Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
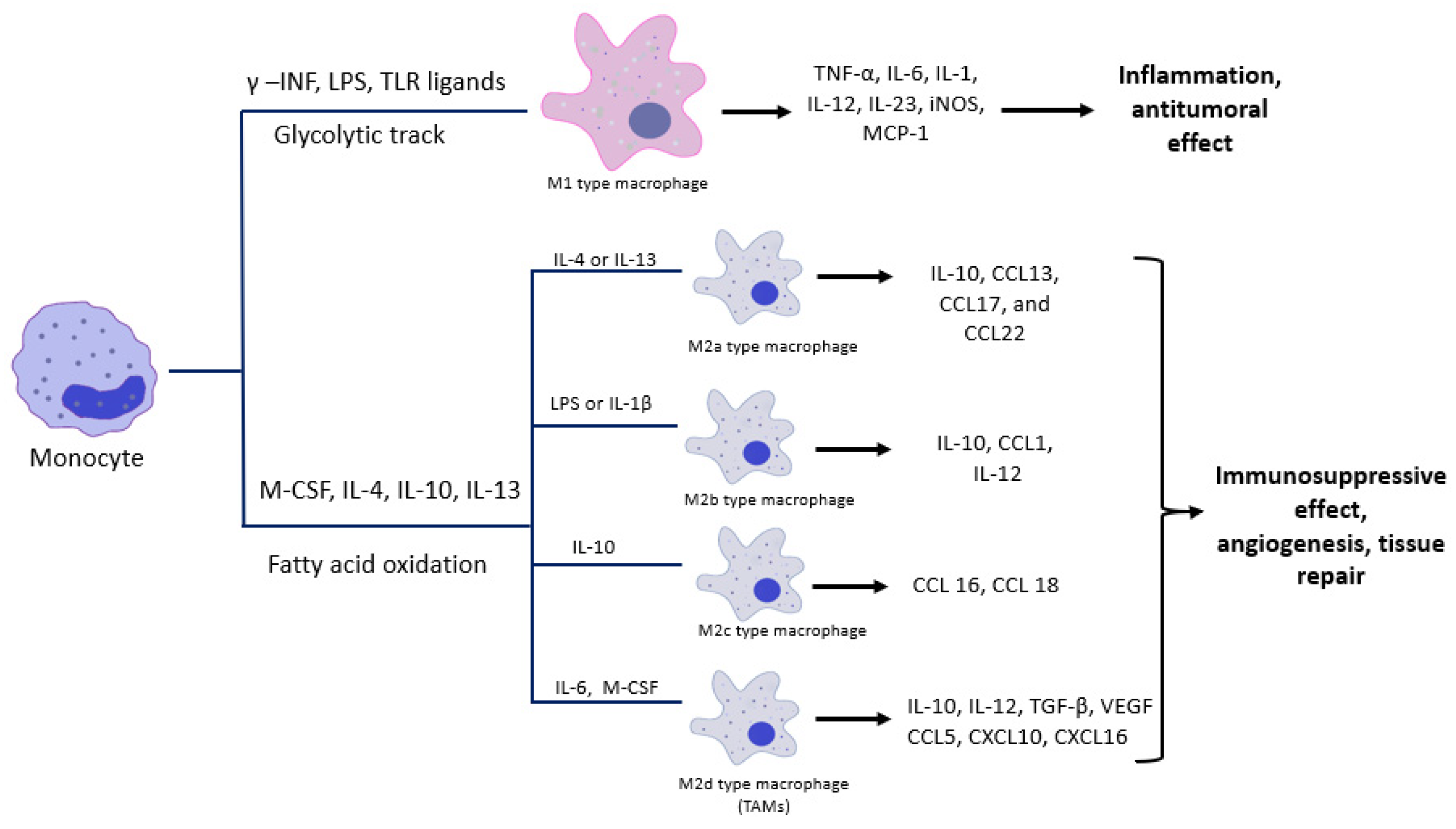
2. What Are Macrophages?
3. Activation of Macrophages
4. The Macrophages and Cancer
5. TAM-Related Cytokines and Chemokines
5.1. TNF-α
5.2. IL-1β
5.3. IL-6
5.4. IL-10
5.5. TGF-β
5.6. CCL2
6. Macrophages in CLL
7. NLC Functions
7.1. Recruiting Cells
7.2. Enhancing Survival and Antiapoptotic Effect
7.3. Proliferation Stimulation
8. Conclusions
Funding
Conflicts of Interest
References
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Yang, X.; Feng, W.L.; Liao, J.F.; Wang, L.N.; Feng, L.; Lin, Y.M.; Ren, Q.; Zheng, G.G. Organ-specific microenvironment modifies diverse functional and phenotypic characteristics of leukemia-associated macrophages in mouse T cell acute lymphoblastic leukemia. J. Immunol. 2015, 194, 2919–2929. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.R.; Sawitsky, A.; Cronkite, E.P.; Chanana, A.D.; Levy, R.N.; Pasternack, B.S. Clinical staging of chronic lymphocytic leukemia. Blood 1975, 46, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef]
- Dohner, H.; Stilgenbauer, S.; Dohner, K.; Bentz, M.; Lichter, P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: Reassessment based on molecular cytogenetic analysis. J. Mol. Med. (Berl.) 1999, 77, 266–281. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordonez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Bea, S.; Gonzalez-Diaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Fangazio, M.; Rasi, S.; Vaisitti, T.; Monti, S.; Cresta, S.; Chiaretti, S.; Del Giudice, I.; Fabbri, G.; Bruscaggin, A.; et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 2012, 119, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; You, M.J.; Yang, Y.; Hu, D.; Tian, C. The Role of Tumor-Associated Macrophages in Leukemia. Acta Haematol. 2020, 143, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Filip, A.A.; Cisel, B.; Wasik-Szczepanek, E. Guilty bystanders: Nurse-like cells as a model of microenvironmental support for leukemic lymphocytes. Clin. Exp. Med. 2015, 15, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Szulczewski, J.M.; Inman, D.R.; Entenberg, D.; Ponik, S.M.; Aguirre-Ghiso, J.; Castracane, J.; Condeelis, J.; Eliceiri, K.W.; Keely, P.J. In Vivo Visualization of Stromal Macrophages via label-free FLIM-based metabolite imaging. Sci. Rep. 2016, 6, 25086. [Google Scholar] [CrossRef] [Green Version]
- Grossman, J.G.; Nywening, T.M.; Belt, B.A.; Panni, R.Z.; Krasnick, B.A.; DeNardo, D.G.; Hawkins, W.G.; Goedegebuure, S.P.; Linehan, D.C.; Fields, R.C. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology 2018, 7, e1470729. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Schridde, A. Origin, Differentiation, and Function of Intestinal Macrophages. Front. Immunol. 2018, 9, 2733. [Google Scholar] [CrossRef]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.I. Metchnikoff and the phagocytosis theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R. Macrophage activity and clinical immunology. Origin and kinetics of mononuclear phagocytes. Ann. N. Y. Acad. Sci. 1976, 278, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Mejia, M.E.; Doseff, A.I. Regulation of monocytes and macrophages cell fate. Front. Biosci. (Landmark Ed) 2009, 14, 2413–2431. [Google Scholar] [CrossRef] [Green Version]
