Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases

MRC Human Genetics Unit, MRC Institute of Genetics & Molecular Medicine, The University of Edinburgh, Western General Hospital, Crewe Road, Edinburgh EH4 2XU, UK
*
Author to whom correspondence should be addressed.
Biomedicines 2019, 7(3), 71; https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030071
Submission received: 4 July 2019
/
Revised: 28 August 2019
/
Accepted: 29 August 2019
/
Published: 16 September 2019
(This article belongs to the Special Issue Zebrafish Models for Development and Disease)
Abstract
:Determining aetiology of genetic disorders caused by damaging mutations in protein-coding genes is well established. However, understanding how mutations in the vast stretches of the noncoding genome contribute to genetic abnormalities remains a huge challenge. Cis-regulatory elements (CREs) or enhancers are an important class of noncoding elements. CREs function as the primary determinants of precise spatial and temporal regulation of their target genes during development by serving as docking sites for tissue-specific transcription factors. Although a large number of potential disease-associated CRE mutations are being identified in patients, lack of robust methods for mechanistically linking these mutations to disease phenotype is currently hampering the understanding of their roles in disease aetiology. Here, we have described the various systems available for testing the CRE potential of stretches of noncoding regions harbouring mutations implicated in human disease. We highlight advances in the field leading to the establishment of zebrafish as a powerful system for robust and cost-effective functional assays of CRE activity, enabling rapid identification of causal variants in regulatory regions and the validation of their role in disruption of appropriate gene expression.
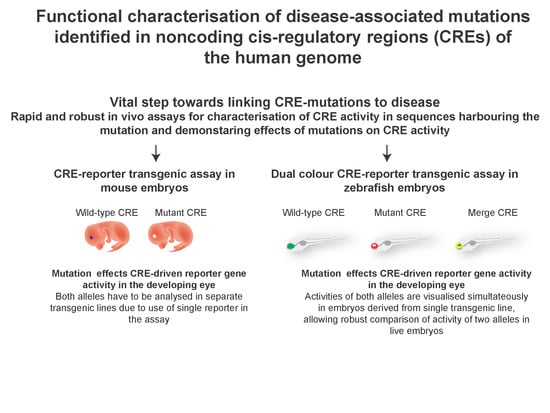
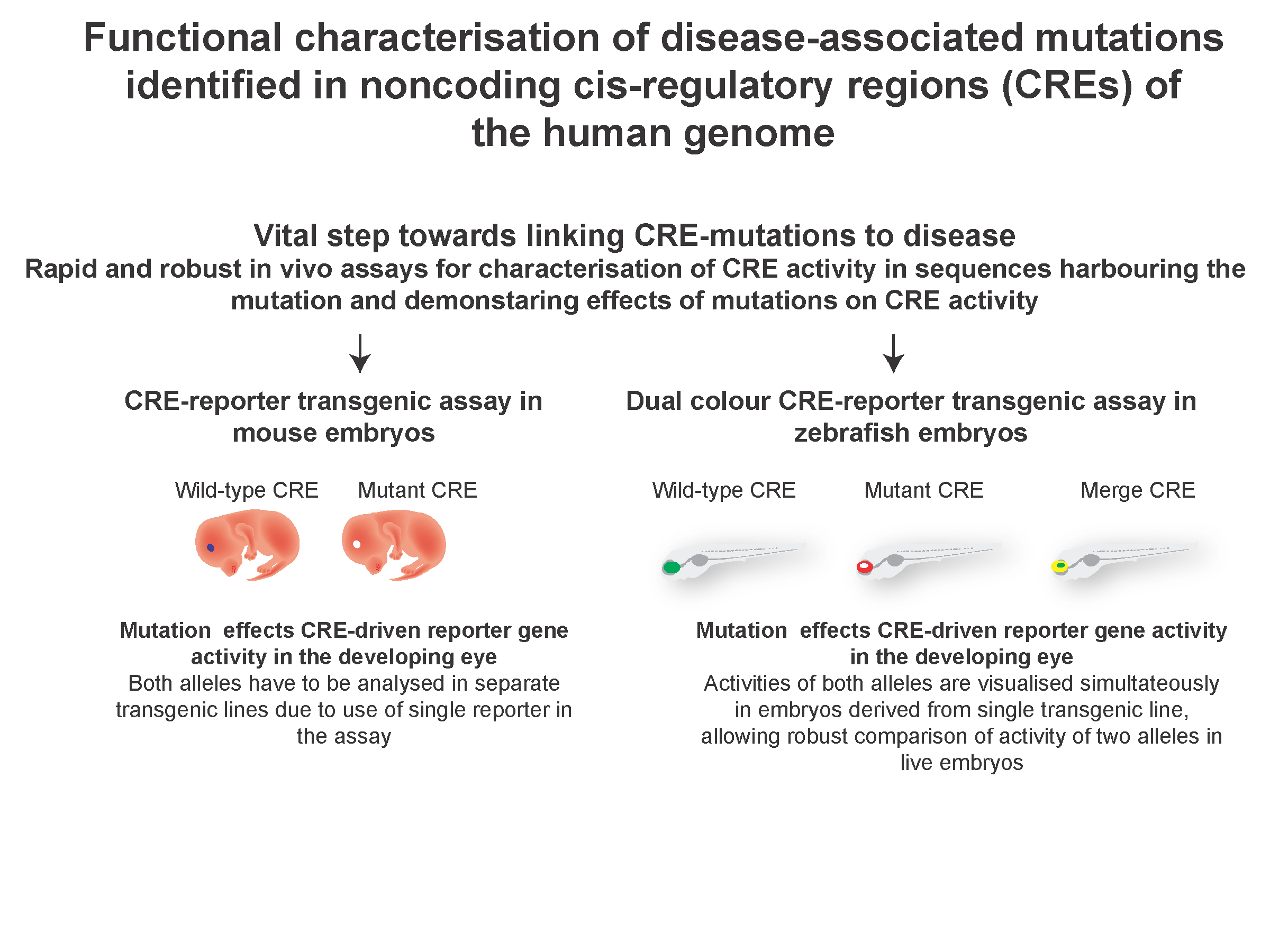
