Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions




 ,
,
Abstract
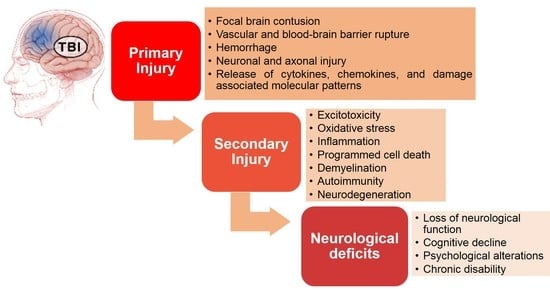
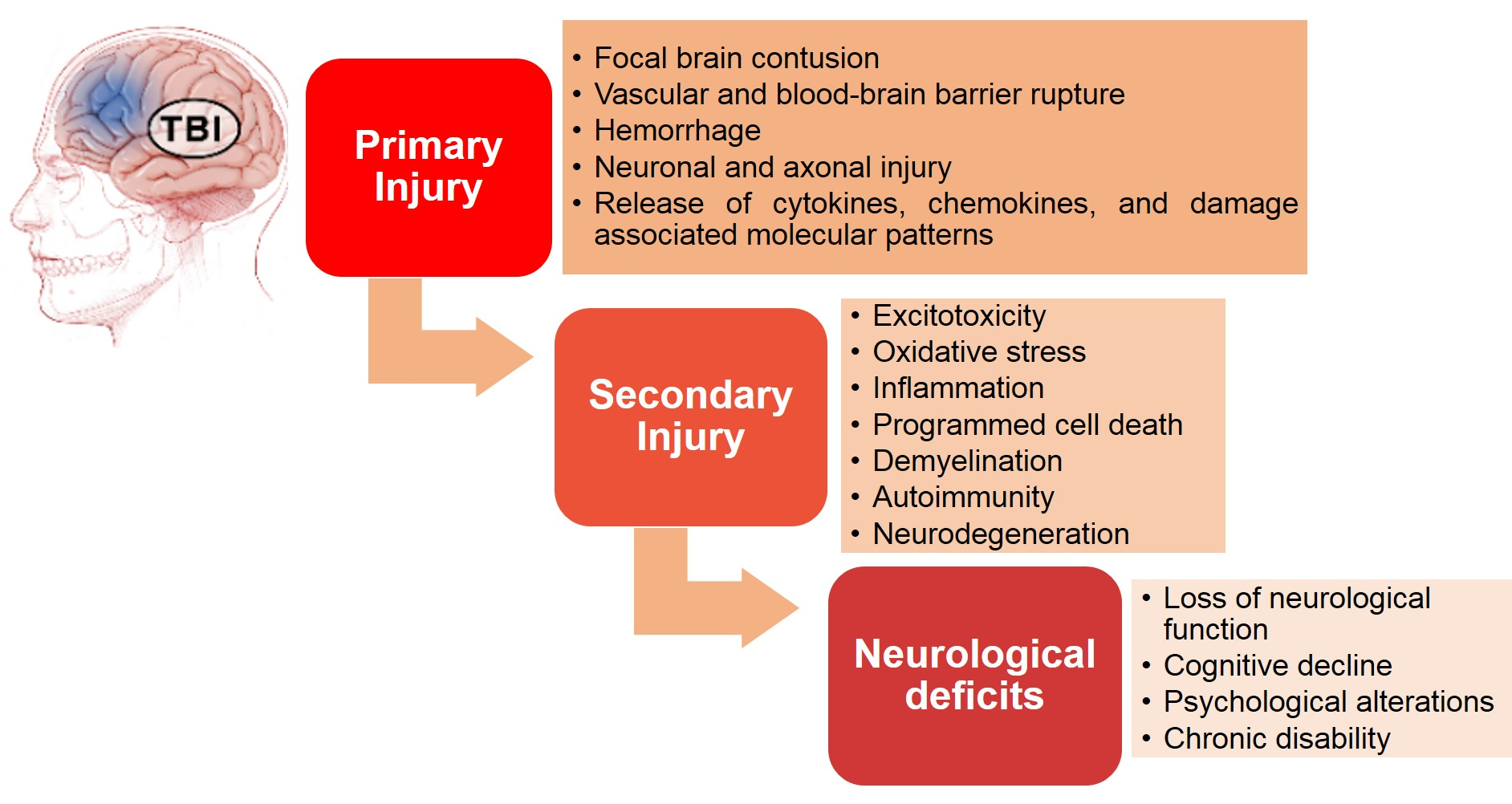
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Excitotoxicity
2.1. Glutamate
2.2. Glutamate Receptors
2.2.1. Synaptic and Extrasynaptic NMDARs
2.2.2. NMDAR Subunits
2.2.3. Therapeutic Strategies Targeting NMDARs
NMDAR Antagonists
NMDAR Subunit Inhibitors
2.3. Postsynaptic Density Protein 95 (PSD-95) and PSD-95 Inhibitors
2.4. mGluR2 Receptors and Gap Junctions
2.5. Glutamate Transporters
2.6. Blood Glutamate Scavengers
2.7. GABAergic Excitotoxicity
3. Oxidative Stress
3.1. Oxidant-Antioxidant Balance
3.2. Superoxide Radicals and Superoxide Scavengers
3.3. Iron, Hydroxyl Radicals, and Iron Chelators
3.4. Nitric oxide Synthase (NOS) and NOS Inhibitors
3.5. Peroxynitrite and Peroxynitrite Scavengers
3.6. Lipid Peroxidation (LP) and LP Inhibitors
3.7. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)-Antioxidant Response Element (ARE) Pathway
3.8. Endothelial Targeted Antioxidant Enzyme Therapy
4. Inflammation
4.1. Inflammatory Mediators
4.2. Cellular (Innate and Adaptive) Responses
4.3. Therapies Targeting Inflammation in TBI
5. Programmed Cell Death (PCD)
5.1. Cell Cycle Activation-Dependent Neuronal Cell Death
5.2. Caspase-Dependent Cell Death
5.3. Caspase-Independent Cell Death Pathways
5.4. Therapies Targeting Cell Death Pathways
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, P.J.; O’Connell, M.T.; Rothwell, N.J.; Hopkins, S.J.; Nortje, J.; Carpenter, K.L.; Timofeev, I.; Al-Rawi, P.G.; Menon, D.K.; Pickard, J.D. Inflammation in human brain injury: Intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J. Neurotrauma 2007, 24, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.A.; Rutland-Brown, W.; Thomas, K.E. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Available online: http://www.ncdsv.org/images/CDC_TBIintheUSEDVisitsHospitalizationsAndDeaths_2006.pdf (accessed on 1 September 2020).
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet (Lond. Engl.) 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Wijdicks, E.F.; Bamlet, W.R.; Maramattom, B.V.; Manno, E.M.; McClelland, R.L. Validation of a new coma scale: The FOUR score. Ann. Neurol. 2005, 58, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saatman, K.E.; Duhaime, A.C.; Bullock, R.; Maas, A.I.; Valadka, A.; Manley, G.T.; Workshop Scientific, T.; Advisory Panel, M. Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 2008, 25, 719–738. [Google Scholar] [CrossRef] [Green Version]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Whalen, M.J.; Dalkara, T.; You, Z.; Qiu, J.; Bermpohl, D.; Mehta, N.; Suter, B.; Bhide, P.G.; Lo, E.H.; Ericsson, M.; et al. Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2008, 28, 490–505. [Google Scholar] [CrossRef] [Green Version]
- Mbye, L.H.; Keles, E.; Tao, L.; Zhang, J.; Chung, J.; Larvie, M.; Koppula, R.; Lo, E.H.; Whalen, M.J. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2012, 32, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.F.; Keles, E.; Tien, L.; Zhang, J.; Kaplan, D.; Lo, E.H.; Whalen, M.J. The pharmacokinetics and pharmacodynamics of Kollidon VA64 dissociate its protective effects from membrane resealing after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2014, 34, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.K.; Michel, M.E.; Ansell, B.; Baethmann, A.; Biegon, A.; Bracken, M.B.; Bullock, M.R.; Choi, S.C.; Clifton, G.L.; Contant, C.F. Clinical trials in head injury. J. Neurotrauma 2002, 19, 503–557. [Google Scholar] [CrossRef]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Bowen, D.M.; DeKosky, S.T. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 1994, 6, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Xinding, Z.; Tianlin, Z.; Liren, C. Excitatory Amino Acids in Cerebrospinal Fluid of Patients with Acute Head Injuries. Clin. Chem. 2001, 47, 1458. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.J.; Moulton, R.J.; MacMillan, V.H.; Shedden, P.M. Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J. Neurosurg. 1993, 79, 369–372. [Google Scholar] [CrossRef]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Greve, M.W.; Zink, B.J. Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. N.Y. 2009, 76, 97–104. [Google Scholar] [CrossRef]
- Tavalin, S.J.; Ellis, E.F.; Satin, L.S. Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J. Neurophysiol. 1995, 74, 2767–2773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA receptor composition: Many regulators, many consequences. Neuroscientist 2013, 19, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Tovar, K.R.; Westbrook, G.L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 1999, 19, 4180–4188. [Google Scholar] [CrossRef] [Green Version]
- Patel, T.P.; Ventre, S.C.; Geddes-Klein, D.; Singh, P.K.; Meaney, D.F. Single-neuron NMDA receptor phenotype influences neuronal rewiring and reintegration following traumatic injury. J. Neurosci. 2014, 34, 4200–4213. [Google Scholar] [CrossRef] [Green Version]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Phillips, L.L.; Lyeth, B.G.; Hamm, R.J.; Reeves, T.M.; Povlishock, J.T. Glutamate antagonism during secondary deafferentation enhances cognition and axo-dendritic integrity after traumatic brain injury. Hippocampus 1998, 8, 390–401. [Google Scholar] [CrossRef]
- Faden, A.I.; O’Leary, D.M.; Fan, L.; Bao, W.; Mullins, P.G.; Movsesyan, V.A. Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improvesoOutcome after brain trauma. Exp. Neurol. 2001, 167, 435–444. [Google Scholar] [CrossRef]
- McIntosh, T.K.; Vink, R.; Soares, H.; Hayes, R.; Simon, R. Effects of the N-methyl-D-aspartate receptor blocker MK-801 on neurologic function after experimental brain injury. J. Neurotrauma 1989, 6, 247–259. [Google Scholar] [CrossRef]
- Muir, K.W. Glutamate-based therapeutic approaches: Clinical trials with NMDA antagonists. Curr. Opin. Pharm. 2006, 6, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Roesler, R.; Quevedo, J.; Schroder, N. Is it time to conclude that NMDA antagonists have failed? Lancet Neurol. 2003, 2, 13. [Google Scholar] [CrossRef]
- Hoyte, L.; Barber, P.A.; Buchan, A.M.; Hill, M.D. The rise and fall of NMDA antagonists for ischemic stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.L.; Dogan, A.; Todd, K.G.; Bowen, K.K.; Dempsey, R.J. Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Res. 2001, 911, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, M.; Nayeb-Aghaei, H.; Kouchek, M.; Miri, M.M.; Goharani, R.; Amoozandeh, A.; Akhavan Salamat, S.; Sistanizad, M. Effect of Memantine on Serum Levels of Neuron-Specific Enolase and on the Glasgow Coma Scale in Patients With Moderate Traumatic Brain Injury. J. Clin. Pharm. 2018, 58, 42–47. [Google Scholar] [CrossRef]
- Wang, C.Q.; Ye, Y.; Chen, F.; Han, W.C.; Sun, J.M.; Lu, X.; Guo, R.; Cao, K.; Zheng, M.J.; Liao, L.C. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience 2017, 343, 30–38. [Google Scholar] [CrossRef]
- Hertle, D.N.; Dreier, J.P.; Woitzik, J.; Hartings, J.A.; Bullock, R.; Okonkwo, D.O.; Shutter, L.A.; Vidgeon, S.; Strong, A.J.; Kowoll, C.; et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain 2012, 135, 2390–2398. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, T.K.; Faden, A.I.; Yamakami, I.; Vink, R. Magnesium deficiency exacerbates and pretreatment improves outcome following traumatic brain injury in rats: 31P magnetic resonance spectroscopy and behavioral studies. J. Neurotrauma 1988, 5, 17–31. [Google Scholar] [CrossRef]
- Arango, M.F.; Bainbridge, D. Magnesium for acute traumatic brain injury. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bai, Y.A.; Li, Y.J.; Liu, K.G.; Wang, M.D.; Xu, G.Z.; Shang, H.L.; Li, Y.F. Magnesium sulfate for acute traumatic brain injury. J. Craniofac. Surg. 2015, 26, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharm. 1993, 44, 851–859. [Google Scholar]
- Dempsey, R.J.; Başkaya, M.K.; Doğan, A. Attenuation of brain edema, blood-brain barrier breakdown, and injury volume by ifenprodil, a polyamine-site N-methyl-D-aspartate receptor antagonist, after experimental traumatic brain injury in rats. Neurosurgery 2000, 47, 399–404; discussion 404–406. [Google Scholar] [CrossRef] [PubMed]
- Maneshi, M.M.; Maki, B.; Gnanasambandam, R.; Belin, S.; Popescu, G.K.; Sachs, F.; Hua, S.Z. Mechanical stress activates NMDA receptors in the absence of agonists. Sci. Rep. 2017, 7, 39610. [Google Scholar] [CrossRef] [Green Version]
- Bigford, G.E.; Alonso, O.F.; Dietrich, D.; Keane, R.W. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J. Neurotrauma 2009, 26, 703–720. [Google Scholar] [CrossRef]
- Spaethling, J.; Le, L.; Meaney, D.F. NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol. Dis. 2012, 46, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.R.; Ndukwe, B.O.; Ren, J.; Satin, L.S.; Goforth, P.B. Stretch injury selectively enhances extrasynaptic, GluN2B-containing NMDA receptor function in cortical neurons. J. Neurophysiol. 2013, 110, 131–140. [Google Scholar] [CrossRef]
- Merchant, R.E.; Bullock, M.R.; Carmack, C.A.; Shah, A.K.; Wilner, K.D.; Ko, G.; Williams, S.A. A double-blind, placebo-controlled study of the safety, tolerability and pharmacokinetics of CP-101,606 in patients with a mild or moderate traumatic brain injury. Ann. N. Y. Acad. Sci. 1999, 890, 42–50. [Google Scholar] [CrossRef]
- Bullock, M.R.; Merchant, R.E.; Carmack, C.A.; Doppenberg, E.; Shah, A.K.; Wilner, K.D.; Ko, G.; Williams, S.A. An open-label study of CP-101,606 in subjects with a severe traumatic head injury or spontaneous intracerebral hemorrhage. Ann. N. Y. Acad. Sci. 1999, 890, 51–58. [Google Scholar] [CrossRef]
