

Evaluating p-tau217 and p-tau231 as Biomarkers for Early Diagnosis and Differentiation of Alzheimer’s Disease: A Narrative Review

 , ,
, ,  and
and 
Abstract
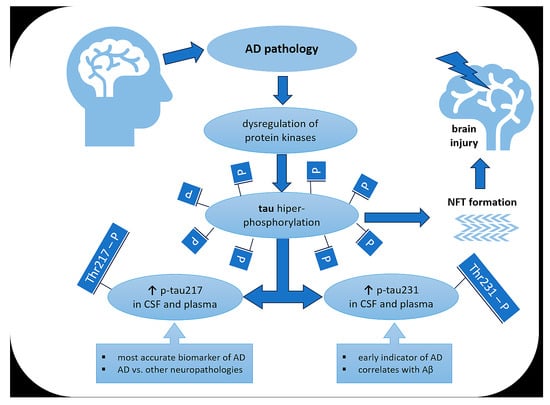
:
1. Introduction
2. Current Diagnosis of AD
3. New ‘Candidates’ for AD Biomarkers
4. Characteristics of tau Protein
5. Characteristics of Hyperphosphorylation Process of tau Protein
6. From tau Phosphorylation to Neurofibrillary Tangle Formation
7. P-tau Isoforms as AD Biomarkers
7.1. P-tau Isoforms Are Effective AD Biomarkers
7.2. Diagnostic Performance of Various p-tau Assays
7.3. P-tau217 Proves to Be Superior to p-tau181 and Other Isoforms
7.4. Longitudinal Effectiveness and Change in p-tau217 Levels
7.5. P-tau231 Is Probably the Earliest AD Biomarker
8. Discussion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| ADNC | AD neuropathological change |
| APOE | apolipoprotein E |
| APP | amyloid protein precursor |
| AUC | area under the curve |
| BACE-1 | β-secretase |
| BMI | body mass index |
| BOLD | resting blood oxygen level-dependent imaging |
| CAA | cerebral amyloid angiopathy |
| CAMK II | calmodulin kinase II |
| CDK-5 | cyclin-dependent kinase 5 |
| CDR | clinical dementia rating |
| CERAD | The Consortium to Establish a Registry for Alzheimer’s Disease |
| CSF | cerebrospinal fluid |
| CTD | C-terminal domain |
| CU | cognitively unimpaired |
| FDG-PET | fluorodeoxyglucose-18 positron resonance imaging |
| fMRI | functional magnetic resonance imaging |
| FTLD | frontotemporal lobar degeneration |
| GFAP | glial fibrillary acid protein |
| GSK-3β | glycogen synthase kinase-3β |
| MAPT | microtubule-associated protein tau |
| MCI | mild cognitive impairment |
| MMSE | Mini-Mental State Examination |
| mPACC | modified Preclinical Alzheimer Cognitive Composite |
| MRI | magnetic resonance imaging |
| MS | mass spectrometry |
| MTBD | microtubule-binding domain |
| MT | Microtubule |
| NAD | non-Alzheimer’s disease condition |
| NFTs | neurofibrillary tangles |
| NTA | N-terminal tau |
| NTPD | N-terminal projection domain |
| PART | primary age-related tauopathy |
| PET | positron emission tomography |
| PP1 | protein phosphatase 1 |
| PP2A | protein phosphatase 2A |
| PP5 | protein phosphatase 5 |
| PRR | proline-rich region |
| PSEN1/2 | presenilin-1/2 |
| p-tau | phosphorylated tau |
| p-tau217 | tau phosphorylated at threonine-217 |
| p-tau181 | tau phosphorylated at threonine-181 |
| p-tau231 | tau phosphorylated at threonine-231 |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| SPECT | single-photon emission computed tomography |
| SUVR | standardized uptake value ratio |
| t-tau | total tau |
| TBI | traumatic brain injury |
References
- Alzheimer, A. Über Einen Eigenartigen Schweren Erkrankungsprozeβ Der Hirnrincle. Neurol Central. Neurol Cent. 1906, 25, 1134. [Google Scholar]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Global Status Report on the Public Health Response to Dementia. Available online: https://www.who.int/publications/i/item/9789240033245 (accessed on 2 February 2024).