1. Introduction
Human genetic diseases affect a wide range of tissues and are caused by numerous types of mutations. Understanding the cause and progression of these diseases relies heavily on the use of animals, including mouse, rat and zebrafish, to generate models mimicking the human condition. The use of these approaches for determining the aetiology of genetic disorders caused by damaging mutations in protein-coding genes is well established. However, functional analysis of mutations in the noncoding regions remains a huge challenge. Rapid technological advances have enabled the widespread application of whole-genome sequencing (WGS) for the identification of putative pathogenic mutations in patient cohorts. It has been firmly established that many of these mutations reside in the noncoding regions of the human genome, most of which are likely to harbour cis-regulatory elements (CREs) [1,2]. CRE sequences are highly enriched for binding sites of tissue-specific transcription factors (TF). The disease-associated sequence variation alters the TF binding sites in CREs, leading to aberrant CRE function and altered target gene expression [3]. Functional analysis of CRE activity, and assessment of the impact of disease-associated sequence changes on this activity, is heavily reliant on the availability of the right TFs in the right stoichiometric concentrations, which is only precisely captured in the context of animal development. Thus, although WGS has the power to identify variants in noncoding regions, the pathogenicity of these variants is much more difficult to assess compared to variants in coding regions. The use of in vivo reporter transgenic assays in mouse or zebrafish for determining their functional potential is indispensable. We describe the techniques and assays available for establishing where and when in embryonic development the enhancers act, and how this activity is affected by the presence of disease-associated mutations.