- Yurkewicz, L.; Weaver, J.; Bullock, M.R.; Marshall, L.F. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J. Neurotrauma 2005, 22, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Biegon, A. Novel approach to the role of NMDA receptors in traumatic brain injury. CNS Neurol. Disord. Drug Targets 2014, 13, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Funke, L.; Dakoji, S.; Bredt, D.S. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu Rev. Biochem. 2005, 74, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [Green Version]
- Arundine, M.; Aarts, M.; Lau, A.; Tymianski, M. Vulnerability of Central Neurons to Secondary Insults after In Vitro Mechanical Stretch. J. Neurosci. 2004, 24, 8106–8123. [Google Scholar] [CrossRef] [Green Version]
- Qu, W.; Liu, N.K.; Wu, X.; Wang, Y.; Xia, Y.; Sun, Y.; Lai, Y.; Li, R.; Shekhar, A.; Xu, X.M. Disrupting nNOS-PSD95 Interaction Improves Neurological and Cognitive Recoveries after Traumatic Brain Injury. Cereb. Cortex 2020, 30, 3859–3871. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.-T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor—PSD-95 Protein Interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; Terbrugge, K.G.; Milot, G.; Clark, W.M.; Macdonald, R.L.; Kelly, M.E.; et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharm. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Sen, T.; Gupta, R.; Kaiser, H.; Sen, N. Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J. Neurosci. 2017, 37, 5900–5911. [Google Scholar] [CrossRef]
- Wakade, C.; Sukumari-Ramesh, S.; Laird, M.D.; Dhandapani, K.M.; Vender, J.R. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J. Neurosurg. 2010, 113, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.; Clausen, B.H.; Moller, M.; Vestergaard, B.; Chi, C.N.; Round, A.; Sorensen, P.L.; Nissen, K.B.; Kastrup, J.S.; Gajhede, M.; et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc. Natl. Acad. Sci. USA 2012, 109, 3317–3322. [Google Scholar] [CrossRef] [Green Version]
- Sommer, J.B.; Bach, A.; Mala, H.; Stromgaard, K.; Mogensen, J.; Pickering, D.S. In vitro and in vivo effects of a novel dimeric inhibitor of PSD-95 on excitotoxicity and functional recovery after experimental traumatic brain injury. Eur. J. Neurosci. 2017, 45, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, J.H.; Denisova, J.V.; Park, W.M.; Fontes, J.D.; Belousov, A.B. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J. Neurosci. 2012, 32, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Hartfield, E.M.; Rinaldi, F.; Glover, C.P.; Wong, L.F.; Caldwell, M.A.; Uney, J.B. Connexin 36 expression regulates neuronal differentiation from neural progenitor cells. Plos ONE 2011, 6, e14746. [Google Scholar] [CrossRef] [Green Version]
- Todd, K.L.; Kristan, W.B., Jr.; French, K.A. Gap junction expression is required for normal chemical synapse formation. J. Neurosci. 2010, 30, 15277–15285. [Google Scholar] [CrossRef]
- Amara, S.G.; Fontana, A.C. Excitatory amino acid transporters: Keeping up with glutamate. Neurochem. Int. 2002, 41, 313–318. [Google Scholar] [CrossRef]
- Suchak, S.K.; Baloyianni, N.V.; Perkinton, M.S.; Williams, R.J.; Meldrum, B.S.; Rattray, M. The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings. J. Neurochem. 2003, 84, 522–532. [Google Scholar] [CrossRef]
- Li, S.; Stys, P.K. Na(+)-K(+)-ATPase inhibition and depolarization induce glutamate release via reverse Na(+)-dependent transport in spinal cord white matter. Neuroscience 2001, 107, 675–683. [Google Scholar] [CrossRef]
- Beschorner, R.; Dietz, K.; Schauer, N.; Mittelbronn, M.; Schluesener, H.J.; Trautmann, K.; Meyermann, R.; Simon, P. Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol. Histopathol. 2007, 22, 515–526. [Google Scholar] [PubMed]
- van Landeghem, F.K.; Weiss, T.; Oehmichen, M.; von Deimling, A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J. Neurotrauma 2006, 23, 1518–1528. [Google Scholar] [CrossRef]
- van Landeghem, F.K.; Stover, J.F.; Bechmann, I.; Bruck, W.; Unterberg, A.; Buhrer, C.; von Deimling, A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia 2001, 35, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.L.; Dogan, A.; Bowen, K.K.; Todd, K.G.; Dempsey, R.J. Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur. J. Neurosci. 2001, 13, 119–128. [Google Scholar]
- Fontana, A.C.; Fox, D.P.; Zoubroulis, A.; Mortensen, O.V.; Raghupathi, R. Neuroprotective Effects of the Glutamate Transporter Activator (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following Traumatic Brain Injury in the Adult Rat. J. Neurotrauma 2016, 33, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, M.; Wang, Y.; Teichberg, V.I. Blood-mediated scavenging of cerebrospinal fluid glutamate. J. Neurochem. 2003, 87, 119–126. [Google Scholar] [CrossRef]
- Helms, H.C.C.; Nielsen, C.U.; Waagepetersen, H.S.; Brodin, B. Glutamate Transporters in the Blood-Brain Barrier. Adv. Neurobiol. 2017, 16, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Gruenbaum, S.E.; Artru, A.A.; Rozet, I.; Dubilet, M.; Tkachov, S.; Brotfain, E.; Klin, Y.; Shapira, Y.; Teichberg, V.I. The neuroprotective effects of oxaloacetate in closed head injury in rats is mediated by its blood glutamate scavenging activity: Evidence from the use of maleate. J. Neurosurg. Anesth. 2009, 21, 235–241. [Google Scholar] [CrossRef]
- Zlotnik, A.; Sinelnikov, I.; Gruenbaum, B.F.; Gruenbaum, S.E.; Dubilet, M.; Dubilet, E.; Leibowitz, A.; Ohayon, S.; Regev, A.; Boyko, M.; et al. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. Anesthesiology 2012, 116, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, M.; Gruenbaum, S.E.; Gruenbaum, B.F.; Shapira, Y.; Zlotnik, A. Brain to blood glutamate scavenging as a novel therapeutic modality: A review. J. Neural. Transm. (Vienna) 2014, 121, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.; Sobrino, T.; Ramos-Cabrer, P.; Argibay, B.; Agulla, J.; Pérez-Mato, M.; Rodríguez-González, R.; Brea, D.; Castillo, J. Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: An experimental study. J. Cereb. Blood Flow Metab. 2011, 31, 1378–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Mato, M.; Ramos-Cabrer, P.; Sobrino, T.; Blanco, M.; Ruban, A.; Mirelman, D.; Menendez, P.; Castillo, J.; Campos, F. Human recombinant glutamate oxaloacetate transaminase 1 (GOT1) supplemented with oxaloacetate induces a protective effect after cerebral ischemia. Cell Death Dis. 2014, 5, e992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva-Candal, A.; Pérez-Díaz, A.; Santamaría, M.; Correa-Paz, C.; Rodríguez-Yáñez, M.; Ardá, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Brea, J.; Azuaje, J.; et al. Clinical validation of blood/brain glutamate grabbing in acute ischemic stroke. Ann. Neurol. 2018, 84, 260–273. [Google Scholar] [CrossRef]
- Hoane, M.R.; Wolyniak, J.G.; Akstulewicz, S.L. Administration of riboflavin improves behavioral outcome and reduces edema formation and glial fibrillary acidic protein expression after traumatic brain injury. J. Neurotrauma 2005, 22, 1112–1122. [Google Scholar] [CrossRef]
- Nilsson, P.; Hillered, L.; Ponten, U.; Ungerstedt, U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J. Cereb. Blood Flow Metab. 1990, 10, 631–637. [Google Scholar] [CrossRef]
- Anderson, K.J.; Miller, K.M.; Fugaccia, I.; Scheff, S.W. Regional distribution of fluoro-jade B staining in the hippocampus following traumatic brain injury. Exp. Neurol. 2005, 193, 125–130. [Google Scholar] [CrossRef]
- Sato, M.; Chang, E.; Igarashi, T.; Noble, L.J. Neuronal injury and loss after traumatic brain injury: Time course and regional variability. Brain Res. 2001, 917, 45–54. [Google Scholar]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Reviews. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef]
- Mongiat, L.A.; Schinder, A.F. Adult neurogenesis and the plasticity of the dentate gyrus network. Eur. J. Neurosci. 2011, 33, 1055–1061. [Google Scholar] [CrossRef]
- Gao, X.; Deng-Bryant, Y.; Cho, W.; Carrico, K.M.; Hall, E.D.; Chen, J. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J. Neurosci. Res. 2008, 86, 2258–2270. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Chen, J. Conditional knockout of brain-derived neurotrophic factor in the hippocampus increases death of adult-born immature neurons following traumatic brain injury. J. Neurotrauma 2009, 26, 1325–1335. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, L.; Gao, X.; Luo, B.; Chen, J. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J. Neuropathol. Exp. Neurol. 2012, 71, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.L.; Xiang, Q.; Shi, Q.Y.; Li, S.Y.; Tan, L.; Wang, J.T.; Jin, X.G.; Luo, A.L. GABAergic excitotoxicity injury of the immature hippocampal pyramidal neurons’ exposure to isoflurane. Anesth. Analg. 2011, 113, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Afshari, D.; Moradian, N.; Rezaei, M. Evaluation of the intravenous magnesium sulfate effect in clinical improvement of patients with acute ischemic stroke. Clin. Neurol. Neurosurg. 2013, 115, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Prata, C.; Vieceli Dalla Sega, F.; Piperno, R.; Hrelia, S. Traumatic Brain Injury and NADPH Oxidase: A Deep Relationship. Oxidative Med. Cell. Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, P.; Szarka, N.; Farkas, E.; Ezer, E.; Czeiter, E.; Amrein, K.; Ungvari, Z.I.; Hartings, J.A.; Buki, A.; Koller, A. Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: Pathomechanism and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Veenith, T.V.; Carter, E.L.; Geeraerts, T.; Grossac, J.; Newcombe, V.F.; Outtrim, J.; Gee, G.S.; Lupson, V.; Smith, R.; Aigbirhio, F.I.; et al. Pathophysiologic Mechanisms of Cerebral Ischemia and Diffusion Hypoxia in Traumatic Brain Injury. JAMA Neurol. 2016, 73, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef] [PubMed]