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global Estimates on the Number of Persons across the Alzheimer’s Disease Continuum. Alzheimer’s Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, K.L.; Hviid Hahn-Pedersen, J.; Eichinger, C.S.; Freeman, C.; Clark, A.; Tarazona, L.R.S.; Cummings, J. Burden of Illness in People with Alzheimer’s Disease: A Systematic Review of Epidemiology, Comorbidities and Mortality. J. Prev. Alzheimer’s Dis. 2023, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; van der Kant, R.; Hansson, O. Tau Biomarkers in Alzheimer’s Disease: Towards Implementation in Clinical Practice and Trials. Lancet Neurol. 2022, 21, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Pedrini, S.; Ashton, N.J.; Tegg, M.; Goozee, K.; Singh, A.K.; Karikari, T.K.; Simrén, J.; Vanmechelen, E.; Armstrong, N.J.; et al. Diagnostic and Prognostic Plasma Biomarkers for Preclinical Alzheimer’s Disease. Alzheimer’s Dement. 2022, 18, 1141–1154. [Google Scholar] [CrossRef]
- Prins, S.; de Kam, M.L.; Teunissen, C.E.; Groeneveld, G.J. Inflammatory Plasma Biomarkers in Subjects with Preclinical Alzheimer’s Disease. Alzheimers. Res. Ther. 2022, 14, 106. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Kirova, A.-M.; Bays, R.B.; Lagalwar, S. Working Memory and Executive Function Decline across Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 748212. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Noh, G.O.; Kim, K. Behavioural and Psychological Symptoms of Dementia in Patients with Alzheimer’s Disease and Family Caregiver Burden: A Path Analysis. BMC Geriatr. 2021, 21, 160. [Google Scholar] [CrossRef] [PubMed]
- Trepson, W.L. Risk Factors for Alzheimer’s Disease. Sci. Insights 2020, 32, 125–132. [Google Scholar] [CrossRef]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimer’s Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef]
- Villaseca, P.; Cisternas, P.; Inestrosa, N.C. Menopause and Development of Alzheimer’s Disease: Roles of Neural Glucose Metabolism and Wnt Signaling. Front. Endocrinol. 2022, 13, 1021796. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Knupp, A.; Szabo, M.P.; Williams, C.A.; Kinoshita, C.; Hailey, D.W.; Wang, Y.; Andersen, O.M.; Young, J.E. The Alzheimer’s Gene SORL1 Is a Regulator of Endosomal Traffic and Recycling in Human Neurons. Cell. Mol. Life Sci. 2022, 79, 162. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s Disease: Advances in Genetics, Pathophysiology, and Therapeutic Approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Litke, R.; Garcharna, L.C.; Jiwani, S.; Neugroschl, J. Modifiable Risk Factors in Alzheimer Disease and Related Dementias: A Review. Clin. Ther. 2021, 43, 953–965. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Zhao, Q.-F.; Li, J.-Q.; Wang, J.; Yu, J.-T. Meta-Analysis of Modifiable Risk Factors for Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, jnnp-2015-310548. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Fossati, S. Hypertension and Hyperhomocysteinemia as Modifiable Risk Factors for Alzheimer’s Disease and Dementia: New Evidence, Potential Therapeutic Strategies, and Biomarkers. Alzheimer’s Dement. 2023, 19, 671–695. [Google Scholar] [CrossRef] [PubMed]
- Elsworthy, R.J.; Aldred, S. Depression in Alzheimer’s Disease: An Alternative Role for Selective Serotonin Reuptake Inhibitors? J. Alzheimer’s Dis. 2019, 69, 651–661. [Google Scholar] [CrossRef] [PubMed]
- West, R.K.; Ravona-Springer, R.; Sharvit-Ginon, I.; Ganmore, I.; Manzali, S.; Tirosh, A.; Golan, S.; Boccara, E.; Heymann, A.; Beeri, M.S. Long-term Trajectories and Current BMI Are Associated with Poorer Cognitive Functioning in Middle-aged Adults at High Alzheimer’s Disease Risk. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2021, 13, e12247. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhong, T.; Zhang, M.; Xu, Y.; Zhang, M.; Chen, L. Alzheimer’s Disease: Causal Effect between Obesity and APOE Gene Polymorphisms. Int. J. Mol. Sci. 2023, 24, 13531. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. NeuroMolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12, 41. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Taneja, V.; Verma, M.; Vats, A. Toxic Species in Amyloid Disorders: Oligomers or Mature Fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138. [Google Scholar] [CrossRef] [PubMed]