2. Prediction of Cis-Regulatory or Enhancer Activity in Noncoding Regions of Human Genome
Advances in genomic sequencing technologies have enabled widespread application of whole genome sequencing technology to patient DNA samples. These studies have led to the identification of clinically-relevant mutations in the noncoding regions of the human genome, most of which are likely disrupting CRE function of these sequences by altering the sequences of transcription factor binding sites [4]. The next step towards linking these sequence changes to the aetiology of the disease is defining the coordinates of CREs which harbour these sequence changes and predicting the target genes whose expression would be affected by the altered CRE function. Identifying stretches of sequence conservation between evolutionarily diverged species or duplicated gene loci using a variety of web- based tools, e.g., PIPmaker, VISTA, ECR browser, UCSC, Ensembl, etc. [5] is a useful way of prioritising elements for further functional studies. Evolutionary conservation of CRE sequence indicates functional roles of the CRE. However, these methods fail to detect evolutionarily diverged or lineage specific CREs. Furthermore, it is difficult to predict the target genes using these methods as CREs may function over large distances and may not necessarily regulate the most proximal gene [4]. A few studies have utilised conservation of sequence and synteny over large evolutionary distances as a measure of predicting putative CREs and the target genes regulated by them [6].
These predictions are further substantiated by looking for enrichments of hallmarks of enhancer function on the predicted CREs. Transcription factor profiling of the CRE sequences helps discern the possible effects of disease-associated mutations in CREs. These profiles can be predicted both computationally (using a variety of web-based tools e.g., UCSC, MEME, TRANSFAC, JASPAR, etc.) and experimentally (using ChIP-Chip, ChIP-seq, yeast one hybrid and protein binding microarrays) [7,8]. Enrichment of transcription factors, like p300 and CBP, are useful indicators of active CRE presence. Techniques like DNaseI hypersensitivity mapping by DNaseI-seq [9], FAIRE (formaldehyde-assisted isolation of regulatory elements) by FAIRE-seq [10] and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) [11] generate genome-wide profiles of nucleosome-free regions available for transcription factor binding, indicative of CRE presence. Histone modification profiling (in particular for H3K4Me1, H3K27Ac) by Chip-seq and ChIP-Chip [12,13] generates genome-wide enrichment profiles for active CRE-associated histone modifications, as indicators of the presence of functional CREs. Chromatin looping studies by a variety of chromosome conformation capture techniques (e.g., 3C, 4C, 5C, Hi-C, ChIA-PET), DAM-ID and 3D FISH [14,15] have been employed to detect interactions between predicted CREs and their target gene promoter, in the context of the entire gene locus.
These assays have been performed on a large number of in vitro cultured cell lines, whole embryos and tissues derived from human, mouse and zebrafish [16]. The information is publicly available on genome browsers like UCSC and Ensembl, enabling rapid analysis of predicted CREs for the presence of these features. However, since a large number of cells are required to perform these assays, they have been mostly restricted to in vitro cultured cell lines or larger tissues (containing mixed cell populations). Biological relevance of these datasets can therefore be limited due to the lack of potentially important developmental context, given the tissue-specific nature of CRE function. Recent advances in single-cell technologies [11] have the potential for overcoming this limitation, provided robust methods of obtaining the precise cell-types where the CREs are active are developed.
3. Testing the Predicted CRE Activity in CRE-Reporter Assays
The putative CREs harbouring the disease-associated mutations are tested in CRE-reporter assays to determine their function and how this is affected by the presence of disease-associated mutations. The most widely used assays, along with their merits and de-merits, are described in Table 1.
4. Zebrafish Dual-Colour CRE-Reporter Assay for Assessing Effects of Mutations on CRE Function
Although WGS has the power to identify variants in noncoding regions, the pathogenicity of these variants is much more difficult to assess compared to variants in coding regions. The use of in vivo reporter transgenic assays in mouse or zebrafish for determining their functional potential is indispensable.
Zebrafish is an excellent in vivo system for characterising putative tissue-specific CREs [19,20]. Large numbers of embryos can be injected. The resulting transgenic lines are ideal for live imaging analysis as zebrafish embryos are transparent and develop rapidly outside the mother, making it feasible to visualise or tag specific cell types in the living embryo [27]. However, the lack of sequence conservation over large portions of the noncoding parts of the human and zebrafish genomes hampered the full exploitation of this powerful system for the functional characterisation of CREs. Research over the past few years has demonstrated the potential of zebrafish models to assess the function of human and mouse CREs, irrespective of their primary sequence conservation in zebrafish [28,29,30,31,32,33]. These studies established that, in spite of significant changes in CRE sequences over the course of evolution, CREs can still capture the transcription factors required for their function in the cell and tissue types they are active in. Based on these studies, a highly robust approach was developed in our laboratory [20 (Figure 1) for testing the in vivo spatial and temporal activity of wild type and putative SNP/mutation bearing human CREs in the same developing embryo, using dual fluorescence CRE-reporter zebrafish transgenics that allow direct comparison of CRE-activity of the two alleles.
