β-Catenin Regulates Wound Healing and IL-6 Expression in Activated Human Astrocytes

 ,
, 
Abstract
:
1. Introduction
2. Experimental Section
2.1. Isolation, Culture and Treatment of Human Astrocytes
2.2. Transfection of Astrocytes
2.3. In Vitro Scratch Assay
2.4. Measures of Cell Viability
2.5. Cell Proliferation/Bromodeoxyuridine (BrdU) Incorporation Assay
2.6. Quantification of Proinflammatory Cytokines by ELISA
2.7. Real Time Gene Expression Analysis
2.8. Western Blot Analyses
2.9. WES Analyses
2.10. Statistical Analysis
2.11. Data Availability
3. Results
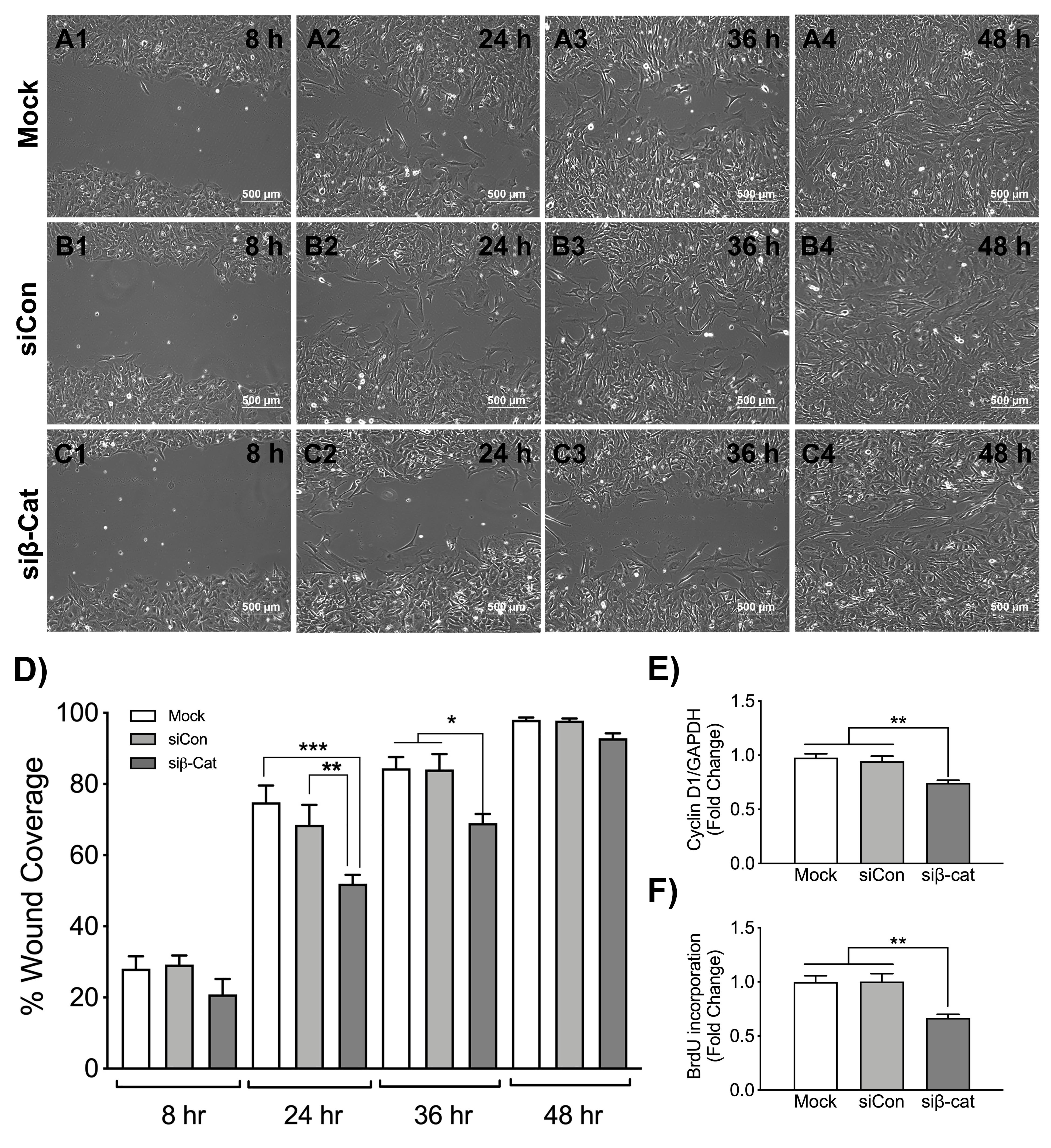
3.1. IL-1β and TNF-α Induce Wnt/β-Catenin and NF-κB Signaling in Astrocytes
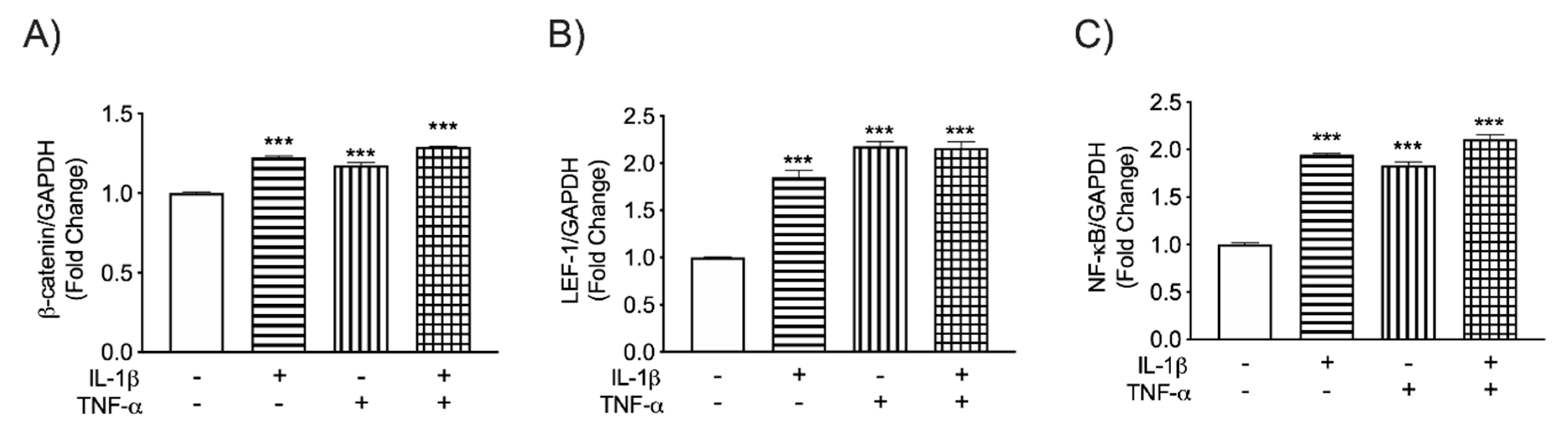
3.2. β-Catenin Differentially Regulates LEF-1 and NF-κB Transcription in IL-1β and TNF-α-Activated Astrocytes
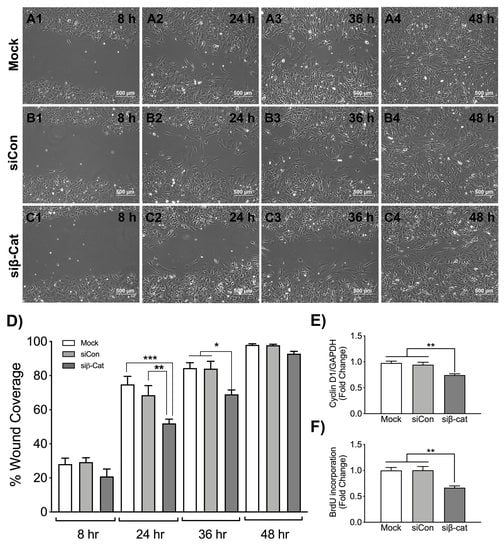
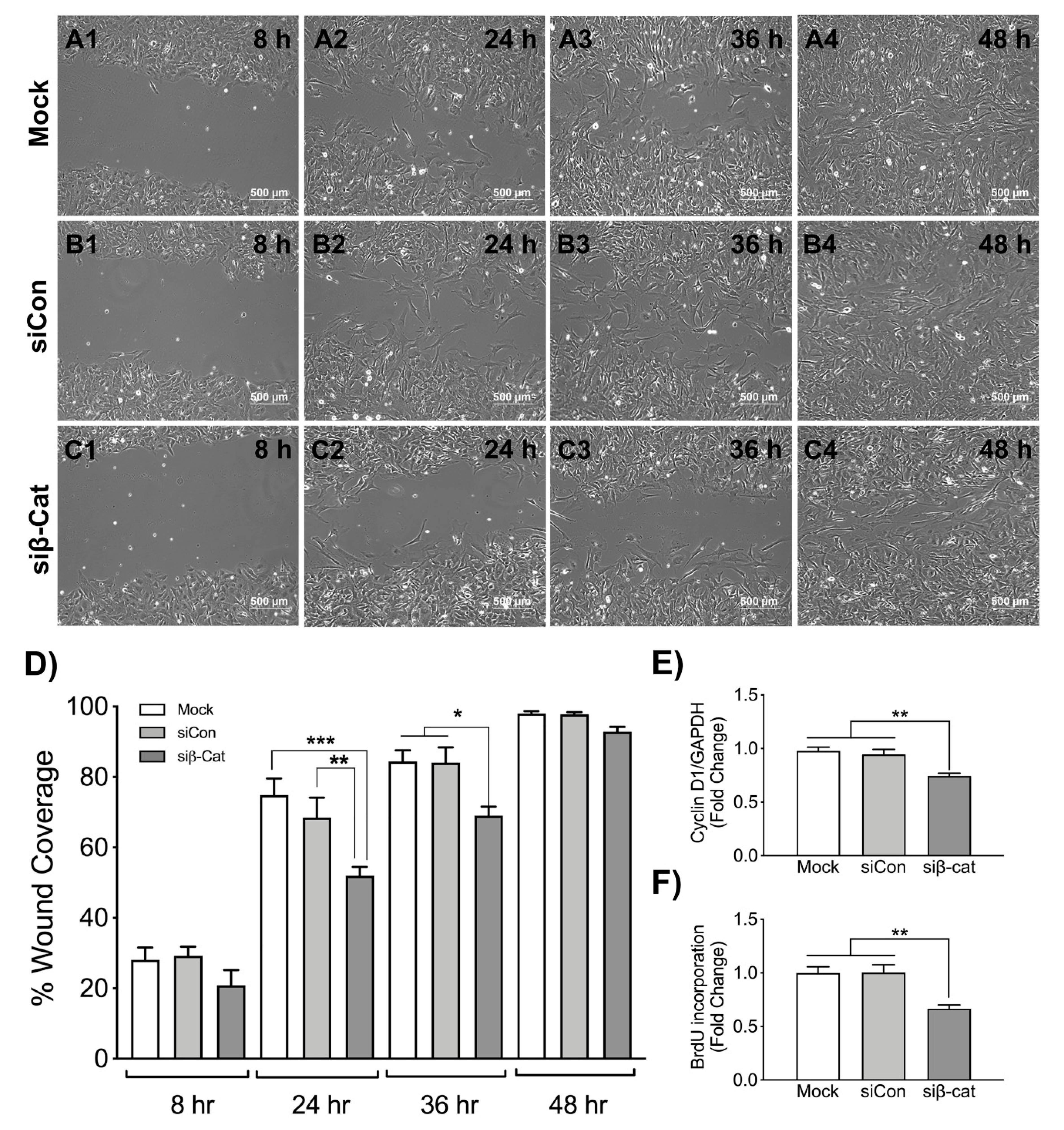
3.3. β-Catenin Regulates Wound Healing and Astrocyte Proliferation
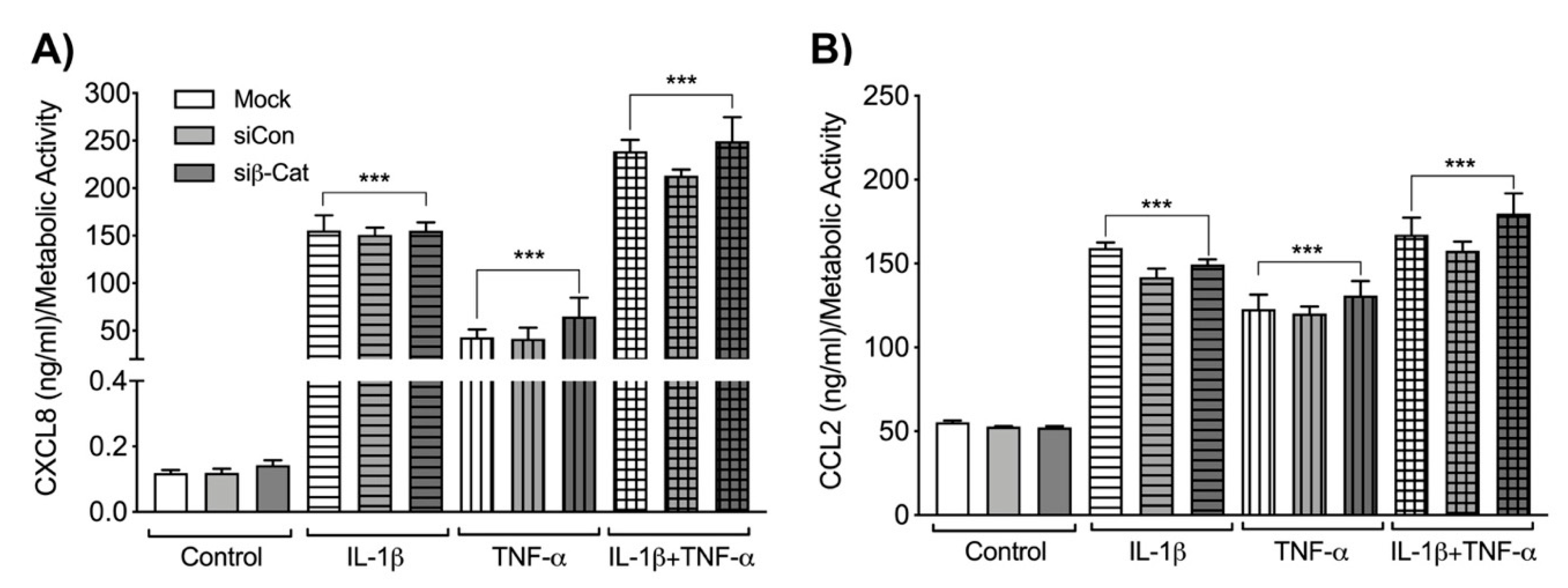
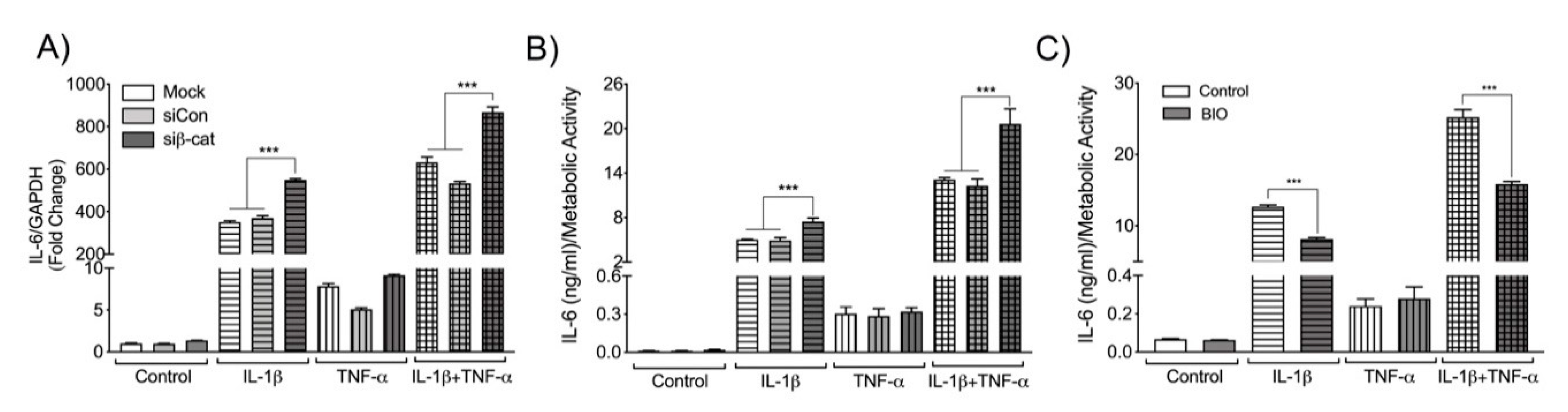
3.4. β-Catenin Negatively Regulates Astrocyte IL-6 Expression
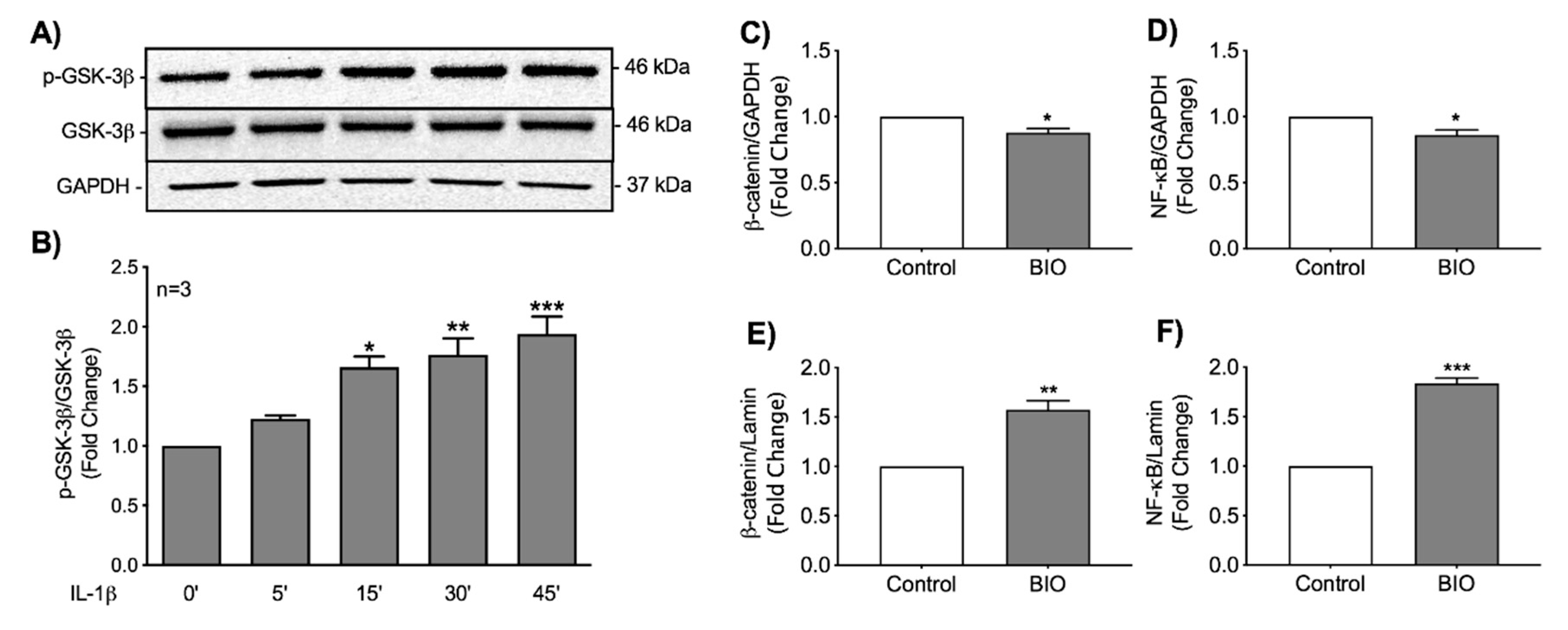
3.5. Stabilization of β-Catenin Increases Nuclear Localization of NF-κB
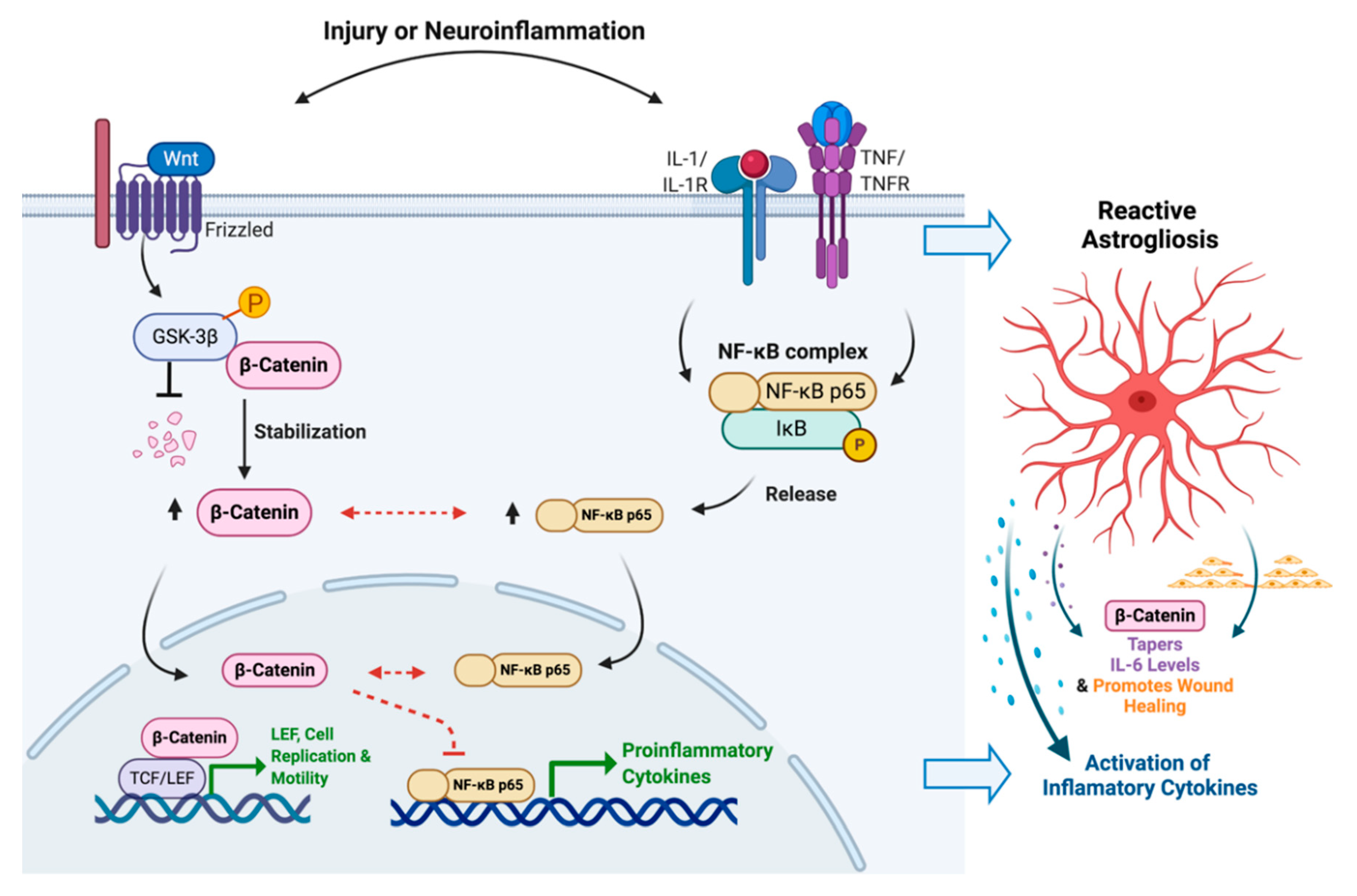
3.6. Molecular Interactions between GSK-3β, β-Catenin, NF-κB and IL-6 during HIVE and HAD
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. Hiv-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: Charter study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spudich, S.; Gonzalez-Scarano, F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef] [Green Version]
- Louboutin, J.P.; Strayer, D. Role of Oxidative Stress in HIV-1-Associated Neurocognitive Disorder and Protection by Gene Delivery of Antioxidant Enzymes. Antioxidants 2014, 3, 770–797. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [Green Version]