- Amin, L.; Harris, D.A. Aβ Receptors Specifically Recognize Molecular Features Displayed by Fibril Ends and Neurotoxic Oligomers. Nat. Commun. 2021, 12, 3451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. The Prion-like Transmission of Tau Oligomers via Exosomes. Front. Aging Neurosci. 2022, 14, 974414. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.M.; Mandelkow, E.-M.; Mandelkow, E.; Buée, L.; Goedert, M.; et al. What Is the Evidence That Tau Pathology Spreads through Prion-like Propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A Crucial Factor in Alzheimer’s Disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The Diagnosis of Mild Cognitive Impairment Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jiang, F.; Cheng, C.; Huang, J.; Chen, Q.; Le, W. Mild Behavioral Impairment: An Early Sign and Predictor of Alzheimer’s Disease Dementia. Curr. Alzheimer Res. 2022, 19, 407–419. [Google Scholar] [CrossRef]
- Mo, M.; Zacarias-Pons, L.; Hoang, M.T.; Mostafaei, S.; Jurado, P.G.; Stark, I.; Johnell, K.; Eriksdotter, M.; Xu, H.; Garcia-Ptacek, S. Psychiatric Disorders Before and After Dementia Diagnosis. JAMA Netw. Open 2023, 6, e2338080. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Pascoal, T.A.; Karikari, T.K.; Benedet, A.L.; Lantero-Rodriguez, J.; Brinkmalm, G.; Snellman, A.; Schöll, M.; Troakes, C.; Hye, A.; et al. Plasma P-Tau231: A New Biomarker for Incipient Alzheimer’s Disease Pathology. Acta Neuropathol. 2021, 141, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, M.; Komatsu, H.; Sengoku, R.; Shibukawa, M.; Morimoto, S.; Matsubara, T.; Arakawa, A.; Orita, M.; Ishibashi, K.; Mitsutake, A.; et al. CSF P-Tau181 and Other Biomarkers in Patients With Neuronal Intranuclear Inclusion Disease. Neurology 2023, 100, e1009–e1019. [Google Scholar] [CrossRef] [PubMed]
- Batzu, L.; Rota, S.; Hye, A.; Heslegrave, A.; Trivedi, D.; Gibson, L.L.; Farrell, C.; Zinzalias, P.; Rizos, A.; Zetterberg, H.; et al. Plasma P-Tau181, Neurofilament Light Chain and Association with Cognition in Parkinson’s Disease. npj Park. Dis. 2022, 8, 154. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Thijssen, E.H.; Verberk, I.M.W. Plasma P-Tau217: From ‘New Kid’ to Most Promising Candidate for Alzheimer’s Disease Blood Test. Brain 2020, 143, 3170–3172. [Google Scholar] [CrossRef] [PubMed]
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; et al. Plasma P-Tau231 and p-Tau217 as State Markers of Amyloid-β Pathology in Preclinical Alzheimer’s Disease. Nat. Med. 2022, 28, 1797–1801. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal Plasma P-Tau217 Is Increased in Early Stages of Alzheimer’s Disease. Brain 2020, 143, 3234–3241. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of Tau Protein in Alzheimer’s Disease: The Prime Pathological Player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau Protein in Neurodegenerative Diseases—A Review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A Current View on Tau Protein Phosphorylation in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau Protein in Health and Disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders11These Authors Contributed Equally to This Work. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-C.; Jiang, T.; Yang, A.-F.; Du, Y.-J.; Wu, M.; Kong, L.-H. Epigenetic Modulation on Tau Phosphorylation in Alzheimer’s Disease. Neural Plast. 2019, 2019, 6856327. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Sakakibara, Y.; Ibaraki, K.; Takei, K.; Iijima, K.M.; Sekiya, M. Distinct Brain Pathologies Associated with Alzheimer’s Disease Biomarker-Related Phospho-Tau 181 and Phospho-Tau 217 in App Knock-in Mouse Models of Amyloid-β Amyloidosis. Brain Commun. 2022, 4, fcac286. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ban, T.; Liu, C.; Chen, Q.; Wang, X.; Yan, M.; Hu, X.; Su, X.; Bao, Y.; Sun, L.; et al. Activation of Cdk5/P25 and Tau Phosphorylation Following Chronic Brain Hypoperfusion in Rats Involves Micro RNA -195 Down-regulation. J. Neurochem. 2015, 134, 1139–1151. [Google Scholar] [CrossRef]
