009, 8, 285–305. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Kim, W.U. Targeted Immunotherapy for Autoimmune Disease. Immune. Netw. 2022, 22, e9. [Google Scholar] [CrossRef]
- Östör, A.J.K. Anti-cytokine biologics. In Oxford Textbook of Rheumatology, 4th ed.; Richard, A., Watts, E.A., Eds.; Oxford Academic: Oxford, UK, 2013; pp. 636–641. [Google Scholar]
- Zakrzewska, M.; Marcinkowska, E.; Wiedlocha, A. FGF-1: From biology through engineering to potential medical applications. Crit. Rev. Clin. Lab. Sci. 2008, 45, 91–135. [Google Scholar] [CrossRef]
- Guerrini, M.M.; Okamoto, K.; Komatsu, N.; Sawa, S.; Danks, L.; Penninger, J.M.; Nakashima, T.; Takayanagi, H. Inhibition of the TNF Family Cytokine RANKL Prevents Autoimmune Inflammation in the Central Nervous System. Immunity 2015, 43, 1174–1185. [Google Scholar] [CrossRef]
- Heng, A.H.S.; Han, C.W.; Abbott, C.; McColl, S.R.; Comerford, I. Chemokine-Driven Migration of Pro-Inflammatory CD4(+) T Cells in CNS Autoimmune Disease. Front. Immunol. 2022, 13, 817473. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Kamimura, D.; Arima, Y.; Murakami, M. Gateway reflexes: A new paradigm of neuroimmune interactions. Clin. Exp. Neuroimmunol. 2017, 8, 23–32. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Schiano, I.; Didsbury, J. Improved Brain Insulin/IGF Signaling and Reduced Neuroinflammation with T3D-959 in an Experimental Model of Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.A.; Panos, J.J.; Sloper, D.; Varma, V.; Sarkar, S. Alzheimer’s disease: A step closer to understanding type 3 diabetes in African Americans. Metab. Brain. Dis. 2021, 36, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A. Oxidative stress hypothesis for Alzheimer’s disease and its potential therapeutic implications. Rinsho Shinkeigaku 2013, 53, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Sheha, M. Pharmacokinetic and ulcerogenic studies of naproxen prodrugs designed for specific brain delivery. Arch. Pharm. Res. 2012, 35, 523–530. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine/Chemokine | Full-Other Names | Systemic Functions | Roles in Neurodegeneration | Citations |
|---|---|---|---|---|
| b-FGF | Basic fibroblast growth factor; FGF2 | Angiogenic and broad-spectrum mitogenic factor; localized in basement membranes and vascular subendothelial extracellular matrix; cytoprotective; role in wound healing | Both a neurotrophin and mediator of neuronal injury; signals through networks involved in neurogenesis (temporal lobe) and post-traumatic injury related neurodegeneration. Increased levels in various neurodegenerative diseases. Immunoreactivity detected in astrocytes, senile plaques, neuropil threads, and neurofibrillary tangles in AD. | [26,27,28,29,30] |
| Eotaxin | Eosinophil chemotactic protein; CCL11 (eotaxin-1), CCL24 (eotaxin-2), and CCL26 (eotaxin-3) | CC chemokine subfamily of proteins chemotactic for eosinophils; binds to CCR2, CCR3, CCR5; levels elevated in aging; active consumption of cannabis and schizophrenia | Elevated in CSF and plasma of aging mice; impairs neurogenesis, cognition, memory; plasma levels elevated in AD and other forms of neurodegeneration | [26,31,32] |
| G-CSF | Granulocyte colony stimulating factor; CSF-3 | Stimulates granulocyte activation, proliferation, survival, and differentiation, produced by endothelium and macrophages | Supports neuroprotection due to anti-apoptotic effects via pAKT activation | [26,33,34] |
| GM-CSF | Granulocyte- macrophage colony stimulating factor; CSF-2 | Cytokine promotes host defenses; stimulates stem cells to generate granulocytes and induces differentiation of monocytes into macrophages or dendritic cells | Neuroprotective. Prevents neurodegeneration in MPTP models of PD; mediates autoimmune encephalitis | [26,33,35,36,37,38,39,40] |