- Readnower, R.D.; Chavko, M.; Adeeb, S.; Conroy, M.D.; Pauly, J.R.; McCarron, R.M.; Sullivan, P.G. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 2010, 88, 3530–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWitt, D.S.; Prough, D.S. Blast-induced brain injury and posttraumatic hypotension and hypoxemia. J. Neurotrauma 2009, 26, 877–887. [Google Scholar] [CrossRef]
- Vuceljic, M.; Zunic, G.; Romic, P.; Jevtic, M. Relation between both oxidative and metabolic-osmotic cell damages and initial injury severity in bombing casualties. Vojnosanit. Pregl. 2006, 63, 545–551. [Google Scholar] [CrossRef]
- Tran, L.V. Understanding the pathophysiology of traumatic brain injury and the mechanisms of action of neuroprotective interventions. J. Trauma Nurs. 2014, 21, 30–35. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Kontos, H.A. Continuing axonal and vascular change following experimental brain trauma. Cent. Nerv. Syst. Trauma 1985, 2, 285–298. [Google Scholar] [CrossRef]
- Kontos, H.A.; Wei, E.P. Superoxide production in experimental brain injury. J. Neurosurg. 1986, 64, 803–807. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine; Oxford University Press: Oxfard, UK, 2007. [Google Scholar]
- Marklund, S.L.; Westman, N.G.; Lundgren, E.; Roos, G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982, 42, 1955–1961. [Google Scholar]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef]
- Kontos, H.A.; Povlishock, J.T. Oxygen radicals in brain injury. Cent. Nerv. Syst. Trauma 1986, 3, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.H.; Epstein, C.J.; Li, Y.; Huang, T.T.; Carlson, E.; Kinouchi, H.; Yang, G.; Kamii, H.; Mikawa, S.; Kondo, T.; et al. Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J. Neurotrauma 1995, 12, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, S.; Kinouchi, H.; Kamii, H.; Gobbel, G.T.; Chen, S.F.; Carlson, E.; Epstein, C.J.; Chan, P.H. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J. Neurosurg. 1996, 85, 885–891. [Google Scholar] [CrossRef]
- Lewen, A.; Fujimura, M.; Sugawara, T.; Matz, P.; Copin, J.C.; Chan, P.H. Oxidative stress-dependent release of mitochondrial cytochrome c after traumatic brain injury. J. Cereb. Blood Flow Metab. 2001, 21, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewen, A.; Sugawara, T.; Gasche, Y.; Fujimura, M.; Chan, P.H. Oxidative cellular damage and the reduction of APE/Ref-1 expression after experimental traumatic brain injury. Neurobiol. Dis. 2001, 8, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Shie, F.S.; Zhang, J.; Lee, C.P.; Ho, Y.S. Prevention of mitochondrial dysfunction in post-traumatic mouse brain by superoxide dismutase. J. Neurochem. 2005, 95, 732–744. [Google Scholar] [CrossRef]
- Pineda, J.A.; Aono, M.; Sheng, H.; Lynch, J.; Wellons, J.C.; Laskowitz, D.T.; Pearlstein, R.D.; Bowler, R.; Crapo, J.; Warner, D.S. Extracellular superoxide dismutase overexpression improves behavioral outcome from closed head injury in the mouse. J. Neurotrauma 2001, 18, 625–634. [Google Scholar] [CrossRef]
- Muizelaar, J.P.; Marmarou, A.; Young, H.F.; Choi, S.C.; Wolf, A.; Schneider, R.L.; Kontos, H.A. Improving the outcome of severe head injury with the oxygen radical scavenger polyethylene glycol-conjugated superoxide dismutase: A phase II trial. J. Neurosurg. 1993, 78, 375–382. [Google Scholar] [CrossRef]
- Muizelaar, J.P.; Kupiec, J.W.; Rapp, L.A. PEG-SOD after head injury. J. Neurosurg. 1995, 83, 942. [Google Scholar] [CrossRef]
- Aoyama, N.; Katayama, Y.; Kawamata, T.; Maeda, T.; Mori, T.; Yamamoto, T.; Kikuchi, T.; Uwahodo, Y. Effects of antioxidant, OPC-14117, on secondary cellular damage and behavioral deficits following cortical contusion in the rat. Brain Res. 2002, 934, 117–124. [Google Scholar] [CrossRef]
- The Dana Consortium. Safety and tolerability of the antioxidant OPC-14117 in HIV-associated cognitive impairment. The Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Neurology 1997, 49, 142–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaleska, M.M.; Floyd, R.A. Regional lipid peroxidation in rat brain in vitro: Possible role of endogenous iron. Neurochem. Res. 1985, 10, 397–410. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [PubMed]
- Sadrzadeh, S.M.; Eaton, J.W. Hemoglobin-mediated oxidant damage to the central nervous system requires endogenous ascorbate. J. Clin. Investig. 1988, 82, 1510–1515. [Google Scholar] [CrossRef] [Green Version]
- Long, D.A.; Ghosh, K.; Moore, A.N.; Dixon, C.E.; Dash, P.K. Deferoxamine improves spatial memory performance following experimental brain injury in rats. Brain Res. 1996, 717, 109–117. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, R.; Li, M.; Li, F.; Meng, H.; Zhu, G.; Lin, J.; Feng, H. Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol. Sci. 2013, 34, 639–645. [Google Scholar] [CrossRef]
- Panter, S.S.; Braughler, J.M.; Hall, E.D. Dextran-coupled deferoxamine improves outcome in a murine model of head injury. J. Neurotrauma 1992, 9, 47–53. [Google Scholar] [CrossRef]
- Daglas, M.; Adlard, P.A. The Involvement of Iron in Traumatic Brain Injury and Neurodegenerative Disease. Front. Neurosci. 2018, 12, 981. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, S.; Ahmad, A.S.; Chamara, K.; Dore, S. Unique Properties Associated with the Brain Penetrant Iron Chelator HBED Reveal Remarkable Beneficial Effects after Brain Trauma. J. Neurotrauma 2018. [Google Scholar] [CrossRef]
- Gahm, C.; Holmin, S.; Mathiesen, T. Temporal profiles and cellular sources of three nitric oxide synthase isoforms in the brain after experimental contusion. Neurosurgery 2000, 46, 169–177. [Google Scholar] [CrossRef]
- Cherian, L.; Hlatky, R.; Robertson, C.S. Nitric oxide in traumatic brain injury. Brain Pathol. (Zur. Switz.) 2004, 14, 195–201. [Google Scholar] [CrossRef]
- Toczylowska, B.; Chalimoniuk, M.; Wodowska, M.; Mayzner-Zawadzk, E. Changes in concentration of cerebrospinal fluid components in patients with traumatic brain injury. Brain Res. 2006, 1104, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.; Kochanek, P.M.; Obrist, W.D.; Wong, H.R.; Billiar, T.R.; Wisniewski, S.R.; Marion, D.W. Cerebrospinal fluid and plasma nitrite and nitrate concentrations after head injury in humans. Crit. Care Med. 1996, 24, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Uzan, M.; Tanriover, N.; Bozkus, H.; Gumustas, K.; Guzel, O.; Kuday, C. Nitric oxide (NO) metabolism in the cerebrospinal fluid of patients with severe head injury. Inflammation as a possible cause of elevated no metabolites. Surg. Neurol. 2001, 56, 350–356. [Google Scholar] [CrossRef]
- Mesenge, C.; Verrecchia, C.; Allix, M.; Boulu, R.R.; Plotkine, M. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J. Neurotrauma 1996, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Chatzipanteli, K.; Busto, R.; Dietrich, W.D. Effects of L-NAME and 7-NI on NOS catalytic activity and behavioral outcome after traumatic brain injury in the rat. J. Neurotrauma 1999, 16, 203–212. [Google Scholar] [CrossRef]
- Lu, Y.C.; Liu, S.; Gong, Q.Z.; Hamm, R.J.; Lyeth, B.G. Inhibition of nitric oxide synthase potentiates hypertension and increases mortality in traumatically brain-injured rats. Mol. Chem. Neuropathol. 1997, 30, 125–137. [Google Scholar] [CrossRef]
- Cherian, L.; Chacko, G.; Goodman, J.C.; Robertson, C.S. Cerebral hemodynamic effects of phenylephrine and L-arginine after cortical impact injury. Crit. Care Med. 1999, 27, 2512–2517. [Google Scholar] [CrossRef]
- Hlatky, R.; Lui, H.; Cherian, L.; Goodman, J.C.; O’Brien, W.E.; Contant, C.F.; Robertson, C.S. The role of endothelial nitric oxide synthase in the cerebral hemodynamics after controlled cortical impact injury in mice. J. Neurotrauma 2003, 20, 995–1006. [Google Scholar] [CrossRef]
- Hlatky, R.; Goodman, J.C.; Valadka, A.B.; Robertson, C.S. Role of nitric oxide in cerebral blood flow abnormalities after traumatic brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Castilla, L.; Ahmed, O.; Goodman, J.C.; Gopinath, S.; Valadka, A.; Robertson, C. L-arginine reactivity in cerebral vessels after severe traumatic brain injury. Neurol. Res. 2010, 32, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.S.; Gopinath, S.P.; Valadka, A.B.; Van, M.; Swank, P.R.; Goodman, J.C. Variants of the endothelial nitric oxide gene and cerebral blood flow after severe traumatic brain injury. J. Neurotrauma 2011, 28, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Giannopoulos, S.; Katsanos, A.H.; Tsivgoulis, G.; Marshall, R.S. Statins and cerebral hemodynamics. J. Cereb. Blood Flow Metab. 2012, 32, 1973–1976. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Yun, C.W.; Park, W.K.; Kong, J.Y.; Kim, K.S.; Park, Y.; Lee, S.; Kim, B.K. Modulation of the activity of pro-inflammatory enzymes, COX-2 and iNOS, by chrysin derivatives. Pharmacol. Res. 2004, 49, 37–43. [Google Scholar] [CrossRef]
- Khan, A.; Vaibhav, K.; Javed, H.; Tabassum, R.; Ahmed, M.E.; Khan, M.M.; Khan, M.B.; Shrivastava, P.; Islam, F.; Siddiqui, M.S.; et al. 1,8-cineole (eucalyptol) mitigates inflammation in amyloid Beta toxicated PC12 cells: Relevance to Alzheimer’s disease. Neurochem. Res. 2014, 39, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Vaibhav, K.; Shrivastava, P.; Khan, A.; Ahmed, M.E.; Ashafaq, M.; Khan, M.B.; Islam, F.; Safhi, M.M.; Islam, F. Perillyl alcohol improves functional and histological outcomes against ischemia-reperfusion injury by attenuation of oxidative stress and repression of COX-2, NOS-2 and NF-kappaB in middle cerebral artery occlusion rats. Eur. J. Pharmacol. 2015, 747, 190–199. [Google Scholar] [CrossRef]
- Vaibhav, K.; Shrivastava, P.; Javed, H.; Khan, A.; Ahmed, M.E.; Tabassum, R.; Khan, M.M.; Khuwaja, G.; Islam, F.; Siddiqui, M.S.; et al. Piperine suppresses cerebral ischemia-reperfusion-induced inflammation through the repression of COX-2, NOS-2, and NF-kappaB in middle cerebral artery occlusion rat model. Mol. Cell. Biochem. 2012, 367, 73–84. [Google Scholar] [CrossRef]