- Lundgaard, I.; Osorio, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White matter astrocytes in health and disease. Neuroscience 2014, 276, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, A.M.; Alberghina, L.; Papa, M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci. Lett. 2014, 565, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Brabers, N.A.; Nottet, H.S. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur. J. Clin. Investig. 2006, 36, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-1-associated dementia. Nature 2001, 410, 988–993. [Google Scholar] [CrossRef]
- Sharief, M.K.; Ciardi, M.; Thompson, E.J.; Sorice, F.; Rossi, F.; Vullo, V.; Cirelli, A. Tumour necrosis factor-alpha mediates blood-brain barrier damage in HIV-1 infection of the central nervous system. Mediat. Inflamm. 1992, 1, 191–196. [Google Scholar] [CrossRef]
- Perrella, O.; Guerriero, M.; Izzo, E.; Soscia, M.; Carrieri, P.B. Interleukin-6 and granulocyte macrophage-CSF in the cerebrospinal fluid from HIV infected subjects with involvement of the central nervous system. Arq. Neuropsiquiatr. 1992, 50, 180–182. [Google Scholar] [CrossRef]
- Achim, C.L.; Heyes, M.P.; Wiley, C.A. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brains. J. Clin. Investig. 1993, 91, 2769–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.; Ghorpade, A. C/EBPbeta regulates multiple IL-1beta-induced human astrocyte inflammatory genes. J. Neuroinflamm. 2012, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Suryadevara, R.; Holter, S.; Borgmann, K.; Persidsky, R.; Labenz-Zink, C.; Persidsky, Y.; Gendelman, H.E.; Wu, L.; Ghorpade, A. Regulation of tissue inhibitor of metalloproteinase-1 by astrocytes: Links to HIV-1 dementia. Glia 2003, 44, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Borgmann, K.; Ghorpade, A. HIV-1, methamphetamine and astrocytes at neuroinflammatory Crossroads. Front. Microbiol. 