| IFN-γ | Interferon-gamma; type II interferon | Pro-inflammatory cytokine and potent activator of macrophages; plays a role in mediating innate and adaptive immune responses; delayed immune response | Mediates delayed post-ischemia neurodegeneration via IFN-γ secreted by splenic macrophages; promotes inflammatory-mediated impairment of neural stem and neuroprogenitor cell maturation and differentiation | [10,41,42,43,44] |
| IL-10 | Interleukin-10; cytokine synthesis inhibitory factor | Anti-inflammatory cytokine; suppresses pro-inflammatory genes and cytokine secretion in macrophages and neutrophil | Neuroprotective; prevents LPS-induce neurodegeneration; expressed in microglia | [26,35,45] |
| IL-12 (p70) | Interleukin-12; p70 is the active heterodimer | Pro-inflammatory cytokine; promotes antigen expression in B cells, macrophages, neutrophils, and dendritic cells. Bolsters production of IFN-γ and TNF-α; stimulates IL-7 in macrophages | Induces excitotoxic neuronal injury in brain by stimulating IL-7 in microglia | [26,35,45,46,47] |
| IL-13 | Interleukin-13 | Cytokine secreted by TH2 T helper cells; effects similar to those of IL-4 but mainly reduces allergic inflammatory responses; reduces TH2 helper cell functions; mediates pro-inflammatory responses such as enhanced secretion of IgE by activated B cells | Potentially neuroprotective for cortical neurons; modulates cortical excitability; expression correlates with Aβ deposition in multiple sclerosis | [48,49] |
| IL-15 | Interleukin-15 | Pleiotropic pro-inflammatory cytokine, structurally similar to IL-2, produced by activated monocytes, macrophages, and dendritic cells. Promotes T cell proliferation and cytotoxicity via NK and cytotoxic T cells | Potential biomarker for AD due to elevated serum levels; produced by activated astrocytes | [17,50] |
| IL-17A | Interleukin-17A | Pro-inflammatory cytokine produced by T helper cells and induced by IL-23. Recruits monocytes and neutrophils to sites of inflammation; role in auto-immune diseases and microbial defenses | T-cell-mediated delayed phase inflammatory injury in ischemic stroke | [51,52,53] |
| IL-1β | Interleukin-1beta; leukocyte pyrogen; leukocyte activating factor | Pro-inflammatory cytokine produced by activated macrophages; promotes p53-mediated apoptosis | Expressed by microglia in response to injury and exacerbates neuronal injury [IL-1]; causes excitotoxic neurodegeneration via increased generation of glutamate and increases MS progression by way of p53-linked apoptosis; causes death of oligodendrocytes; positive effects include enhanced synaptic transmission | [54,55,56] |
| IL-1RA | Interleukin-1 receptor antagonist; IL-1 inhibitor | Increases adhesion molecule expression; induces metalloproteinases and prostaglandins | Neuroprotective: inhibits cytotoxic, ischemic, excitotoxic, and traumatic injury in the brain. | [56] |
| IL-2 | Interleukin-2 | Cytokine-signaling regulator of activities in leukocytes responsible for immunity; increases T cell proliferation; activates B cells | Neuroprotective for maintaining septal–hippocampal cholinergic neurons; however, high levels cause cognitive dysfunction | [33,57] |
| IL-4 | Interleukin-4 | Cytokine induces differentiation of naïve T cells; regulates immune responses, both adaptive and humoral; reduces Th1, IFN-γ, macrophages, and dendritic cell IL-12 via anti-inflammatory actions | May regulate dopaminergic functions in neuron; similar effects as those associated with IL-13. | [26,49,58,59,60] |