- Wada, K.; Chatzipanteli, K.; Kraydieh, S.; Busto, R.; Dietrich, W.D. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery 1998, 43, 1427–1436. [Google Scholar] [CrossRef]
- Moochhala, S.M.; Md, S.; Lu, J.; Teng, C.H.; Greengrass, C. Neuroprotective role of aminoguanidine in behavioral changes after blast injury. J. Trauma 2004, 56, 393–403. [Google Scholar] [CrossRef]
- Louin, G.; Marchand-Verrecchia, C.; Palmier, B.; Plotkine, M.; Jafarian-Tehrani, M. Selective inhibition of inducible nitric oxide synthase reduces neurological deficit but not cerebral edema following traumatic brain injury. Neuropharmacology 2006, 50, 182–190. [Google Scholar] [CrossRef]
- Stover, J.F.; Belli, A.; Boret, H.; Bulters, D.; Sahuquillo, J.; Schmutzhard, E.; Zavala, E.; Ungerstedt, U.; Schinzel, R.; Tegtmeier, F.; et al. Nitric oxide synthase inhibition with the antipterin VAS203 improves outcome in moderate and severe traumatic brain injury: A placebo-controlled randomized Phase IIa trial (NOSTRA). J. Neurotrauma 2014, 31, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeier, F.; Schinzel, R.; Beer, R.; Bulters, D.; LeFrant, J.Y.; Sahuquillo, J.; Unterberg, A.; Andrews, P.; Belli, A.; Ibanez, J.; et al. Efficacy of Ronopterin (VAS203) in Patients with Moderate and Severe Traumatic Brain Injury (NOSTRA phase III trial): Study protocol of a confirmatory, placebo-controlled, randomised, double blind, multi-centre study. Trials 2020, 21, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Kupina, N.C.; Althaus, J.S. Peroxynitrite scavengers for the acute treatment of traumatic brain injury. Ann. N. Y. Acad. Sci. 1999, 890, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shohami, E.; Beit-Yannai, E.; Bass, R.; Trembovler, V.; Samuni, A. Mechanism of brain protection by nitroxide radicals in experimental model of closed-head injury. Free Radic. Biol. Med. 1998, 24, 332–340. [Google Scholar] [CrossRef]
- Bonini, M.G.; Mason, R.P.; Augusto, O. The Mechanism by which 4-hydroxy-2,2,6,6-tetramethylpiperidene-1-oxyl (tempol) diverts peroxynitrite decomposition from nitrating to nitrosating species. Chem. Res. Toxicol. 2002, 15, 506–511. [Google Scholar] [CrossRef]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurother. J. Am. Soc. Exp. Neurother. 2010, 7, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Marklund, N.; Clausen, F.; Lewen, A.; Hovda, D.A.; Olsson, Y.; Hillered, L. alpha-Phenyl-tert-N-butyl nitrone (PBN) improves functional and morphological outcome after cortical contusion injury in the rat. Acta Neurochir. 2001, 143, 73–81. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- Hall, E.D.; Yonkers, P.A.; McCall, J.M.; Braughler, J.M. Effects of the 21-aminosteroid U74006F on experimental head injury in mice. J. Neurosurg. 1988, 68, 456–461. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, T.K.; Thomas, M.; Smith, D.; Banbury, M. The novel 21-aminosteroid U74006F attenuates cerebral edema and improves survival after brain injury in the rat. J. Neurotrauma 1992, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Dimlich, R.V.; Tornheim, P.A.; Kindel, R.M.; Hall, E.D.; Braughler, J.M.; McCall, J.M. Effects of a 21-aminosteroid (U-74006F) on cerebral metabolites and edema after severe experimental head trauma. Adv. Neurol. 1990, 52, 365–375. [Google Scholar]
- Marshall, L.F.; Maas, A.I.; Marshall, S.B.; Bricolo, A.; Fearnside, M.; Iannotti, F.; Klauber, M.R.; Lagarrigue, J.; Lobato, R.; Persson, L.; et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J. Neurosurg. 1998, 89, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Galvani, S.; Coatrieux, C.; Elbaz, M.; Grazide, M.H.; Thiers, J.C.; Parini, A.; Uchida, K.; Kamar, N.; Rostaing, L.; Baltas, M.; et al. Carbonyl scavenger and antiatherogenic effects of hydrazine derivatives. Free Radic. Biol. Med. 2008, 45, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Wang, J.A.; Miller, D.M.; Cebak, J.E.; Hill, R.L. Newer pharmacological approaches for antioxidant neuroprotection in traumatic brain injury. Neuropharmacology 2019, 145, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Gilmer, L.K.; Miller, D.M.; Cebak, J.E.; Wang, J.A.; Hall, E.D. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J. Cereb. Blood Flow Metab. 2013, 33, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebak, J.E.; Singh, I.N.; Hill, R.L.; Wang, J.A.; Hall, E.D. Phenelzine Protects Brain Mitochondrial Function In Vitro and In Vivo following Traumatic Brain Injury by Scavenging the Reactive Carbonyls 4-Hydroxynonenal and Acrolein Leading to Cortical Histological Neuroprotection. J. Neurotrauma 2017, 34, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.; Matveychuk, D.; MacKenzie, E.M.; Holt, A.; Wang, Y.; Kar, S. Attenuation of the effects of oxidative stress by the MAO-inhibiting antidepressant and carbonyl scavenger phenelzine. Chem. Biol. Interact. 2019, 304, 139–147. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharm. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.; Yan, W.; Zhu, L.; Hu, Z.; Ding, Y.; Tang, K. Role of Nrf2 in protection against traumatic brain injury in mice. J. Neurotrauma 2009, 26, 131–139. [Google Scholar] [CrossRef]
- Hong, Y.; Yan, W.; Chen, S.; Sun, C.R.; Zhang, J.M. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharm. Sin. 2010, 31, 1421–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Kong, J.; Wang, H.; Wu, J.; Lu, T.; Jiang, J.; Ni, H.; Liang, W. Protective effect of tert-butylhydroquinone on cerebral inflammatory response following traumatic brain injury in mice. Injury 2011, 42, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Wang, H.D.; Xu, J.G.; Ding, K.; Li, T. Pretreatment with tert-butylhydroquinone attenuates cerebral oxidative stress in mice after traumatic brain injury. J. Surg. Res. 2014, 188, 206–212. [Google Scholar] [CrossRef]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp. Neurol. 2015, 264, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Maynard, M.E.; Underwood, E.L.; Redell, J.B.; Zhao, J.; Kobori, N.; Hood, K.N.; Moore, A.N.; Dash, P.K. Carnosic Acid Improves Outcome after Repetitive Mild Traumatic Brain Injury. J. Neurotrauma 2019, 36, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef]
- Ding, K.; Xu, J.; Wang, H.; Zhang, L.; Wu, Y.; Li, T. Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice. Neurochem. Int. 2015, 91, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shao, A.; Zhao, M.; Chen, S.; Yu, J.; Zhou, J.; Liang, F.; Shi, L.; Dixon, B.J.; Wang, Z.; et al. Melatonin attenuates neuronal apoptosis through up-regulation of K(+) -Cl(−) cotransporter KCC2 expression following traumatic brain injury in rats. J. Pineal Res. 2016, 61, 241–250. [Google Scholar] [CrossRef]
- Luo, C.; Yang, Q.; Liu, Y.; Zhou, S.; Jiang, J.; Reiter, R.J.; Bhattacharya, P.; Cui, Y.; Yang, H.; Ma, H.; et al. The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic. Biol. Med. 2019, 130, 215–233. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2-ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef]
- Barlow, K.M.; Brooks, B.L.; MacMaster, F.P.; Kirton, A.; Seeger, T.; Esser, M.; Crawford, S.; Nettel-Aguirre, A.; Zemek, R.; Angelo, M.; et al. A double-blind, placebo-controlled intervention trial of 3 and 10 mg sublingual melatonin for post-concussion syndrome in youths (PLAYGAME): Study protocol for a randomized controlled trial. Trials 2014, 15, 271. [Google Scholar] [CrossRef] [Green Version]
- Pandya, J.D.; Readnower, R.D.; Patel, S.P.; Yonutas, H.M.; Pauly, J.R.; Goldstein, G.A.; Rabchevsky, A.G.; Sullivan, P.G. N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp. Neurol. 2014, 257, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, H.D.; Zhou, X.M.; Fang, J.; Zhu, L.; Ding, K. N-acetylcysteine amide provides neuroprotection via Nrf2-ARE pathway in a mouse model of traumatic brain injury. Drug Des. Devel. 2018, 12, 4117–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.; Nascimento, B.; Akhtar, U.; Rhind, S.G.; Tien, H.; Nathens, A.; da Luz, L.T. Systematic Review of Human and Animal Studies Examining the Efficacy and Safety of N-Acetylcysteine (NAC) and N-Acetylcysteine Amide (NACA) in Traumatic Brain Injury: Impact on Neurofunctional Outcome and Biomarkers of Oxidative Stress and Inflammation. Front. Neurol. 2017, 8, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutton, E.M.; Farney, S.K.; Andrews, A.M.; Shuvaev, V.V.; Chuang, G.Y.; Muzykantov, V.R.; Ramirez, S.H. Endothelial Targeted Strategies to Combat Oxidative Stress: Improving Outcomes in Traumatic Brain Injury. Front. Neurol. 2019, 10, 582. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Otto, V.I.; Stahel, P.F.; Kossmann, T. Role of cerebral inflammation after traumatic brain injury: A revisited concept. Shock (Augustaga.) 2001, 16, 165–177. [Google Scholar] [CrossRef]
- Bye, N.; Habgood, M.D.; Callaway, J.K.; Malakooti, N.; Potter, A.; Kossmann, T.; Morganti-Kossmann, M.C. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 2007, 204, 220–233. [Google Scholar] [CrossRef]