- Kim, B.M.; You, M.-H.; Chen, C.-H.; Lee, S.; Hong, Y.; Hong, Y.; Kimchi, A.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 Has a Critical Role in Aberrant Tau Protein Regulation and Function. Cell Death Dis. 2014, 5, e1237. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau Protein Phosphatases in Alzheimer’s Disease: The Leading Role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; He, X.; Han, F.; Cai, L.; Wang, L.; Niu, T.; Zhai, J.; Ji, B.; Gao, J.; Wang, J. Phosphorylation at Ser289 Enhances the Oligomerization of Tau Repeat R2. J. Chem. Inf. Model. 2023, 63, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Troquier, L.; Burnouf, S.; Belarbi, K.; Van der Jeugd, A.; Ahmed, T.; Fernandez-Gomez, F.; Caillierez, R.; Grosjean, M.-E.; Begard, S.; et al. From Tau Phosphorylation to Tau Aggregation: What about Neuronal Death? Biochem. Soc. Trans. 2010, 38, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; He, X.; Gao, J.; Wang, J. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2023, 14, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Oseid, D.E.; Wells, E.A.; Robinson, A.S. The Interplay between GSK3β and Tau Ser262 Phosphorylation during the Progression of Tau Pathology. Int. J. Mol. Sci. 2022, 23, 11610. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Saito, T.; Asada, A.; Shimizu, S.; Iijima, K.M.; Ando, K. Microtubule Affinity–Regulating Kinase 4 with an Alzheimer’s Disease-Related Mutation Promotes Tau Accumulation and Exacerbates Neurodegeneration. J. Biol. Chem. 2020, 295, 17138–17147. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Gorion, K.-M.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 Phosphorylation Promotes Aggregation and Reveals Neuropathologic Diversity in Alzheimer’s Disease and Other Tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.-P.; Anderton, B.H.; Authelet, M.; Dayanandan, R.; Leroy, K.; Lovestone, S.; Octave, J.-N.; Pradier, L.; Touchet, N.; Tremp, G. Neurofibrillary Tangles and Tau Phosphorylation. Biochem. Soc. Symp. 2001, 67, 81–88. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated Tau Interactome in the Human Alzheimer’s Disease Brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Jack, C.R.; Wiste, H.J.; Algeciras-Schimnich, A.; Figdore, D.J.; Schwarz, C.G.; Lowe, V.J.; Ramanan, V.K.; Vemuri, P.; Mielke, M.M.; Knopman, D.S.; et al. Predicting Amyloid PET and Tau PET Stages with Plasma Biomarkers. Brain 2023, 146, 2029–2044. [Google Scholar] [CrossRef]
- Xiao, Z.; Wu, W.; Ma, X.; Wu, J.; Liang, X.; Zhou, X.; Cao, Y.; Zhao, Q.; Ding, D. Plasma P-tau217, P-tau181, and NfL as Early Indicators of Dementia Risk in a Community Cohort: The Shanghai Aging Study. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2023, 15, e12514. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M.; Manly, J.J.; Honig, L.S.; Sanchez, D.; Reyes-Dumeyer, D.; Lantigua, R.A.; Lao, P.J.; Stern, Y.; Vonsattel, J.P.; Teich, A.F.; et al. Plasma P-tau181, P-tau217, and Other Blood-based Alzheimer’s Disease Biomarkers in a Multi-ethnic, Community Study. Alzheimer’s Dement. 2021, 17, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Karikari, T.K.; Ashton, N.J.; Lantero Rodríguez, J.; Milà-Alomà, M.; Gispert, J.D.; Salvadó, G.; Minguillon, C.; Fauria, K.; Shekari, M.; et al. Novel Tau Biomarkers Phosphorylated at T181, T217 or T231 Rise in the Initial Stages of the Preclinical Alzheimer’s Continuum When Only Subtle Changes in Aβ Pathology Are Detected. EMBO Mol. Med. 2020, 12, e12921. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Therriault, J.; Pascoal, T.; Rosa-Neto, P. Impact of P-Tau181 and p-Tau217 Levels on Enrollment for Randomized Clinical Trials and Future Use of Anti-Amyloid and Anti-Tau Drugs. Expert Rev. Neurother. 