| IL-5 | Interleukin-5 | Pro-inflammatory cytokine; produced by Th2 T helper cells; promotes activated B cell proliferation, maturation, and immunoglobulin secretion | Induces proliferation and activation of microglia; increases nitrite production and probably nitrosative stress; serum levels elevated in major depressive disorders; mediates its effects on CNS plasticity by utilizing neural-plasticity-related RAS GTPase-extracellular signal-regulated kinase (Ras-ERK) pathway | [61,62] |
| IL-6 | Interleukin-6 | Pro-inflammatory cytokine and anti-inflammatory myokine; induces B and T cell proliferation; induces expression of protease inhibitors; macrophages and T cells secreted to enhance immune responses | Expressed in microglia; accumulates around amyloid beta cortical senile plaques; increased levels elevated in PBMCs from AD subjects; MPTP models of PD induce IL-6, but paradoxically neuroprotective. | [18,63,64,65,66,67] |
| IL-7 | Interleukin-7 | Hematopoietic growth factor made by stromal, neuronal, dendritic, hepatocellular, and epithelial cells. Positive regulator of B and T cell development and differentiation | CNS and peripherally increased in association with CNS autoimmune diseases (MS/EAE); promoted by elevated levels of TNF-α, IL6, and IFN-γ; increases proliferation of myelin-activated T cells | [26,46,68,69] |
| IL-8 | Interleukin-8 | Chemokine ligand (C-X-C motif); regulates neutrophil migration by signaling through CXCR2; induces expression of proinflammatory proteases MMP-2 and MMP-9; induces proapoptotic protein Bim (Bcl-2-interacting mediator of cell death) and cell death | Levels increased by brain injury; higher levels propagate secondary injury | [26,43,54,67,70,71,72] |
| IL-9 | Interleukin-9 | Cytokine cellular signaling molecule that modulates pro-inflammatory responses, stimulating proliferation and inhibiting apoptosis; roles in autoimmune disease and asthma | Increased production in AD brain cells. Promotes T cell migration into the CNS | [66,73,74,75] |
| IP-10 | Interferon gamma induced protein 10; CXCL10 | Chemokine binds to cell-surface CXCR3 receptors to activate monocyte /macrophage chemoattraction of dendritic cells, NK cells, and T cells. Promotes adhesion of T cells to endothelial cells, antitumor activity, and angiogenesis | Elevated in several neurodegenerative diseases and in MS; mediates stroke-induced neurodegeneration | [41,76,77,78] |
| MCP-1 | Monocyte chemoattractant protein 1; CCL2 (chemokine motif ligand 2) | Chemokine anchored in the plasma membrane and secreted by monocytes, macrophages and dendritic cells, mainly in response to PDGF and CCR2 and CCR4 surface receptors; attracts monocytes | Induced in astrocytes by PDGF-BB; attracts monocytes, promoting their transmigration through a disrupted blood–brain barrier. Increased levels impair attention, executive function, and psychomotor speed. | [26,78,79,80,81,82] |
| MIP-1α | Macrophage inflammatory protein 1 alpha; chemokine motif ligand 3 (CCL3) | Chemokine with chemoattraction for T cells, NK cells, monocytes, and immature dendritic cells; induces release and synthesis of as IL-1, IL-6 and TNF-α, i.e., pro-inflammatory cytokines from macrophages and fibroblasts. | Promotes neurodegeneration by attracting infiltration of microglia and macrophages; increased expression associated with spongiform neurodegeneration caused by oncornavirus | [18,36,71,80,82,83,84] |
| MIP-1β | Macrophage inflammatory protein 1 beta; chemokine motif ligand 4 (CCL4) | Chemokine with chemoattraction for NK and T cells with actions similar to MIP-1α. Interacts with CCL3. | Impairs attention, executive function, and psychomotor speed; increased expression with oncornavirus-induced spongiform neurodegeneration | [26,36,71,83] |