- Kubes, P.; Ward, P.A. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. (Zur. Switz.) 2000, 10, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S232–S240. [Google Scholar] [CrossRef] [Green Version]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- King, M.D.; Laird, M.D.; Ramesh, S.S.; Youssef, P.; Shakir, B.; Vender, J.R.; Alleyne, C.H.; Dhandapani, K.M. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: An emerging role for neuroproteomics. Neurosurg. Focus 2010, 28, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Vaibhav, K.; Saad, N.M.; Fatima, S.; Vender, J.R.; Baban, B.; Hoda, M.N.; Dhandapani, K.M. White matter damage after traumatic brain injury: A role for damage associated molecular patterns. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2017, 1863, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J. Peripheral immune cells in the pathology of traumatic brain injury? Curr. Opin. Crit. Care 2011, 17, 122–130. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, N.J. Annual review prize lecture cytokines - killers in the brain? J. Physiol. 1999, 514 Pt 1, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Shuaib, A. Involvement of inflammatory cytokines in central nervous system injury. Prog. Neurobiol. 2002, 67, 161–172. [Google Scholar] [CrossRef]
- Lu, W.; Gersting, J.A.; Maheshwari, A.; Christensen, R.D.; Calhoun, D.A. Developmental expression of chemokine receptor genes in the human fetus. Early Hum. Dev. 2005, 81, 489–496. [Google Scholar] [CrossRef]
- Dalgard, C.L.; Cole, J.T.; Kean, W.S.; Lucky, J.J.; Sukumar, G.; McMullen, D.C.; Pollard, H.B.; Watson, W.D. The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci. 2012, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Shein, S.L.; Shellington, D.K.; Exo, J.L.; Jackson, T.C.; Wisniewski, S.R.; Jackson, E.K.; Vagni, V.A.; Bayir, H.; Clark, R.S.; Dixon, C.E.; et al. Hemorrhagic shock shifts the serum cytokine profile from pro- to anti-inflammatory after experimental traumatic brain injury in mice. J. Neurotrauma 2014, 31, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Redell, J.B.; Moore, A.N.; Grill, R.J.; Johnson, D.; Zhao, J.; Liu, Y.; Dash, P.K. Analysis of functional pathways altered after mild traumatic brain injury. J. Neurotrauma 2013, 30, 752–764. [Google Scholar] [CrossRef]
- White, T.E.; Ford, G.D.; Surles-Zeigler, M.C.; Gates, A.S.; Laplaca, M.C.; Ford, B.D. Gene expression patterns following unilateral traumatic brain injury reveals a local pro-inflammatory and remote anti-inflammatory response. BMC Genom. 2013, 14, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaibhav, K.; Braun, M.; Alverson, K.; Khodadadi, H.; Kutiyanawalla, A.; Ward, A.; Banerjee, C.; Sparks, T.; Malik, A.; Rashid, M.H.; et al. Neutrophil extracellular traps exacerbate neurological deficts after traumatic brain injury. Sci. Adv. 2020. [Google Scholar] [CrossRef]
- Braun, M.; Khan, Z.T.; Khan, M.B.; Kumar, M.; Ward, A.; Achyut, B.R.; Arbab, A.S.; Hess, D.C.; Hoda, M.N.; Baban, B.; et al. Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav. Immun. 2018, 68, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Karnati, H.K.; Mullins, R.; Pick, C.G.; Hoffer, B.J.; Goetzl, E.J.; Kapogiannis, D.; Greig, N.H. Time-dependent cytokine and chemokine changes in mouse cerebral cortex following a mild traumatic brain injury. ELife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A.; Carpenter, K.L.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J. The cytokine response to human traumatic brain injury: Temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. 2011, 31, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmy, A.; Antoniades, C.A.; Guilfoyle, M.R.; Carpenter, K.L.; Hutchinson, P.J. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. Plos ONE 2012, 7, e39677. [Google Scholar] [CrossRef]
- Braun, M.; Vaibhav, K.; Saad, N.; Fatima, S.; Brann, D.W.; Vender, J.R.; Wang, L.P.; Hoda, M.N.; Baban, B.; Dhandapani, K.M. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. J. Immunol. 2017, 198, 3615–3626. [Google Scholar] [CrossRef] [Green Version]
- Vaibhav, K.; Braun, M.; Khan, M.B.; Fatima, S.; Saad, N.; Shankar, A.; Khan, Z.T.; Harris, R.B.S.; Yang, Q.; Huo, Y.; et al. Remote ischemic post-conditioning promotes hematoma resolution via AMPK-dependent immune regulation. J. Exp. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Soares, H.D.; Hicks, R.R.; Smith, D.; McIntosh, T.K. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 1995, 15, 8223–8233. [Google Scholar] [CrossRef]
- Carlos, T.M.; Clark, R.S.; Franicola-Higgins, D.; Schiding, J.K.; Kochanek, P.M. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J. Leukoc. Biol. 1997, 61, 279–285. [Google Scholar] [CrossRef]
- Holmin, S.; Mathiesen, T.; Shetye, J.; Biberfeld, P. Intracerebral inflammatory response to experimental brain contusion. Acta Neurochir. 1995, 132, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, R.; Kaiser, A.; Lang, C.; Bohnert, M.; Betz, P. A quantitative immunohistochemical study on the time-dependent course of acute inflammatory cellular response to human brain injury. Int. J. Leg. Med. 1999, 112, 227–232. [Google Scholar]
- Hsieh, C.L.; Kim, C.C.; Ryba, B.E.; Niemi, E.C.; Bando, J.K.; Locksley, R.M.; Liu, J.; Nakamura, M.C.; Seaman, W.E. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 2013, 43, 2010–2022. [Google Scholar] [CrossRef] [Green Version]
- Kelley, B.J.; Lifshitz, J.; Povlishock, J.T. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J. Neuropathol. Exp. Neurol. 2007, 66, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Thomas, T.C.; Ziebell, J.M.; Pauly, J.R.; Lifshitz, J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 2012, 225, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Holmin, S.; Schalling, M.; Hojeberg, B.; Nordqvist, A.C.; Skeftruna, A.K.; Mathiesen, T. Delayed cytokine expression in rat brain following experimental contusion. J. Neurosurg. 1997, 86, 493–504. [Google Scholar] [CrossRef]
- Oehmichen, M.; Jakob, S.; Mann, S.; Saternus, K.S.; Pedal, I.; Meissner, C. Macrophage subsets in mechanical brain injury (MBI)--a contribution to timing of MBI based on immunohistochemical methods: A pilot study. Leg. Med. 2009, 11, 118–124. [Google Scholar] [CrossRef]
- Walsh, J.T.; Zheng, J.; Smirnov, I.; Lorenz, U.; Tung, K.; Kipnis, J. Regulatory T Cells in Central Nervous System Injury: A Double-Edged Sword. J. Immunol. 2014, 193, 5013–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzolla, A.; Gelderman, K.A.; Hultqvist, M.; Vestberg, M.; Gustafsson, K.; Mattsson, R.; Holmdahl, R. CD68-expressing cells can prime T cells and initiate autoimmune arthritis in the absence of reactive oxygen species. Eur. J. Immunol. 2011, 41, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergelli, M.; Pinet, V.; Vogt, A.B.; Kalbus, M.; Malnati, M.; Riccio, P.; Long, E.O.; Martin, R. HLA-DR-restricted presentation of purified myelin basic protein is independent of intracellular processing. Eur. J. Immunol. 1997, 27, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Mukherjee, S.; Kain, J.M.; Rogers, S.K.; Henderson, S.K.; Motal, H.L.; Newell Rogers, M.K.; Shapiro, L.A. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol. Commun. 2014, 2, 143. [Google Scholar] [CrossRef] [Green Version]
- Mosley, R.L.; Hutter-Saunders, J.A.; Stone, D.K.; Gendelman, H.E. Inflammation and Adaptive Immunity in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Hickey, W.F.; Hsu, B.L.; Kimura, H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991, 28, 254–260. [Google Scholar] [CrossRef]
- Holmin, S.; Söderlund, J.; Biberfeld, P.; Mathiesen, T. Intracerebral Inflammation after Human Brain Contusion. Neurosurgery 1998, 42, 291–298. [Google Scholar] [CrossRef]
- Hua, R.; Mao, S.S.; Zhang, Y.M.; Chen, F.X.; Zhou, Z.H.; Liu, J.Q. Effects of pituitary adenylate cyclase activating polypeptide on CD4(+)/CD8(+) T cell levels after traumatic brain injury in a rat model. World J. Emerg. Med. 2012, 3, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Gutcher, I.; Becher, B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Investig. 2007, 117, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Kabelitz, D.; Medzhitov, R. Innate immunity-cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr. Opin. Immunol. 2007, 19, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, A.; Mauro, C.; Marelli-Berg, F. T lymphocyte trafficking: Molecules and mechanisms. Front. Biosci. 2013, 18, 422–440. [Google Scholar]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef]
- Rostami, A.; Ciric, B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 2013, 333, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, Y.-Y.; Bhaumik, S.K.; Li, Y.; Baumann, J.M.; Lin, X.; Zhang, Y.; Lin, S.-H.; Dunn, R.S.; Liu, C.-Y.; et al. Blocking Lymphocyte Trafficking with FTY720 Prevents Inflammation-Sensitized Hypoxic–Ischemic Brain Injury in Newborns. J. Neurosci. 2014, 34, 16467–16481. [Google Scholar] [CrossRef]
- Baxi, E.G.; DeBruin, J.; Tosi, D.M.; Grishkan, I.V.; Smith, M.D.; Kirby, L.A.; Strasburger, H.J.; Fairchild, A.N.; Calabresi, P.A.; Gocke, A.R. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci. 2015, 35, 8626–8639. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra274. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Segal, B.M. T(H)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 2011, 23, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Grifka-Walk, H.M.; Lalor, S.J.; Segal, B.M. Highly polarized Th17 cells induce EAE via a T-bet independent mechanism. Eur. J. Immunol. 2013, 43, 2824–2831. [Google Scholar] [CrossRef] [Green Version]