2015, 6, 1143. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M. HIV-1 associated dementia: Update on pathological mechanisms and therapeutic approaches. Curr. Opin. Neurol. 2009, 22, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Gendelman, H.E.; Ghorpade, A.; Persidsky, Y. The neuropathogenesis of HIV-1 associated dementia. In In Defense of the Brain: Current Concepts in the Immunopathogenesis and Clinical Aspects of CNS Infections; Peterson, P.K., Remington, J.S., Eds.; Blackwell Science: Maiden, MA, USA, 1997; pp. 290–304. [Google Scholar]
- Ghorpade, A.; Holter, S.; Borgmann, K.; Persidsky, R.; Wu, L. HIV-1 and IL-1beta regulate Fas ligand expression in human astrocytes through the NF-kappaB pathway. J. Neuroimmunol. 2003, 141, 141–149. [Google Scholar] [CrossRef]
- Li, W.; Henderson, L.J.; Major, E.O.; Al-Harthi, L. IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J. Immunol. 2011, 186, 6771–6778. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt signaling: Role in Alzheimer disease and schizophrenia. J. Neuroimmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Arenas, E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010, 11, 77–86. [Google Scholar] [CrossRef]
- Al-Harthi, L. Interplay between Wnt/beta-catenin signaling and HIV: Virologic and biologic consequences in the CNS. J. Neuroimmune Pharmacol. 2012, 7, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Caricasole, A.; Bakker, A.; Copani, A.; Nicoletti, F.; Gaviraghi, G.; Terstappen, G.C. Two sides of the same coin: Wnt signaling in neurodegeneration and neuro-oncology. Biosci. Rep. 2005, 25, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Narasipura, S.D.; Henderson, L.J.; Fu, S.W.; Chen, L.; Kashanchi, F.; Al-Harthi, L. Role of beta-Catenin and TCF/LEF Family Members in Transcriptional Activity of HIV in Astrocytes. J. Virol. 