The functional output from each CRE version (wild type or SNP/mutation bearing) is visualised simultaneously as eGFP or mCherry signal within a live developing embryo bearing both transgenes. This enables unambiguous comparison of the activity of both wild type and mutant CREs in a developmental context, simultaneous assessment of multiple separate elements for subtle differences in spatio-temporal overlap, and the validation of putative TFs by analysing the effect of morpholino-mediated depletion of the putative TF on CRE activity. We established proof of principle for the robustness of the assay using known mutations associated with holoprosencephaly, identified in a CRE from the SHH locus [20]. This approach has been subsequently used for rapid functional screening of putative CRE mutations in various disease cohorts, including Aniridia [29] and Pierre Robin Sequence [31]. This assay has clear advantages over other conventional CRE-reporter transgenic assays. It provides a rapid, unambiguous detection of subtle differences in CRE activities while using a very low number of animals compared to previous single reporter assays in mouse and zebrafish (described in Table 1). Furthermore, embryos obtained from these transgenic lines could be subjected to fluorescence activated cell sorting techniques to isolate the precise cell-types where the CREs are active. These cells would serve as the ideal biological material for investigating the predicted CREs for hallmarks of CRE function by techniques described in Section 1. The current design of the assay relies on integration of the CRE-reporter constructs in the zebrafish genome mediated by Tol2 recombination sites, which are randomly distributed in the zebrafish genome [34]. CRE activities are assessed in embryos derived from multiple independent transgenic lines to rule out any bias arising in the analysis due to site of integration of the constructs. An alternate system of integrating CRE-reporter cassettes in the zebrafish genome is mediated by recombination via phiC31 recombination sites [22,23]. This allows targeted integration of the transgene into pre-defined sites in the zebrafish genome, thus removing any bias in the analysis arising from site of integration of the transgene. An assay combining the advantages of simultaneous dual-colour imaging of CRE activity of the two alleles with targeted integration of the CRE-reporter cassette in the zebrafish genome would be highly suitable for robust qualitative and quantitative assessment of the effects of disease-associated mutations on CRE activities.
The approaches described in this review enable rapid and confident identification of the stretches of noncoding DNA harbouring CREs, likely to be crucial for regulating the expression of target genes implicated in the particular disease cohort. However, since all the methods described here study the function of CREs in isolation and outside the genomic context of their native locus, these predictions would need further validation [35]. Confident assignment of pathogenicity to mutations harboured in CREs would require the use of genome-editing techniques for generating mouse or zebrafish models bearing deletion of disease-associated CRE or knock-in of the disease-associated CRE variants. The choice of the model system in these studies would be based on the CRE region being investigated. Zebrafish could be the method of choice where the CREs and the predicted target gene affected by the mutation are conserved in the zebrafish genome. Genome-editing experiments in zebrafish would also prove to be extremely powerful for genome-wide screens for identification of CREs implicated in a specific disease condition where the phenotyping assays are well-defined in zebrafish [32,33]. These studies will improve our ability to confidently and rapidly discern pathogenic vs. non-pathogenic CRE variants, and will enhance understanding of the genetics of human disorders. They will also enable confident diagnosis and genetic counselling for patients, particularly in cases where no coding region mutations in candidate genes have been identified.
Author Contributions
Writing—review and editing, A.M. and S.B.