- Laird, M.D.; Sukumari-Ramesh, S.; Swift, A.E.; Meiler, S.E.; Vender, J.R.; Dhandapani, K.M. Curcumin attenuates cerebral edema following traumatic brain injury in mice: A possible role for aquaporin-4? J. Neurochem. 2010, 113, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Ma, Z.; Fu, Q.; Ma, S. Curcumin attenuates allergic airway inflammation by regulation of CD4+CD25+ regulatory T cells (Tregs)/Th17 balance in ovalbumin-sensitized mice. Fitoterapia 2013, 87, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Moon, S.J.; Lee, S.H.; Yang, E.J.; Min, J.K.; Cho, S.G.; Yang, C.W.; Park, S.H.; Kim, H.Y.; Cho, M.L. Curcumin attenuates acute graft-versus-host disease severity via in vivo regulations on Th1, Th17 and regulatory T cells. Plos ONE 2013, 8, e67171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Li, X.K.; Funeshima-Fuji, N.; Kimura, H.; Matsumoto, Y.; Isaka, Y.; Takahara, S. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int. Immunopharmacol. 2009, 9, 575–581. [Google Scholar] [CrossRef]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive Neurodegeneration after Experimental Brain Trauma: Association with Chronic Microglial Activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- McKee, C.A.; Lukens, J.R. Emerging Roles for the Immune System in Traumatic Brain Injury. Front. Immunol. 2016, 7, 556. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Correale, J.; Villa, A. The neuroprotective role of inflammation in nervous system injuries. J. Neurol. 2004, 251, 1304–1316. [Google Scholar] [CrossRef]
- Roberts, I.; Yates, D.; Sandercock, P.; Farrell, B.; Wasserberg, J.; Lomas, G.; Cottingham, R.; Svoboda, P.; Brayley, N.; Mazairac, G.; et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial. Lancet (Lond. Engl.) 2004, 364, 1321–1328. [Google Scholar] [CrossRef]
- Edwards, P.; Arango, M.; Balica, L.; Cottingham, R.; El-Sayed, H.; Farrell, B.; Fernandes, J.; Gogichaisvili, T.; Golden, N.; Hartzenberg, B.; et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet (Lond. Engl.) 2005, 365, 1957–1959. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Dev. 2014, 8, 2221–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, C.C.; Lin, J.W.; Chang, M.W.; Wang, C.C.; Kuo, J.R.; Yang, C.Z.; Chang, C.P. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J. Neurochem. 2010, 115, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Cheong, C.U.; Chang, C.P.; Chao, C.M.; Cheng, B.C.; Yang, C.Z.; Chio, C.C. Etanercept attenuates traumatic brain injury in rats by reducing brain TNF- alpha contents and by stimulating newly formed neurogenesis. Mediat. Inflamm. 2013, 2013, 620837. [Google Scholar] [CrossRef] [Green Version]
- Chio, C.C.; Chang, C.H.; Wang, C.C.; Cheong, C.U.; Chao, C.M.; Cheng, B.C.; Yang, C.Z.; Chang, C.P. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-alpha. BMC Neurosci. 2013, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Tobinick, E.; Rodriguez-Romanacce, H.; Levine, A.; Ignatowski, T.A.; Spengler, R.N. Immediate neurological recovery following perispinal etanercept years after brain injury. Clin. Drug Investig. 2014, 34, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. Immediate Resolution of Hemispatial Neglect and Central Post-Stroke Pain After Perispinal Etanercept: Case Report. Clin. Drug Investig. 2020, 40, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Weissenberger, A.; Bonev, V.; King, L.D.; Bonham, M.D.; Ferguson, S.; Smith, A.D.; Goodman-Jones, A.A.; Espinet, A.J. Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: Improved mobility and pain alleviation. Expert Opin. Investig. Drugs 2020, 29, 311–326. [Google Scholar] [CrossRef]
- Ignatowski, T.A.; Spengler, R.N.; Dhandapani, K.M.; Folkersma, H.; Butterworth, R.F.; Tobinick, E. Perispinal etanercept for post-stroke neurological and cognitive dysfunction: Scientific rationale and current evidence. CNS Drugs 2014, 28, 679–697. [Google Scholar] [CrossRef] [Green Version]
- Tobinick, E.; Kim, N.M.; Reyzin, G.; Rodriguez-Romanacce, H.; DePuy, V. Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 2012, 26, 1051–1070. [Google Scholar] [CrossRef]
- Baratz, R.; Tweedie, D.; Rubovitch, V.; Luo, W.; Yoon, J.S.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Tumor necrosis factor-alpha synthesis inhibitor, 3,6′-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J. Neurochem. 2011, 118, 1032–1042. [Google Scholar] [CrossRef]
- Baratz, R.; Tweedie, D.; Wang, J.Y.; Rubovitch, V.; Luo, W.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Transiently lowering tumor necrosis factor-alpha synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflamm. 2015, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahel, P.F.; Shohami, E.; Younis, F.M.; Kariya, K.; Otto, V.I.; Lenzlinger, P.M.; Grosjean, M.B.; Eugster, H.P.; Trentz, O.; Kossmann, T.; et al. Experimental closed head injury: Analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J. Cereb. Blood Flow Metab. 2000, 20, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, J.G.; Reeder, E.L.; Collins, S.M.; Gopalan, P.; Robson, M.J. Contributions of Interleukin-1 Receptor Signaling in Traumatic Brain Injury. Front. Behav. Neurosci. 2019, 13, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehranian, R.; Andell-Jonsson, S.; Beni, S.M.; Yatsiv, I.; Shohami, E.; Bartfai, T.; Lundkvist, J.; Iverfeldt, K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. Neurotrauma 2002, 19, 939–951. [Google Scholar] [CrossRef]
- Bergold, P.J. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp. Neurol. 2016, 275 Pt. 3, 367–380. [Google Scholar] [CrossRef]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase II randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, F.; Hanell, A.; Bjork, M.; Hillered, L.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Clausen, F.; Hanell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Brann, D.W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2017, 2017, 6057609. [Google Scholar] [CrossRef]
- Ismael, S.; Ahmed, H.A.; Adris, T.; Parveen, K.; Thakor, P.; Ishrat, T. The NLRP3 inflammasome: A potential therapeutic target for traumatic brain injury. Neural Regen. Res. 2020, 16, 49–57. [Google Scholar] [CrossRef]
- Kerr, N.; Lee, S.W.; Perez-Barcena, J.; Crespi, C.; Ibañez, J.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome proteins as biomarkers of traumatic brain injury. Plos ONE 2018, 13, e0210128. [Google Scholar] [CrossRef] [PubMed]
- Desu, H.L.; Plastini, M.; Illiano, P.; Bramlett, H.M.; Dietrich, W.D.; de Rivero Vaccari, J.P.; Brambilla, R.; Keane, R.W. IC100: A novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Denes, A. Interleukin-1alpha and brain inflammation. IUBMB Life 2015, 67, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.A.; Todd, B.P.; Mahoney, J.; Pieper, A.A.; Ferguson, P.J.; Bassuk, A.G. Combined Blockade of Interleukin-1alpha and -1beta Signaling Protects Mice from Cognitive Dysfunction after Traumatic Brain Injury. ENeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergenroeder, G.W.; Moore, A.N.; McCoy, J.P., Jr.; Samsel, L.; Ward, N.H., 3rd; Clifton, G.L.; Dash, P.K. Serum IL-6: A candidate biomarker for intracranial pressure elevation following isolated traumatic brain injury. J. Neuroinflamm. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock (Augustaga.) 2013, 40, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.J.; Xu, J.; Ma, B.Y.; Chen, G.; Gu, P.Y.; Wei, D.; Hu, W.X. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin. J. Traumatol. 2014, 17, 1–7. [Google Scholar]
- Yang, L.; Wang, F.; Yang, L.; Yuan, Y.; Chen, Y.; Zhang, G.; Fan, Z. HMGB1 a-Box Reverses Brain Edema and Deterioration of Neurological Function in a Traumatic Brain Injury Mouse Model. Cell. Physiol. Biochem. 2018, 46, 2532–2542. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, B.; Cao, S.; Wang, Y. Epigallocatechin-3-Gallate (EGCG) Attenuates Traumatic Brain Injury by Inhibition of Edema Formation and Oxidative Stress. Korean J. Physiol. Pharmacol. 2015, 19, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.; Grogan, D.; Ahluwalia, M.; Salles, É.L.; Ahluwalia, P.; Khodadadi, H.; Alverson, K.; Nguyen, A.; Raju, S.P.; Gaur, P.; et al. Targeting the endocannabinoid system: A predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. History of cannabis as medicine: Nineteenth century irish physicians and correlations of their observations to modern research. In Cannabis Sativa L.: Botany and Biotechnology; Chanda, S., Lata, H., Elsohly, M.., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 63–78. [Google Scholar]
- Russo, E.B. Clinical Endocannabinoid Deficiency Reconsidered: Current Research Supports the Theory in Migraine, Fibromyalgia, Irritable Bowel, and Other Treatment-Resistant Syndromes. Cannabis Cannabinoid Res. 2016, 1, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Rhyne, D.N.; Anderson, S.L.; Gedde, M.; Borgelt, L.M. Effects of Medical Marijuana on Migraine Headache Frequency in an Adult Population. Pharmacotherapy 2016, 36, 505–510. [Google Scholar] [CrossRef]
- Russo, E.B.; Hohmann, A.G. Role of cannabinoids in pain management. In Comprehensive Treatment of Chronic Pain by Medical, Interventional and Behavioral Approaches; Deer, T., Gordin, V., Eds.; Springer: New York, NY, USA, 2013; pp. 181–197. [Google Scholar]
- Serpell, M.; Ratcliffe, S.; Hovorka, J.; Schofield, M.; Taylor, L.; Lauder, H.; Ehler, E. A double-blind, randomized, placebo-controlled, parallel group study of THC/CBD spray in peripheral neuropathic pain treatment. Eur. J. Pain (Lond. Engl.) 2014, 18, 999–1012. [Google Scholar] [CrossRef]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharm. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef]
- Rog, D.J.; Nurmikko, T.J.; Friede, T.; Young, C.A. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology 2005, 65, 812–819. [Google Scholar] [CrossRef]
- Johnson, J.R.; Burnell-Nugent, M.; Lossignol, D.; Ganae-Motan, E.D.; Potts, R.; Fallon, M.T. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J. Pain Symptom Manag. 2010, 39, 167–179. [Google Scholar] [CrossRef]
- Benyo, Z.; Ruisanchez, E.; Leszl-Ishiguro, M.; Sandor, P.; Pacher, P. Endocannabinoids in cerebrovascular regulation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H785–H801. [Google Scholar] [CrossRef] [Green Version]