2012, 86, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Wortman, B.; Darbinian, N.; Sawaya, B.E.; Khalili, K.; Amini, S. Evidence for regulation of long terminal repeat transcription by Wnt transcription factor TCF-4 in human astrocytic cells. J. Virol. 2002, 76, 11159–11165. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Shu, J.; Gelman, B.B.; Lisinicchia, J.G.; Tang, S.J. Wnt signaling in the pathogenesis of human HIV-associated pain syndromes. J. Neuroimmune Pharmacol. 2013, 8, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.B.; Ji, G.; Li, B.; Andersson, T.; Neugebauer, V.; Tang, S.J. A Wnt5a signaling pathway in the pathogenesis of HIV-1 gp120-induced pain. Pain 2015, 156, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 beta (GSK-3beta) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Bhakar, A.L.; Tannis, L.L.; Zeindler, C.; Russo, M.P.; Jobin, C.; Park, D.S.; MacPherson, S.; Barker, P.A. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J. Neurosci. 2002, 22, 8466–8475. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.; Memet, S.; Lilienbaum, A.; Feuillard, J.; Raphael, M.; Israel, A. NF-kappaB activity in transgenic mice: Developmental regulation and tissue specificity. Development 1996, 122, 2117–2128. [Google Scholar]
- Nitkiewicz, J.; Borjabad, A.; Morgello, S.; Murray, J.; Chao, W.; Emdad, L.; Fisher, P.B.; Potash, M.J.; Volsky, D.J. HIV induces expression of complement component C3 in astrocytes by NF-kappaB-dependent activation of interleukin-6 synthesis. J. Neuroinflamm. 2017, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.S.; Waters, M.R.; Biswas, D.D.; Brown, L.N.; Surace, M.J.; Floros, C.; Siebenlist, U.; Kordula, T. RelB controls adaptive responses of astrocytes during sterile inflammation. Glia 2019, 67, 1449–1461. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Gardner, J.; Ghorpade, A. Tissue inhibitor of metalloproteinase (TIMP)-1: The TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 2003, 74, 801–806. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Borgmann, K.; Edara, V.V.; Stacy, S.; Ghorpade, A.; Ikezu, T. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J. Extracell. Vesicles 2020, 9, 1706801. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, K.; Gendelman, H.E.; Ghorpade, A. Isolation and HIV-1 infection of primary human microglia from fetal and adult tissue. Methods Mol. Biol. 2005, 304, 49–70. [Google Scholar]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Manthorpe, M.; Fagnani, R.; Skaper, S.D.; Varon, S. An automated colorimetric microassay for neuronotrophic factors. Brain Res. 1986, 390, 191–198. [Google Scholar] [CrossRef]
- Edara, V.V.; Ghorpade, A.; Borgmann, K. Insights into the Gene Expression Profiles of Active and Restricted Red/Green-HIV(+) Human Astrocytes: Implications for Shock or Lock Therapies in the Brain. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2005, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Kafri, P.; Hasenson, S.E.; Kanter, I.; Sheinberger, J.; Kinor, N.; Yunger, S.; Shav-Tal, Y. Quantifying beta-catenin subcellular dynamics and cyclin D1 mRNA transcription during Wnt signaling in single living cells. Elife 2016, 5. [Google Scholar] [CrossRef]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.H.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; He, T.; Huang, D.R.; Pardo, C.A.; Ransohoff, R.M. TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J. Clin. Investig. 2001, 108, 425–435. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, M.; Morgello, S.; Lee, S.C. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J. Neuroimmunol. 2001, 115, 182–191. [Google Scholar] [CrossRef]