Funding
This research was funded by a project grant from Newlife charity for disabled children (632WBI/R43399) and a personal fellowship to SB from the Royal society of Edinburgh and Caledonian research fund (632WBI/R45412).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lee, T.I.; Young, R.A. Transcriptional Regulation and its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfelder, S.; Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Kleinjan, D.A. Disruption of long-range gene regulation in human genetic disease: A kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Qual. Life Res. 2014, 133, 815–845. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G. Genomic Identification of Regulatory Elements by Evolutionary Sequence Comparison and Functional Analysis. Nonviral Vectors Gene Ther. Phys. Methods Med Transl. 2008, 61, 269–293. [Google Scholar]
- Naville, M.; Ishibashi, M.; Ferg, M.; Bengani, H.; Rinkwitz, S.; Krecsmarik, M.; Hawkins, T.A.; Wilson, S.W.; Manning, E.; Chilamakuri, C.S.R.; et al. Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nat. Commun. 2015, 6, 6904. [Google Scholar] [CrossRef]
- Newburger, D.E.; Bulyk, M.L. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009, 37, D77–D82. [Google Scholar] [CrossRef]
- Zinzen, R.P.; Girardot, C.; Gagneur, J.; Braun, M.; Furlong, E.E.M. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 2009, 462, 65–70. [Google Scholar] [CrossRef]
- John, S.; Sabo, P.J.; Canfield, T.K.; Lee, K.; Vong, S.; Weaver, M.; Wang, H.; Vierstra, J.; Reynolds, A.P.; Thurman, R.E.; et al. Genome-scale Mapping of DNaseI Hypersensitivity. Curr. Protoc. Mol. Boil. 2013, 103, 21–27. [Google Scholar]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. The gateway to transcription: Identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol. Life Sci. 2007, 64, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Dekker, J. Genomics tools for unraveling chromosome architecture. Nat. Biotechnol 2010, 28, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H.; et al. ENCODE whole-genome data in the UCSC Genome Browser: Update 2012. Nucleic Acids Res. 2012, 40, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Goode, D.K.; Elgar, G. Capturing the regulatory interactions of eukaryote genomes. Brief Funct. Genom. 2013, 12, 142–160. [Google Scholar] [CrossRef]
- Bhatia, S.; Gordon, C.T.; Foster, R.G.; Melin, L.; Abadie, V.; Baujat, G.; Vazquez, M.-P.; Amiel, J.; Lyonnet, S.; Van Heyningen, V.; et al. Functional Assessment of Disease-Associated Regulatory Variants In Vivo Using a Versatile Dual Colour Transgenesis Strategy in Zebrafish. PLoS Genet. 2015, 11, e1005193. [Google Scholar] [CrossRef]
- Walker, S.L.; Ariga, J.; Mathias, J.R.; Coothankandaswamy, V.; Xie, X.; Distel, M.; Köster, R.W.; Parsons, M.J.; Bhalla, K.N.; Saxena, M.T.; et al. Automated Reporter Quantification In Vivo: High-Throughput Screening Method for Reporter-Based Assays in Zebrafish. PLoS ONE 2012, 7, e29916. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Miguel-Escalada, I.; Slovik, K.J.; Walsh, K.T.; Hadzhiev, Y.; Sanges, R.; Stupka, E.; Marsh, E.K.; Balciuniene, J.; Balciunas, D.; et al. Targeted transgene integration overcomes variability of position effects in zebrafish. Development 2014, 141, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadzhiev, Y.; Miguel-Escalada, I.; Balciunas, D.; Müller, F. Testing of Cis-Regulatory Elements by Targeted Transgene Integration in Zebrafish Using PhiC31 Integrase. Adv. Struct. Saf. Stud. 2016, 1451, 81–91. [Google Scholar]