2020, 20, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Kac, P.R.; Gonzalez-Ortiz, F.; Simrén, J.; Dewit, N.; Vanmechelen, E.; Zetterberg, H.; Blennow, K.; Ashton, N.J.; Karikari, T.K. Diagnostic Value of Serum versus Plasma Phospho-Tau for Alzheimer’s Disease. Alzheimers. Res. Ther. 2022, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Tissot, C.; Therriault, J.; Kunach, P.; L Benedet, A.; Pascoal, T.A.; Ashton, N.J.; Karikari, T.K.; Servaes, S.; Lussier, F.Z.; Chamoun, M.; et al. Comparing Tau Status Determined via Plasma PTau181, PTau231 and [18F]MK6240 Tau-PET. eBioMedicine 2022, 76, 103837. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Benedet, A.L.; Pascoal, T.A.; Karikari, T.K.; Lantero-Rodriguez, J.; Brum, W.S.; Mathotaarachchi, S.; Therriault, J.; Savard, M.; Chamoun, M.; et al. Cerebrospinal Fluid P-Tau231 as an Early Indicator of Emerging Pathology in Alzheimer’s Disease. eBioMedicine 2022, 76, 103836. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood Plasma Phosphorylated-Tau Isoforms Track CNS Change in Alzheimer’s Disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J.; Vermeiren, M.; Servaes, S.; Tissot, C.; Ashton, N.J.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Brum, W.S.; Lussier, F.Z.; et al. Association of Phosphorylated Tau Biomarkers With Amyloid Positron Emission Tomography vs Tau Positron Emission Tomography. JAMA Neurol. 2023, 80, 188. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-Tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772. [Google Scholar] [CrossRef] [PubMed]
- Yakoub, Y.; Ashton, N.J.; Strikwerda-Brown, C.; Montoliu-Gaya, L.; Karikari, T.K.; Kac, P.R.; Gonzalez-Ortiz, F.; Gallego-Rudolf, J.; Meyer, P.; St-Onge, F.; et al. Longitudinal Blood Biomarker Trajectories in Preclinical Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 5620–5631. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.S.; Tissot, C.; Lantero-Rodriguez, J.; Snellman, A.; Therriault, J.; Rahmouni, N.; Macedo, A.C.; Servaes, S.; Wang, Y.; Arias, J.F.; et al. Plasma PTau-217 and N-terminal Tau (NTA) Enhance Sensitivity to Identify Tau PET Positivity in Amyloid-β Positive Individuals. Alzheimer’s Dement. 2024, 20, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of Plasma Phospho-Tau217 Levels With Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021, 78, 149. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; Hansson, O. Prediction of Future Alzheimer’s Disease Dementia Using Plasma Phospho-Tau Combined with Other Accessible Measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Dage, J.L.; Frank, R.D.; Algeciras-Schimnich, A.; Knopman, D.S.; Lowe, V.J.; Bu, G.; Vemuri, P.; Graff-Radford, J.; Jack, C.R.; et al. Performance of Plasma Phosphorylated Tau 181 and 217 in the Community. Nat. Med. 2022, 28, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Palmqvist, S.; Leuzy, A.; Stomrud, E.; Verberk, I.M.W.; Zetterberg, H.; Ashton, N.J.; Pesini, P.; Sarasa, L.; Allué, J.A.; et al. Detecting Amyloid Positivity in Early Alzheimer’s Disease Using Combinations of Plasma Aβ42/Aβ40 and P-tau. Alzheimer’s Dement. 2022, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal Fluid P-Tau217 Performs Better than p-Tau181 as a Biomarker of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef]
- Mundada, N.S.; Rojas, J.C.; Vandevrede, L.; Thijssen, E.H.; Iaccarino, L.; Okoye, O.C.; Shankar, R.; Soleimani-Meigooni, D.N.; Lago, A.L.; Miller, B.L.; et al. Head-to-Head Comparison between Plasma p-Tau217 and Flortaucipir-PET in Amyloid-Positive Patients with Cognitive Impairment. Alzheimers. Res. Ther. 2023, 15, 157. [Google Scholar] [CrossRef]
- Jonaitis, E.M.; Janelidze, S.; Cody, K.A.; Langhough, R.; Du, L.; Chin, N.A.; Mattsson-Carlgren, N.; Hogan, K.J.; Christian, B.T.; Betthauser, T.J.; et al. Plasma Phosphorylated Tau 217 in Preclinical Alzheimer’s Disease. Brain Commun. 2023, 5, 1–11. [Google Scholar] [CrossRef]