| PDGF-BB | Platelet-derived growth Factor-BB | Chemokine for monocytes and neutrophils; mitogenic for cells of mesenchymal origin; Two forms of PDGF-B dimerization exist: PDGF-BB (homodimer) or PDGF-AB (heterodimer with PDGF-A). | Neuroprotective; promotes neuronal survival; induces neurogenesis in dopaminergic neurons; however, also induces MCP-1 in astrocytes | [26,82,85,86,87,88,89] |
| RANTES | Regulated upon activation, normal T-cell expressed, and secreted | Chemokine with chemoattraction for T cells and leukocytes, promotes monocyte adhesion to endothelial cells. Binds to CCR1, CCR3, and CCR5 receptors | Major chemokine expressed in brain; including reactive astrocytes in mouse brains infected with scrapie virus; potential for neuroprotection post-ischemic stroke via neuronal induction of neurotrophic factors within peri-infarct zones, leading to enhanced neuronal survival via autocrine or paracrine mechanisms | [90,91,92] |
| TNF-α | Tumor necrosis factor-alpha; cachexin | Pro-inflammatory cytokine of activated macrophages; binds to TNFR1; induces expression of other cytokines, chemokines (RANTES), metalloproteinases, and adhesion molecules in the setting of acute phase responses; pathogenic role in cachexia, fever (hyperpyrexia), inflammatory responses, and cellular apoptosis. Anti-tumor and anti-viral effects. Dysregulated expression in cancer, psoriasis, and inflammatory bowel disease. | Dysregulated expression in neurodegeneration including AD and in major depression. In neurodegeneration, TNF-α induces neuronal excitotoxic injury (via glutamate); accumulates around senile plaques; induced by MPTP; neuronal excitoxicity; can also increase synaptic transmission | [54,64,65,77,93] |
| VEGF | Vascular endothelial growth factor; vascular permeability factor | Trophic factor in the PDGF subfamily; stimulates de novo vasculogenesis and angiogenesis, fibroblast proliferation, and monocyte/macrophage migration; restores oxygen supply to tissues injured by deprivation; increases microvascular permeability; levels elevated in diabetes and cancer. | CSF levels elevated in normal brain aging; may be neuroprotective, reduced CNS/CSF levels correlate with hippocampal atrophy and loss of executive functions and memory; interactive effect with Aβ | [18,94,95,96,97,98,99] |
| Control | MCI | AD | |
|---|---|---|---|
| Number Subjects | 21 | 8 | 10 |
| Age: Years ± S.D [Range] | 45.6 ± 11.8 [28–77] | 69.1 ± 7.2 [59–77] | 67.5 + 11.3 [49–83] |
| Sex: M/F | 11/10 | 7/1 | 5/5 |
| MMSE Score ± SD [Range] | N.D. | 26.4 ± 3.1 [22–30] | 21.9 ± 5.5 [14–28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Monte, S.M.; Tong, M.; Hapel, A.J. Concordant and Discordant Cerebrospinal Fluid and Plasma Cytokine and Chemokine Responses in Mild Cognitive Impairment and Early-Stage Alzheimer’s Disease. Biomedicines 2023, 11, 2394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092394
de la Monte SM, Tong M, Hapel AJ. Concordant and Discordant Cerebrospinal Fluid and Plasma Cytokine and Chemokine Responses in Mild Cognitive Impairment and Early-Stage Alzheimer’s Disease. Biomedicines. 2023; 11(9):2394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092394
Chicago/Turabian Stylede la Monte, Suzanne M., Ming Tong, and Andrew J. Hapel. 2023. "Concordant and Discordant Cerebrospinal Fluid and Plasma Cytokine and Chemokine Responses in Mild Cognitive Impairment and Early-Stage Alzheimer’s Disease" Biomedicines 11, no. 9: 2394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines11092394