- Kleinjan, D.A.; Seawright, A.; Schedl, A.; A Quinlan, R.; Danes, S.; Van Heyningen, V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 2001, 10, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Kokubu, C.; Horie, K.; Abe, K.; Ikeda, R.; Mizuno, S.; Uno, Y.; Ogiwara, S.; Ohtsuka, M.; Isotani, A.; Okabe, M.; et al. A transposon-based chromosomal engineering method to survey a large cis-regulatory landscape in mice. Nat. Genet. 2009, 41, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Symmons, O.; Uslu, V.V.; Dolle, D.; Hot, C.; Ettwiller, L.; Spitz, F. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat. Genet. 2011, 43, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. Dis. Model. Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef]
- Bhatia, S.; Monahan, J.M.; Ravi, V.; Gautier, P.; Murdoch, E.; Brenner, S.; Van Heyningen, V.; Venkatesh, B.; Kleinjan, D.A. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev. Boil. 2014, 387, 214–228. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; De Marco, R.; Damante, G.; Grainger, R.; Van Heyningen, V.; Kleinjan, D.A. Disruption of Autoregulatory Feedback by a Mutation in a Remote, Ultraconserved PAX6 Enhancer Causes Aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Ravi, V.; Bhatia, S.; Gautier, P.; Loosli, F.; Tay, B.-H.; Tay, A.; Murdoch, E.; Coutinho, P.; Van Heyningen, V.; Brenner, S.; et al. Sequencing of Pax6 Loci from the Elephant Shark Reveals a Family of Pax6 Genes in Vertebrate Genomes, Forged by Ancient Duplications and Divergences. PLoS Genet. 2013, 9, e1003177. [Google Scholar] [CrossRef]
- Rainger, J.K.; Bhatia, S.; Bengani, H.; Gautier, P.; Rainger, J.; Pearson, M.; Ansari, M.; Crow, J.; Mehendale, F.; Palinkasova, B.; et al. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum. Mol. Genet. 2014, 23, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Song, M.; Devine, P.; Bruneau, B.G.; Scott, I.C.; Wilson, M.D. Heart enhancers with deeply conserved regulatory activity are established early in zebrafish development. Nat. Commun. 2018, 9, 4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, G.; Tyagi, S.; Ramialison, M. Navigating the non-coding genome in heart development and Congenital Heart Disease. Differentiation. 2019, 107, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Mechaly, A.S.; Becker, T.S.; Rinkwitz, S. Using zebrafish transgenesis to test human genomic sequences for specific enhancer activity. Methods 2013, 62, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Lancman, J.J.; Berenguer, M.; Dong, P.D.S.; Duester, G. Genomic Knockout of Two Presumed Forelimb Tbx5 Enhancers Reveals They Are Nonessential for Limb Development. Cell Rep. 2018, 23, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
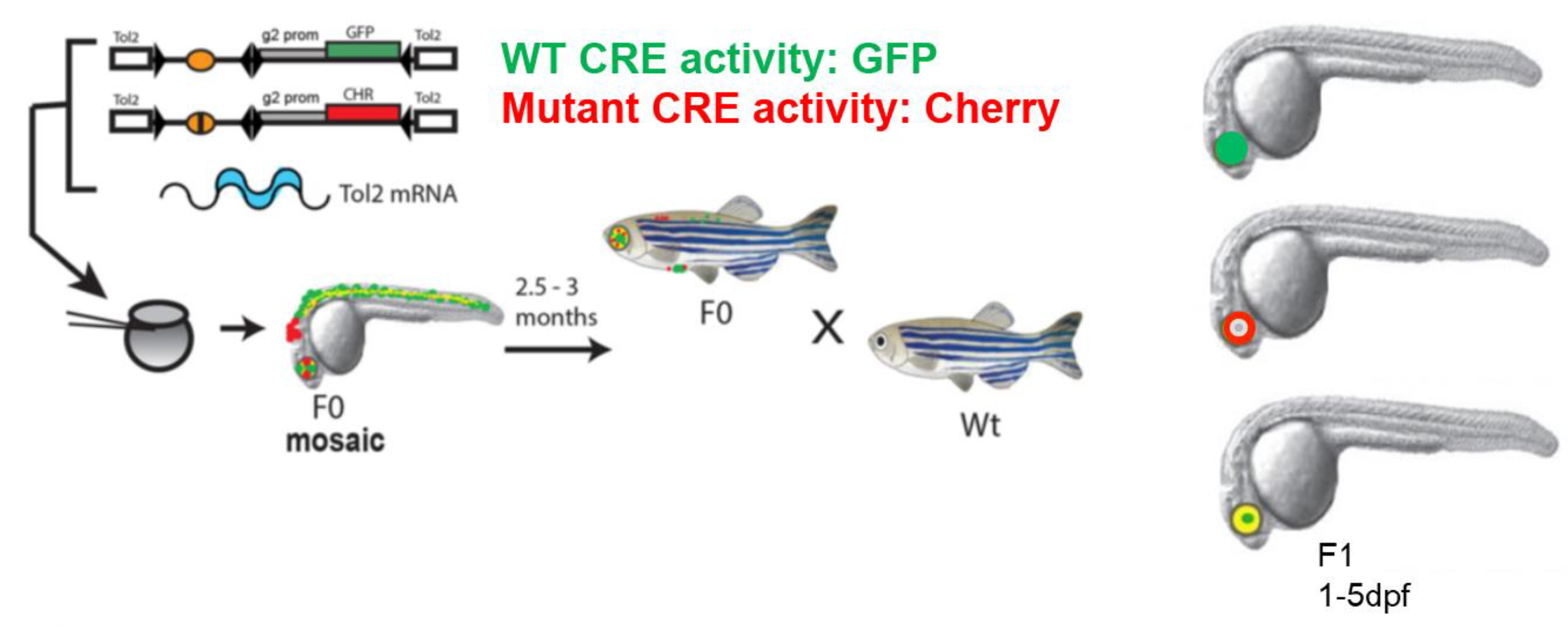