- Yu, L.; Boyle, P.A.; Janelidze, S.; Petyuk, V.A.; Wang, T.; Bennett, D.A.; Hansson, O.; Schneider, J.A. Plasma P-Tau181 and p-Tau217 in Discriminating PART, AD and Other Key Neuropathologies in Older Adults. Acta Neuropathol. 2023, 146, 1–11. [Google Scholar] [CrossRef]
- Salvadó, G.; Ossenkoppele, R.; Ashton, N.J.; Beach, T.G.; Serrano, G.E.; Reiman, E.M.; Zetterberg, H.; Mattsson-Carlgren, N.; Janelidze, S.; Blennow, K.; et al. Specific Associations between Plasma Biomarkers and Postmortem Amyloid Plaque and Tau Tangle Loads. EMBO Mol. Med. 2023, 15, e17123. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Salvadó, G.; Schindler, S.E.; He, Y.; Janelidze, S.; Collij, L.E.; Saef, B.; Henson, R.L.; Chen, C.D.; Gordon, B.A.; et al. Highly Accurate Blood Test for Alzheimer’s Disease Is Similar or Superior to Clinical Cerebrospinal Fluid Tests. Nat. Med. 2024; in press. [Google Scholar] [CrossRef]
- Karikari, T.K.; Ashton, N.J.; Brinkmalm, G.; Brum, W.S.; Benedet, A.L.; Montoliu-Gaya, L.; Lantero-Rodriguez, J.; Pascoal, T.A.; Suárez-Calvet, M.; Rosa-Neto, P.; et al. Blood Phospho-Tau in Alzheimer Disease: Analysis, Interpretation, and Clinical Utility. Nat. Rev. Neurol. 2022, 18, 400–418. [Google Scholar] [CrossRef]
- Bayoumy, S.; Verberk, I.M.W.; den Dulk, B.; Hussainali, Z.; Zwan, M.; van der Flier, W.M.; Ashton, N.J.; Zetterberg, H.; Blennow, K.; Vanbrabant, J.; et al. Clinical and Analytical Comparison of Six Simoa Assays for Plasma P-Tau Isoforms P-Tau181, P-Tau217, and P-Tau231. Alzheimers. Res. Ther. 2021, 13, 198. [Google Scholar] [CrossRef]
- Janelidze, S.; Bali, D.; Ashton, N.J.; Barthélemy, N.R.; Vanbrabant, J.; Stoops, E.; Vanmechelen, E.; He, Y.; Dolado, A.O.; Triana-Baltzer, G.; et al. Head-to-Head Comparison of 10 Plasma Phospho-Tau Assays in Prodromal Alzheimer’s Disease. Brain 2023, 146, 1592–1601. [Google Scholar] [CrossRef]
- Ashton, N.J.; Puig-Pijoan, A.; Milà-Alomà, M.; Fernández-Lebrero, A.; García-Escobar, G.; González-Ortiz, F.; Kac, P.R.; Brum, W.S.; Benedet, A.L.; Lantero-Rodriguez, J.; et al. Plasma and CSF Biomarkers in a Memory Clinic: Head-to-head Comparison of Phosphorylated Tau Immunoassays. Alzheimer’s Dement. 2023, 19, 1913–1924. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A Soluble Phosphorylated Tau Signature Links Tau, Amyloid and the Evolution of Stages of Dominantly Inherited Alzheimer’s Disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Mielke, M.M.; Frank, R.D.; Dage, J.L.; Jeromin, A.; Ashton, N.J.; Blennow, K.; Karikari, T.K.; Vanmechelen, E.; Zetterberg, H.; Algeciras-Schimnich, A.; et al. Comparison of Plasma Phosphorylated Tau Species With Amyloid and Tau Positron Emission Tomography, Neurodegeneration, Vascular Pathology, and Cognitive Outcomes. JAMA Neurol. 2021, 78, 1108. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Bateman, R.J.; Hirtz, C.; Marin, P.; Becher, F.; Sato, C.; Gabelle, A.; Lehmann, S. Cerebrospinal Fluid Phospho-Tau T217 Outperforms T181 as a Biomarker for the Differential Diagnosis of Alzheimer’s Disease and PET Amyloid-Positive Patient Identification. Alzheimers. Res. Ther. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma Phosphorylated Tau 217 and Phosphorylated Tau 181 as Biomarkers in Alzheimer’s Disease and Frontotemporal Lobar Degeneration: A Retrospective Diagnostic Performance Study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef]
- Salvadó, G.; Horie, K.; Barthélemy, N.R.; Vogel, J.W.; Binette, A.P.; Chen, C.D.; Aschenbrenner, A.J.; Gordon, B.A.; Benzinger, T.L.S.; Holtzman, D.M.; et al. Novel CSF Tau Biomarkers Can Be Used for Disease Staging of Sporadic Alzheimer’s. Alzheimer’s Dement. 2023, 19, e075367. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Salvadó, G.; Ashton, N.J.; Tideman, P.; Stomrud, E.; Zetterberg, H.; Ossenkoppele, R.; Betthauser, T.J.; Cody, K.A.; Jonaitis, E.M.; et al. Prediction of Longitudinal Cognitive Decline in Preclinical Alzheimer Disease Using Plasma Biomarkers. JAMA Neurol. 2023, 80, 360. [Google Scholar] [CrossRef]