- Dragon, S.; Rahman, M.S.; Yang, J.; Unruh, H.; Halayko, A.J.; Gounni, A.S. IL-17 enhances IL-1beta-mediated CXCL-8 release from human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1023–L1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Li, F.; Mahavadi, S.; Murthy, K.S. Upregulation of RGS4 expression by IL-1beta in colonic smooth muscle is enhanced by ERK1/2 and p38 MAPK and inhibited by the PI3K/Akt/GSK3beta pathway. Am. J. Physiol. Cell Physiol. 2009, 296, C1310–C1320. [Google Scholar] [CrossRef] [Green Version]
- Nejak-Bowen, K.; Kikuchi, A.; Monga, S.P. Beta-catenin-NF-kappaB interactions in murine hepatocytes: A complex to die for. Hepatology 2013, 57, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Ha, J.H.; Chung, S.I.; Yoon, Y. Beta-catenin regulates NF-kappaB activity and inflammatory cytokine expression in bronchial epithelial cells treated with lipopolysaccharide. Int. J. Mol. Med. 2014, 34, 632–638. [Google Scholar] [CrossRef]
- Schon, S.; Flierman, I.; Ofner, A.; Stahringer, A.; Holdt, L.M.; Kolligs, F.T.; Herbst, A. beta-catenin regulates NF-kappaB activity via TNFRSF19 in colorectal cancer cells. Int. J. Cancer 2014, 135, 1800–1811. [Google Scholar] [CrossRef]
- Vines, A.; Cahoon, S.; Goldberg, I.; Saxena, U.; Pillarisetti, S. Novel anti-inflammatory role for glycogen synthase kinase-3beta in the inhibition of tumor necrosis factor-alpha- and interleukin-1beta-induced inflammatory gene expression. J. Biol. Chem. 2006, 281, 16985–16990. [Google Scholar] [CrossRef] [Green Version]
- Robinson, K.F.; Narasipura, S.D.; Wallace, J.; Ritz, E.M.; Al-Harthi, L. β-Catenin and TCFs/LEF signaling discordantly regulate IL-6 expression in astrocytes. Cell Commun. Signal. 2020, 18, 93. [Google Scholar] [CrossRef]
- Norris, J.G.; Tang, L.P.; Sparacio, S.M.; Benveniste, E.N. Signal transduction pathways mediating astrocyte IL-6 induction by IL-1 beta and tumor necrosis factor-alpha. J. Immunol. 1994, 152, 841–850. [Google Scholar]