Figure 1.
In vivo characterisation of disease-associated cis-regulatory variants by dual fluorescence reporter transgenic analysis in zebrafish. Schematic representation of the assay pipeline (modified from [20]). Both alleles (wild type and mutant) of potential disease-associated CREs are cloned in a construct with a reporter cassette of choice, to create the cis-element-reporter cassette flanked by Tol2 sites. Gata2-promoter (g2 prom) derived from mouse genome is included in the CRE-reporter constructs to serve as a minimal promoter in the assay. Co-injection of reporter constructs containing wild type (Wt) and mutant (Mut) versions of the CRE with Tol2 transposase-encoding RNA into early zebrafish embryos results in independent integration of the reporter cassettes. Transgenic founders (F0) are bred to establish transgenic lines. Expression patterns are examined in fish of F1 or later generations. Differences between Wt and Mut elements can be compared directly in the same fish using the GFP and mCherry fluorescent reporters.
Figure 1.
In vivo characterisation of disease-associated cis-regulatory variants by dual fluorescence reporter transgenic analysis in zebrafish. Schematic representation of the assay pipeline (modified from [20]). Both alleles (wild type and mutant) of potential disease-associated CREs are cloned in a construct with a reporter cassette of choice, to create the cis-element-reporter cassette flanked by Tol2 sites. Gata2-promoter (g2 prom) derived from mouse genome is included in the CRE-reporter constructs to serve as a minimal promoter in the assay. Co-injection of reporter constructs containing wild type (Wt) and mutant (Mut) versions of the CRE with Tol2 transposase-encoding RNA into early zebrafish embryos results in independent integration of the reporter cassettes. Transgenic founders (F0) are bred to establish transgenic lines. Expression patterns are examined in fish of F1 or later generations. Differences between Wt and Mut elements can be compared directly in the same fish using the GFP and mCherry fluorescent reporters.


{kind=link}
{kind=link}
Table 1.
Characterisation and functional validation approaches of predicted/putative cis-regulatory elements (CREs).
Table 1.
Characterisation and functional validation approaches of predicted/putative cis-regulatory elements (CREs).
| CRE-reporter assays in in vitro cultured cell lines Description:
|
| CRE-reporter assays in zebrafish Description:
|
| CRE-reporter assays in mouse Description:
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mann, A.; Bhatia, S. Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases. Biomedicines 2019, 7, 71. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030071
AMA Style
Mann A, Bhatia S. Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases. Biomedicines. 2019; 7(3):71. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030071
Chicago/Turabian StyleMann, Anita, and Shipra Bhatia. 2019. "Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases" Biomedicines 7, no. 3: 71. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030071
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.