- Palmqvist, S.; Stomrud, E.; Cullen, N.; Janelidze, S.; Manuilova, E.; Jethwa, A.; Bittner, T.; Eichenlaub, U.; Suridjan, I.; Kollmorgen, G.; et al. An Accurate Fully Automated Panel of Plasma Biomarkers for Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 1204–1215. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Janelidze, S.; Bateman, R.J.; Smith, R.; Stomrud, E.; Serrano, G.E.; Reiman, E.M.; Palmqvist, S.; Dage, J.L.; Beach, T.G.; et al. Soluble P-tau217 Reflects Amyloid and Tau Pathology and Mediates the Association of Amyloid with Tau. EMBO Mol. Med. 2021, 13, e14022. [Google Scholar] [CrossRef]
- Therriault, J.; Servaes, S.; Tissot, C.; Rahmouni, N.; Ashton, N.J.; Benedet, A.L.; Karikari, T.K.; Macedo, A.C.; Lussier, F.Z.; Stevenson, J.; et al. Equivalence of Plasma P-tau217 with Cerebrospinal Fluid in the Diagnosis of Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 4967–4977. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Benedet, A.L.; Tissot, C.; Vrillon, A.; Ashton, N.J.; Brum, W.S.; Lantero-Rodriguez, J.; Stevenson, J.; Nilsson, J.; Sauer, M.; et al. Mass Spectrometric Simultaneous Quantification of Tau Species in Plasma Shows Differential Associations with Amyloid and Tau Pathologies. Nat. Aging 2023, 3, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Horie, K.; Salvadó, G.; Barthélemy, N.R.; Janelidze, S.; Li, Y.; He, Y.; Saef, B.; Chen, C.D.; Jiang, H.; Strandberg, O.; et al. CSF MTBR-Tau243 Is a Specific Biomarker of Tau Tangle Pathology in Alzheimer’s Disease. Nat. Med. 2023, 29, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Moms, J.C.; Heyman, A.; Mohs, R.C.; Hughes, J.P.; van Belle, G.; Fillenbaum, G.; Mellits, E.D.; Clark, C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and Neuropsychological Assesment of Alzheimer’s Disease. Neurology 1989, 39, 1159. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Smith, R.; Cullen, N.C.; Strandberg, O.; Vogel, J.W.; Binette, A.P.; Borroni, E.; Janelidze, S.; Ohlsson, T.; Jögi, J.; et al. Biomarker-Based Prediction of Longitudinal Tau Positron Emission Tomography in Alzheimer Disease. JAMA Neurol. 2022, 79, 149. [Google Scholar] [CrossRef]
- Ashton, N.J.; Janelidze, S.; Mattsson-Carlgren, N.; Binette, A.P.; Strandberg, O.; Brum, W.S.; Karikari, T.K.; González-Ortiz, F.; Di Molfetta, G.; Meda, F.J.; et al. Differential Roles of Aβ42/40, p-Tau231 and p-Tau217 for Alzheimer’s Trial Selection and Disease Monitoring. Nat. Med. 2022, 28, 2555–2562. [Google Scholar] [CrossRef]
- Lilek, J.; Ajroud, K.; Feldman, A.Z.; Krishnamachari, S.; Ghourchian, S.; Gefen, T.; Spencer, C.L.; Kawles, A.; Mao, Q.; Tranovich, J.F.; et al. Accumulation of PTau231 at the Postsynaptic Density in Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 92, 241–260. [Google Scholar] [CrossRef]
- Smirnov, D.S.; Ashton, N.J.; Blennow, K.; Zetterberg, H.; Simrén, J.; Lantero-Rodriguez, J.; Karikari, T.K.; Hiniker, A.; Rissman, R.A.; Salmon, D.P.; et al. Plasma Biomarkers for Alzheimer’s Disease in Relation to Neuropathology and Cognitive Change. Acta Neuropathol. 2022, 143, 487–503. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T.A.; Lussier, F.Z.; Tissot, C.; Chamoun, M.; Bezgin, G.; Servaes, S.; Benedet, A.L.; Ashton, N.J.; Karikari, T.K.; et al. Biomarker Modeling of Alzheimer’s Disease Using PET-Based Braak Staging. Nat. Aging 2022, 2, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Dubarbie, F.; Guerra-Ruiz, A.; López-García, S.; Irure-Ventura, J.; Lage, C.; Fernández-Matarrubia, M.; Pozueta-Cantudo, A.; García-Martínez, M.; Corrales-Pardo, A.; Bravo, M.; et al. Influence of Physiological Variables and Comorbidities on Plasma Aβ40, Aβ42, and p-Tau181 Levels in Cognitively Unimpaired Individuals. Int. J. Mol. Sci. 2024, 25, 1481. [Google Scholar] [CrossRef]