- Hunt, P.W.; Sinclair, E.; Rodriguez, B.; Shive, C.; Clagett, B.; Funderburg, N.; Robinson, J.; Huang, Y.; Epling, L.; Martin, J.N.; et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J. Infect. Dis. 2014, 210, 1228–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008, 5, e203. [Google Scholar] [CrossRef]
- Tenorio, A.R.; Zheng, Y.; Bosch, R.J.; Krishnan, S.; Rodriguez, B.; Hunt, P.W.; Plants, J.; Seth, A.; Wilson, C.C.; Deeks, S.G.; et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J. Infect. Dis. 2014, 210, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Bressler, P.; Kinter, A.; Duh, E.; Timmer, W.; Rabson, A.; Justement, S.; Stanley, S.; Fauci, A. Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor-alpha by transcriptional and post-transcriptional mechanisms. J. Exp. Med. 1990, 172, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G.; Fauci, A.S. The effect of cytokines and pharmacologic agents on chronic HIV infection. AIDS Res. Hum. Retrovir. 1992, 8, 191–197. [Google Scholar] [CrossRef]
- Lynch, H.E.; Goldberg, G.L.; Chidgey, A.; Van den Brink, M.R.; Boyd, R.; Sempowski, G.D. Thymic involution and immune reconstitution. Trends Immunol. 2009, 30, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Airoldi, M.; Bandera, A.; Trabattoni, D.; Tagliabue, B.; Arosio, B.; Soria, A.; Rainone, V.; Lapadula, G.; Annoni, G.; Clerici, M.; et al. Neurocognitive impairment in HIV-infected naive patients with advanced disease: The role of virus and intrathecal immune activation. Clin. Dev. Immunol. 2012, 2012, 467154. [Google Scholar] [CrossRef] [Green Version]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The Role of Interleukin 6 During Viral Infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [Green Version]
- Laurenzi, M.A.; Siden, A.; Persson, M.A.; Norkrans, G.; Hagberg, L.; Chiodi, F. Cerebrospinal fluid interleukin-6 activity in HIV infection and inflammatory and noninflammatory diseases of the nervous system. Clin. Immunol. Immunopathol. 1990, 57, 233–241. [Google Scholar] [CrossRef]