- Schurman, L.D.; Lichtman, A.H. Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Front. Pharmacol. 2017, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in Oxidative Stress-Related Neurodegenerative Diseases: Role of Endocannabinoid System Modulation. Antioxid. Redox Signal. 2018, 29, 75–108. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Moro, M.A.; Martinez-Orgado, J. Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Letteron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018. [Google Scholar] [CrossRef]
- Kho, D.T.; Glass, M.; Graham, E.S. Is the Cannabinoid CB2 Receptor a Major Regulator of the Neuroinflammatory Axis of the Neurovascular Unit in Humans? Adv. Pharmacol. (San Diegocalif.) 2017, 80, 367–396. [Google Scholar] [CrossRef]
- Nozaki, C.; Markert, A.; Zimmer, A. Inhibition of FAAH reduces nitroglycerin-induced migraine-like pain and trigeminal neuronal hyperactivity in mice. Eur. Neuropsychopharmacol. 2015, 25, 1388–1396. [Google Scholar] [CrossRef]
- Maas, A.I.; Murray, G.; Henney, H., 3rd; Kassem, N.; Legrand, V.; Mangelus, M.; Muizelaar, J.P.; Stocchetti, N.; Knoller, N. Efficacy and safety of dexanabinol in severe traumatic brain injury: Results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006, 5, 38–45. [Google Scholar] [CrossRef]
- Latorre, J.G.; Schmidt, E.B. Cannabis, Cannabinoids, and Cerebral Metabolism: Potential Applications in Stroke and Disorders of the Central Nervous System. Curr. Cardiol. Rep. 2015, 17, 627. [Google Scholar] [CrossRef]
- Russo, E.B. Synthetic and natural cannabinoids: The cardiovascular risk. Br. J. Cardiol. 2015, 22, 7–9. [Google Scholar]
- Pacher, P.; Steffens, S.; Hasko, G.; Schindler, T.H.; Kunos, G. Cardiovascular effects of marijuana and synthetic cannabinoids: The good, the bad, and the ugly. Nat. Rev. Cardiol. 2018, 15, 151–166. [Google Scholar] [CrossRef]
- Magid, L.; Heymann, S.; Elgali, M.; Avram, L.; Cohen, Y.; Liraz-Zaltsman, S.; Mechoulam, R.; Shohami, E. Role of CB2 Receptor in the Recovery of Mice after Traumatic Brain Injury. J. Neurotrauma 2019, 36, 1836–1846. [Google Scholar] [CrossRef]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat. Rev. Neurol. 2015, 11, 698–710. [Google Scholar] [CrossRef]
- Saxena, P.; Newman, M.A.; Shehatha, J.S.; Redington, A.N.; Konstantinov, I.E. Remote ischemic conditioning: Evolution of the concept, mechanisms, and clinical application. J. Card. Surg. 2010, 25, 127–134. [Google Scholar] [CrossRef]
- Hoda, M.N.; Fagan, S.C.; Khan, M.B.; Vaibhav, K.; Chaudhary, A.; Wang, P.; Dhandapani, K.M.; Waller, J.L.; Hess, D.C. A 2 × 2 factorial design for the combination therapy of minocycline and remote ischemic perconditioning: Efficacy in a preclinical trial in murine thromboembolic stroke model. Exp. Transl. Stroke Med. 2014, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.C.; Hoda, M.N.; Khan, M.B. Humoral Mediators of Remote Ischemic Conditioning: Important Role of eNOS/NO/Nitrite. Acta Neurochir. Suppl. 2016, 121, 45–48. [Google Scholar] [CrossRef]
- Loukogeorgakis, S.P.; Williams, R.; Panagiotidou, A.T.; Kolvekar, S.K.; Donald, A.; Cole, T.J.; Yellon, D.M.; Deanfield, J.E.; MacAllister, R.J. Transient Limb Ischemia Induces Remote Preconditioning and Remote Postconditioning in Humans by a KATP Channel–Dependent Mechanism. Circulation 2007, 116, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Wang, Y.; Ayub, A.; Ashraf, M. Mitochondrial K(ATP) channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1295–H1303. [Google Scholar] [CrossRef] [Green Version]
- Vaibhav, K.; Baban, B.; Khan, M.B.; Liu, J.Y.; Huo, Y.; Hess, D.C.; Dhandapani, K.M.; Hoda, M.N. Remote ischemic preconditioning protects from traumatic brain injury (TBI). J. Neurotrauma 2014, 31, A87. [Google Scholar]
- Vaibhav, K.; Baban, B.; Wang, P.; Khan, M.B.; Pandya, C.; Ahmed, H.; Chaudhary, A.; Ergul, A.; Heger, I.; Hess, D.C.; et al. Remote Ischemic Conditioning (RIC) Attenuates Post-TBI Ischemic Injury and Improves Behavioral Outcomes. Stroke A J. Cereb. Circ. 2015, 46, ATP92. [Google Scholar]
- Joseph, B.; Pandit, V.; Zangbar, B.; Kulvatunyou, N.; Khalil, M.; Tang, A.; O’Keeffe, T.; Gries, L.; Vercruysse, G.; Friese, R.S.; et al. Secondary brain injury in trauma patients: The effects of remote ischemic conditioning. J. Trauma Acute Care Surg. 2015, 78, 698–703; discussion 703–705. [Google Scholar] [CrossRef] [Green Version]
- Pandit, V.; Khan, M.; Zakaria, E.R.; Largent-Milnes, T.M.; Hamidi, M.; O’Keeffe, T.; Vanderah, T.W.; Joseph, B. Continuous remote ischemic conditioning attenuates cognitive and motor deficits from moderate traumatic brain injury. J. Trauma Acute Care Surg. 2018, 85, 48–53. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Azim, A.; Ibraheem, K.; Largent-Milnes, T.M.; Rhee, P.; Vanderah, T.W.; Joseph, B. Remote ischemic conditioning preserves cognition and motor coordination in a mouse model of traumatic brain injury. J. Trauma Acute Care Surg. 2017, 83, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Minambres, E.; Ballesteros, M.A.; Mayorga, M.; Marin, M.J.; Munoz, P.; Figols, J.; Lopez-Hoyos, M. Cerebral apoptosis in severe traumatic brain injury patients: An in vitro, in vivo, and postmortem study. J. Neurotrauma 2008, 25, 581–591. [Google Scholar] [CrossRef]
- Bredesen, D.E. Key note lecture: Toward a mechanistic taxonomy for cell death programs. Stroke A J. Cereb. Circ. 2007, 38, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredesen, D.E. Programmed cell death mechanisms in neurological disease. Curr. Mol. Med. 2008, 8, 173–186. [Google Scholar] [CrossRef]
- Stoica, B.A.; Byrnes, K.R.; Faden, A.I. Cell cycle activation and CNS injury. Neurotox. Res. 2009, 16, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, S.; Movsesyan, V.; Ahmed, F.; Cernak, I.; Schinelli, S.; Stoica, B.; Faden, A.I. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernak, I.; Stoica, B.; Byrnes, K.R.; Di Giovanni, S.; Faden, A.I. Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle (Georget. Tex.) 2005, 4, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Stoica, B.; Byrnes, K.; Faden, A.I. Multifunctional Drug Treatment in Neurotrauma. Neurother. J. Am. Soc. Exp. Neurother. 2009, 6, 14–27. [Google Scholar] [CrossRef]
- Hilton, G.D.; Stoica, B.A.; Byrnes, K.R.; Faden, A.I. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J. Cereb. Blood Flow Metab. 2008, 28, 1845–1859. [Google Scholar] [CrossRef] [Green Version]
- Yakovlev, A.G.; Faden, A.I. Caspase-dependent apoptotic pathways in CNS injury. Mol. Neurobiol. 2001, 24, 131–144. [Google Scholar] [CrossRef]
- Wan, J.; Wang, J.; Cheng, H.; Yu, Y.; Xing, G.; Oiu, Z.; Qian, X.; He, F. Proteomic analysis of apoptosis initiation induced by all-trans retinoic acid in human acute promyelocytic leukemia cells. Electrophoresis 2001, 22, 3026–3037. [Google Scholar] [CrossRef]
- Zhang, X.; Alber, S.; Watkins, S.C.; Kochanek, P.M.; Marion, D.W.; Graham, S.H.; Clark, R.S. Proteolysis consistent with activation of caspase-7 after severe traumatic brain injury in humans. J. Neurotrauma 2006, 23, 1583–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakovlev, A.G.; Knoblach, S.M.; Fan, L.; Fox, G.B.; Goodnight, R.; Faden, A.I. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997, 17, 7415–7424. [Google Scholar] [CrossRef] [Green Version]
- Larner, S.F.; Hayes, R.L.; McKinsey, D.M.; Pike, B.R.; Wang, K.K. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J. Neurochem. 2004, 88, 78–90. [Google Scholar] [CrossRef]
- Knoblach, S.M.; Nikolaeva, M.; Huang, X.; Fan, L.; Krajewski, S.; Reed, J.C.; Faden, A.I. Multiple caspases are activated after traumatic brain injury: Evidence for involvement in functional outcome. J. Neurotrauma 2002, 19, 1155–1170. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Nathoo, N.; Narotam, P.K.; Agrawal, D.K.; Connolly, C.A.; van Dellen, J.R.; Barnett, G.H.; Chetty, R. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J. Neurosurg. 2004, 101, 233–240. [Google Scholar] [CrossRef]
- Brophy, G.M.; Pineda, J.A.; Papa, L.; Lewis, S.B.; Valadka, A.B.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Tepas, J.J., 3rd; et al. alphaII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. J. Neurotrauma 2009, 26, 471–479. [Google Scholar] [CrossRef] [Green Version]
- McGinn, M.J.; Kelley, B.J.; Akinyi, L.; Oli, M.W.; Liu, M.C.; Hayes, R.L.; Wang, K.K.; Povlishock, J.T. Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J. Neuropathol. Exp. Neurol. 2009, 68, 241–249. [Google Scholar] [CrossRef]
- Mondello, S.; Robicsek, S.A.; Gabrielli, A.; Brophy, G.M.; Papa, L.; Tepas, J.; Robertson, C.; Buki, A.; Scharf, D.; Jixiang, M.; et al. alphaII-spectrin breakdown products (SBDPs): Diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 2010, 27, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Polster, B.M.; Basanez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005, 280, 6447–6454. [Google Scholar] [CrossRef] [Green Version]
- Takano, J.; Tomioka, M.; Tsubuki, S.; Higuchi, M.; Iwata, N.; Itohara, S.; Maki, M.; Saido, T.C. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: Evidence from calpastatin mutant mice. J. Biol. Chem. 2005, 280, 16175–16184. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Dou, Q.P. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J. Cell. Biochem. 2000, 80, 53–72. [Google Scholar] [CrossRef]
- Mandic, A.; Viktorsson, K.; Strandberg, L.; Heiden, T.; Hansson, J.; Linder, S.; Shoshan, M.C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: Two separate pathways in cisplatin-induced apoptosis. Mol. Cell. Biol. 2002, 22, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef]
- Daugas, E.; Nochy, D.; Ravagnan, L.; Loeffler, M.; Susin, S.A.; Zamzami, N.; Kroemer, G. Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000, 476, 118–123. [Google Scholar] [CrossRef]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Ishihara, Y.; Shimamoto, N. Involvement of endonuclease G in nucleosomal DNA fragmentation under sustained endogenous oxidative stress. J. Biol. Chem. 2006, 281, 6726–6733. [Google Scholar] [CrossRef] [Green Version]