- Pan, F.; Lu, Y.; Huang, Q.; Xie, F.; Yang, J.; Guo, Q. The Potential Impact of Clinical Factors on Blood-Based Biomarkers for Alzheimer’s Disease. Transl. Neurodegener. 2023, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Zenuni, H.; Grillo, P.; Sancesario, G.M.; Bernardini, S.; Mercuri, N.B.; Schirinzi, T. How Comorbidity Reflects on Cerebrospinal Fluid Biomarkers of Neurodegeneration in Aging. J. Alzheimer’s Dis. Reports 2021, 5, 87–92. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Leuzy, A.; Cho, H.; Sudre, C.H.; Strandberg, O.; Smith, R.; Palmqvist, S.; Mattsson-Carlgren, N.; Olsson, T.; Jögi, J.; et al. The Impact of Demographic, Clinical, Genetic, and Imaging Variables on Tau PET Status. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2245–2258. [Google Scholar] [CrossRef]
- Dang, M.; Chen, Q.; Zhao, X.; Chen, K.; Li, X.; Zhang, J.; Lu, J.; Ai, L.; Chen, Y.; Zhang, Z. Tau as a Biomarker of Cognitive Impairment and Neuropsychiatric Symptom in Alzheimer’s Disease. Hum. Brain Mapp. 2023, 44, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; McQuillan, L.; Wang, K.K.W.; Robertson, C.; Chang, B.; Yang, Z.; Xu, H.; Williamson, J.; Wagner, A.K. Temporal Profiles of P-Tau, T-Tau, and P-Tau:Tau Ratios in Cerebrospinal Fluid and Blood from Moderate-Severe Traumatic Brain Injury Patients and Relationship to 6–12 Month Global Outcomes. J. Neurotrauma 2024, 41, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Sfera, A.; Rahman, L.; Zapata-Martín del Campo, C.M.; Kozlakidis, Z. Long COVID as a Tauopathy: Of “Brain Fog” and “Fusogen Storms”. Int. J. Mol. Sci. 2023, 24, 12648. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Assay | Fold Increase in CSF | AUC (95% CI) in CSF | Fold Increase in Plasma | AUC (95% CI) in Plasma | Ref. |
|---|---|---|---|---|---|
| p-tau 181 Lilly | - | - | 1.8 | 0.938 (0.872–1.000) | [96] |
| - | - | 1.2–1.4 | 0.759 (0.676–0.841) | [97] | |
| 3.29 | 0.95 (0.91–0.98) | 2.59 | 0.91 (0.86–0.96) | [98] | |
| p-tau217 Lilly | - | - | 4.1 | 0.995 (0.987–1.000) | [96] |
| - | - | 2.0 | 0.886 (0.827–0.944) | [97] | |
| 7.18 | 0.98 (0.96–1.00) | 3.27 | 0.94 (0.90–0.98) | [98] | |
| t-tau Lilly | 1.97 | 0.85 (0.79–0.90) | 1.36 | 0.73 (0.65–0.81) | [98] |
| p-tau181 ADx | - | - | 2.9 | 0.988 (0.969–1.000) | [96] |
| - | - | 1.8 | 0.841 (0.768–0.913) | [97] | |
| 4.77 | 0.96 (0.93–0.98) | 3.48 | 0.94 (0.91–0.97) | [98] | |
| p-tau231 ADx | - | - | 1.3 | 0.719 (0.607–0.831) | [96] |
| 3.59 | 0.93 (0.88–0.97) | 1.39 | 0.66 (0.58–0.74) | [98] | |
| p-tau181 UGot | - | - | 1.2–1.4 | 0.743 (0.652–0.833) | [97] |
| 2.1 | 0.94 (0.90–0.97) | 1.38 | 0.80 (0.73–0.87) | [98] | |
| p-tau231 UGot | - | - | 1.5 | 0.943 (0.896–0.991) | [96] |
| - | - | 1.2–1.4 | 0.784 (0.703–0.864) | [97] | |
| - | 0.91 (0.87–0.95) | 1.95 | 0.88 (0.83–0.93) | [98] | |
| p-tau181 WashU | - | - | 1.2–1.4 | 0.835 (0.765–0.906) | [97] |
| p-tau217 WashU | - | - | 3.6 | 0.947 (0.907–0.987) | [97] |
| p-tau181 Fuji | - | - | 1.2–1.4 | 0.694 (0.604–0.784) | [97] |
| p-tau181 Splex | - | - | 1.2–1.4 | 0.642 (0.533–0.751) | [97] |
| p-tau181 Quanterix | - | - | 1.9 | 0.936 (0.885–0.987) | [96] |
| 5.02 | 0.96 (0.93–0.99) | 1.66 | 0.80 (0.73–0.87) | [98] | |
| p-tau217 Janss | - | - | 2.7 | 0.858 (0.795–0.920) | [97] |
| 8.53 | 0.98 (0.96–1.00) | 5.22 | 0.96 (0.93–0.99) | [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarek, D.J.; Mizerka, H.; Nuszkiewicz, J.; Szewczyk-Golec, K. Evaluating p-tau217 and p-tau231 as Biomarkers for Early Diagnosis and Differentiation of Alzheimer’s Disease: A Narrative Review. Biomedicines 2024, 12, 786. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines12040786
Jarek DJ, Mizerka H, Nuszkiewicz J, Szewczyk-Golec K. Evaluating p-tau217 and p-tau231 as Biomarkers for Early Diagnosis and Differentiation of Alzheimer’s Disease: A Narrative Review. Biomedicines. 2024; 12(4):786. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines12040786
Chicago/Turabian StyleJarek, Dorian Julian, Hubert Mizerka, Jarosław Nuszkiewicz, and Karolina Szewczyk-Golec. 2024. "Evaluating p-tau217 and p-tau231 as Biomarkers for Early Diagnosis and Differentiation of Alzheimer’s Disease: A Narrative Review" Biomedicines 12, no. 4: 786. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines12040786