- Shah, A.; Verma, A.S.; Patel, K.H.; Noel, R.; Rivera-Amill, V.; Silverstein, P.S.; Chaudhary, S.; Bhat, H.K.; Stamatatos, L.; Singh, D.P.; et al. HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: Suppression by gp120 specific small interfering RNA. PLoS ONE 2011, 6, e21261. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kumar, A. Differential signaling mechanism for HIV-1 Nef-mediated production of IL-6 and IL-8 in human astrocytes. Sci. Rep. 2015, 5, 9867. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
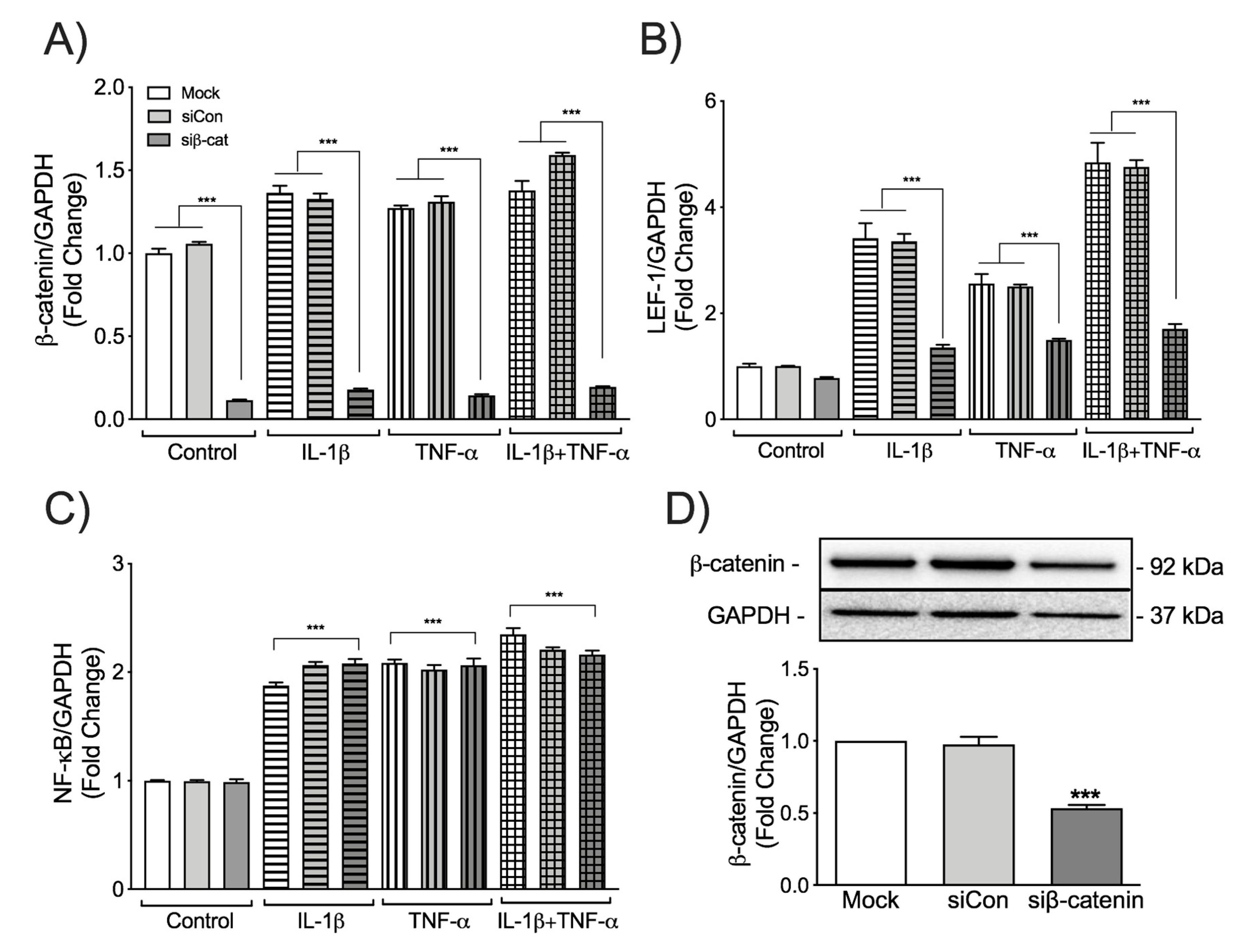{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Assay Number (Thermo Fisher) | Dye |
|---|---|---|
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | 4310884E | (VIC/TAMRA) |
| β-catenin | Hs00355049_m1 | (FAM/MGB) |
| Lymphoid enhancement-binding factor (LEF-1) | Hs01547250_m1 | (FAM/MGB) |
| Interluekin-6 (IL-6) | Hs00985639_m1 | (FAM/MGB) |
| C-C motif chemokine ligand 2 (CCL2) | Hs002341140_m1 | (FAM/MGB) |
| C-X-C motif chemokine ligand 8 (CXCL8) | Hs00174103_m1 | (FAM/MGB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edara, V.V.; Nooka, S.; Proulx, J.; Stacy, S.; Ghorpade, A.; Borgmann, K. β-Catenin Regulates Wound Healing and IL-6 Expression in Activated Human Astrocytes. Biomedicines 2020, 8, 479. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110479
Edara VV, Nooka S, Proulx J, Stacy S, Ghorpade A, Borgmann K. β-Catenin Regulates Wound Healing and IL-6 Expression in Activated Human Astrocytes. Biomedicines. 2020; 8(11):479. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110479
Chicago/Turabian StyleEdara, Venkata Viswanadh, Shruthi Nooka, Jessica Proulx, Satomi Stacy, Anuja Ghorpade, and Kathleen Borgmann. 2020. "β-Catenin Regulates Wound Healing and IL-6 Expression in Activated Human Astrocytes" Biomedicines 8, no. 11: 479. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110479