- Artus, C.; Boujrad, H.; Bouharrour, A.; Brunelle, M.N.; Hoos, S.; Yuste, V.J.; Lenormand, P.; Rousselle, J.C.; Namane, A.; England, P.; et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. Embo J. 2010, 29, 1585–1599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, J.; Graham, S.H.; Du, L.; Kochanek, P.M.; Draviam, R.; Guo, F.; Nathaniel, P.D.; Szabo, C.; Watkins, S.C.; et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J. Neurochem. 2002, 82, 181–191. [Google Scholar] [CrossRef]
- Bano, D.; Munarriz, E.; Chen, H.L.; Ziviani, E.; Lippi, G.; Young, K.W.; Nicotera, P. The plasma membrane Na+/Ca2+ exchanger is cleaved by distinct protease families in neuronal cell death. Ann. N. Y. Acad. Sci. 2007, 1099, 451–455. [Google Scholar] [CrossRef]
- Nur, E.K.A.; Gross, S.R.; Pan, Z.; Balklava, Z.; Ma, J.; Liu, L.F. Nuclear translocation of cytochrome c during apoptosis. J. Biol. Chem. 2004, 279, 24911–24914. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Aviles, E.R., Jr.; Fujikawa, D.G. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 min of generalized seizures. J. Neurosci. Res. 2010, 88, 1727–1737. [Google Scholar] [CrossRef]
- Fujikawa, D.G.; Shinmei, S.S.; Cai, B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: Implications for programmed cell death mechanisms. Neuroscience 2000, 98, 41–53. [Google Scholar] [CrossRef]
- Cregan, S.P.; Dawson, V.L.; Slack, R.S. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23, 2785–2796. [Google Scholar] [CrossRef] [Green Version]
- Cande, C.; Vahsen, N.; Garrido, C.; Kroemer, G. Apoptosis-inducing factor (AIF): Caspase-independent after all. Cell Death Differ. 2004, 11, 591–595. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Whalen, M.J.; Clark, R.S.; Dixon, C.E.; Robichaud, P.; Marion, D.W.; Vagni, V.; Graham, S.H.; Virag, L.; Hasko, G.; Stachlewitz, R.; et al. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J. Cereb. Blood Flow Metab. 1999, 19, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005, 79, 216–223. [Google Scholar] [CrossRef]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Blenn, C.; Wyrsch, P.; Bader, J.; Bollhalder, M.; Althaus, F.R. Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cell. Mol. Life Sci. Cmls 2011, 68, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shichinohe, H.; Kuroda, S.; Ishikawa, T.; Iwasaki, Y. Neuroprotective effect of a heat shock protein inducer, geranylgeranylacetone in permanent focal cerebral ischemia. Brain Res. 2005, 1032, 176–182. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, H.M.; Kim, Y.J.; Lee, K.M.; Kim, M.; Yoon, B.W. Effects of hsp70.1 gene knockout on the mitochondrial apoptotic pathway after focal cerebral ischemia. Stroke A J. Cereb. Circ. 2004, 35, 2195–2199. [Google Scholar] [CrossRef] [Green Version]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Parcellier, A.; Gurbuxani, S.; Schmitt, E.; Solary, E.; Garrido, C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem. Biophys. Res. Commun. 2003, 304, 505–512. [Google Scholar] [CrossRef]
- Gurbuxani, S.; Schmitt, E.; Cande, C.; Parcellier, A.; Hammann, A.; Daugas, E.; Kouranti, I.; Spahr, C.; Pance, A.; Kroemer, G.; et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 2003, 22, 6669–6678. [Google Scholar] [CrossRef] [Green Version]
- Matsumori, Y.; Hong, S.M.; Aoyama, K.; Fan, Y.; Kayama, T.; Sheldon, R.A.; Vexler, Z.S.; Ferriero, D.M.; Weinstein, P.R.; Liu, J. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J. Cereb. Blood Flow Metab. 2005, 25, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, B.; Kimbler, D.E.; Pang, J.; Choi, J.; Moskophidis, D.; Yanasak, N.; Dhandapani, K.M.; Mivechi, N.F. Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J. Neurochem. 2014, 130, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Volbracht, C.; Leist, M.; Kolb, S.A.; Nicotera, P. Apoptosis in caspase-inhibited neurons. Mol. Med. (Camb. Mass.) 2001, 7, 36–48. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, X.; Huang, Z.; Qiu, L.; Xu, F.; Vahsen, N.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; Culmsee, C.; et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007, 14, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabadi, S.V.; Faden, A.I. Selective CDK inhibitors: Promising candidates for future clinical traumatic brain injury trials. Neural Regen. Res. 2014, 9, 1578–1580. [Google Scholar] [CrossRef]
- Tikka, T.; Fiebich, B.L.; Goldsteins, G.; Keinanen, R.; Koistinaho, J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. 2001, 21, 2580–2588. [Google Scholar] [CrossRef]
- Siopi, E.; Llufriu-Daben, G.; Fanucchi, F.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: The effect of minocycline. Neurosci. Lett. 2012, 511, 110–115. [Google Scholar] [CrossRef]
- Homsi, S.; Piaggio, T.; Croci, N.; Noble, F.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: A twelve-week follow-up study. J. Neurotrauma 2010, 27, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Kovesdi, E.; Kamnaksh, A.; Wingo, D.; Ahmed, F.; Grunberg, N.; Long, J.; Kasper, C.; Agoston, D. Acute Minocycline Treatment Mitigates the Symptoms of Mild Blast-Induced Traumatic Brain Injury. Front. Neurol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, S.; Drozda, M.; Zhang, W.; Stavrovskaya, I.G.; Cattaneo, E.; Ferrante, R.J.; Kristal, B.S.; Friedlander, R.M. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10483–10487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Suh, Y.H. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009, 196, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Casha, S.; Zygun, D.; McGowan, M.D.; Bains, I.; Yong, V.W.; John Hurlbert, R. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 2012, 135, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Xiao, G.-M. The neuroprotective effects of progesterone on traumatic brain injury: Current status and future prospects. Acta Pharm. Sin. 2013, 34, 1485–1490. [Google Scholar] [CrossRef]
- Guo, Q.; Sayeed, I.; Baronne, L.M.; Hoffman, S.W.; Guennoun, R.; Stein, D.G. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp. Neurol. 2006, 198, 469–478. [Google Scholar] [CrossRef]
- Moorthy, K.; Sharma, D.; Basir, S.F.; Baquer, N.Z. Administration of estradiol and progesterone modulate the activities of antioxidant enzyme and aminotransferases in naturally menopausal rats. Exp. Gerontol. 2005, 40, 295–302. [Google Scholar] [CrossRef]
- Robertson, C.L.; Saraswati, M. Progesterone protects mitochondrial function in a rat model of pediatric traumatic brain injury. J. Bioenerg. Biomembr. 2015, 47, 43–51. [Google Scholar] [CrossRef]
- Djebaili, M.; Hoffman, S.W.; Stein, D.G. Allopregnanolone and progesterone decrease cell death and cognitive deficits after a contusion of the rat pre-frontal cortex. Neuroscience 2004, 123, 349–359. [Google Scholar] [CrossRef]
- Skolnick, B.E.; Maas, A.I.; Narayan, R.K.; van der Hoop, R.G.; MacAllister, T.; Ward, J.D.; Nelson, N.R.; Stocchetti, N. A Clinical Trial of Progesterone for Severe Traumatic Brain Injury. N. Engl. J. Med. 2014, 371, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.G. Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015, 29, 1259–1272. [Google Scholar] [CrossRef]
- Xiong, Y.; Chopp, M.; Lee, C.P. Erythropoietin improves brain mitochondrial function in rats after traumatic brain injury. Neurol. Res. 2009, 31, 496–502. [Google Scholar] [CrossRef]
- Nichol, A.; French, C.; Little, L.; Haddad, S.; Presneill, J.; Arabi, Y.; Bailey, M.; Cooper, D.J.; Duranteau, J.; Huet, O.; et al. Erythropoietin in traumatic brain injury (EPO-TBI): A double-blind randomised controlled trial. Lancet (Lond. Engl.) 2015, 386, 2499–2506. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Lingsma, H.F.; Pineda, J.A.; Sandel, M.E.; Manley, G.T. Re-Orientation of Clinical Research in Traumatic Brain Injury: Report of an International Workshop on Comparative Effectiveness Research. J. Neurotrauma 2012, 29, 32–46. [Google Scholar] [CrossRef] [Green Version]
- High, W.M., Jr.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar] [CrossRef]
- Moreau, O.K.; Cortet-Rudelli, C.; Yollin, E.; Merlen, E.; Daveluy, W.; Rousseaux, M. Growth hormone replacement therapy in patients with traumatic brain injury. J. Neurotrauma 2013, 30, 998–1006. [Google Scholar] [CrossRef]
- Powner, D.J.; Boccalandro, C.; Alp, M.S.; Vollmer, D.G. Endocrine failure after traumatic brain injury in adults. Neurocritical Care 2006, 5, 61–70. [Google Scholar] [CrossRef]
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Stoica, B.A.; Zhao, Z.; Loane, D.J.; Wu, J.; Dorsey, S.G.; Faden, A.I. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016, 23, 654–668. [Google Scholar] [CrossRef]
- Ge, X.T.; Lei, P.; Wang, H.C.; Zhang, A.L.; Han, Z.L.; Chen, X.; Li, S.H.; Jiang, R.C.; Kang, C.S.; Zhang, J.N. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci. Rep. 2014, 4, 6718. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Chen, F.; Ge, X.; Tan, J.; Lei, P.; Zhang, J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res. 2014, 1582, 12–20. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8100389
Jarrahi A, Braun M, Ahluwalia M, Gupta RV, Wilson M, Munie S, Ahluwalia P, Vender JR, Vale FL, Dhandapani KM, et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines. 2020; 8(10):389. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8100389
Chicago/Turabian StyleJarrahi, Abbas, Molly Braun, Meenakshi Ahluwalia, Rohan V. Gupta, Michael Wilson, Stephanie Munie, Pankaj Ahluwalia, John R. Vender, Fernando L. Vale, Krishnan M. Dhandapani, and et al. 2020. "Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions" Biomedicines 8, no. 10: 389. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8100389