- Sieweke, M.H.; Allen, J.E. Beyond stem cells: Self-renewal of differentiated macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Garceau, V.; Smith, J.; Paton, I.R.; Davey, M.; Fares, M.A.; Sester, D.P.; Burt, D.W.; Hume, D.A. Pivotal Advance: Avian colony-stimulating factor 1 (CSF-1), interleukin-34 (IL-34), and CSF-1 receptor genes and gene products. J. Leukoc. Biol. 2010, 87, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, B.; Lee, K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Tugal, D.; Liao, X.; Jain, M.K. Transcriptional control of macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Zhao, J.; Liu, W. Macrophage stimulating 1-induced inflammation response promotes aortic aneurysm formation through triggering endothelial cells death and activating the NF-kappaB signaling pathway. J. Recept. Signal Transduct. Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Srivastava, M.; Saqib, U.; Naim, A.; Roy, A.; Liu, D.; Bhatnagar, D.; Ravinder, R.; Baig, M.S. The TLR4-NOS1-AP1 signaling axis regulates macrophage polarization. Inflamm. Res. 2017, 66, 323–334. [Google Scholar] [CrossRef]
- Schneider, A.; Weier, M.; Herderschee, J.; Perreau, M.; Calandra, T.; Roger, T.; Giannoni, E. IRF5 Is a Key Regulator of Macrophage Response to Lipopolysaccharide in Newborns. Front. Immunol. 2018, 9, 1597. [Google Scholar] [CrossRef] [Green Version]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Cassetta, L.; Rizzi, C.; Alfano, M.; Poli, G. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 2009, 182, 6237–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Li, H.; Jiang, T.; Li, M.Q.; Zheng, X.L.; Zhao, G.J. Transcriptional Regulation of Macrophages Polarization by MicroRNAs. Front. Immunol. 2018, 9, 1175. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Myasoedova, V.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018, 223, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Lamkin, D.M.; Srivastava, S.; Bradshaw, K.P.; Betz, J.E.; Muy, K.B.; Wiese, A.M.; Yee, S.K.; Waggoner, R.M.; Arevalo, J.M.G.; Yoon, A.J.; et al. C/EBPbeta regulates the M2 transcriptome in beta-adrenergic-stimulated macrophages. Brain Behav. Immun. 2019, 80, 839–848. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Soto, A.; Usategui, A.; Casas-Engel, M.L.; Simon-Fuentes, M.; Nieto, C.; Cuevas, V.D.; Vega, M.A.; Luis Pablos, J.; Corbi, A.L. Serotonin drives the acquisition of a profibrotic and anti-inflammatory gene profile through the 5-HT7R-PKA signaling axis. Sci. Rep. 2017, 7, 14761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, M.D.; Tack, J. The serotonin signaling system: From basic understanding to drug development for functional GI disorders. Gastroenterology 2007, 132, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Denna, T.H.; Storkersen, J.N.; Gerriets, V.A. Beyond a neurotransmitter: The role of serotonin in inflammation and immunity. Pharmacol. Res. 2019, 140, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Morrison, C. Immuno-oncologists eye up macrophage targets. Nat. Rev. Drug Discov. 2016, 15, 373–374. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [Green Version]
- Ohtaki, Y.; Ishii, G.; Nagai, K.; Ashimine, S.; Kuwata, T.; Hishida, T.; Nishimura, M.; Yoshida, J.; Takeyoshi, I.; Ochiai, A. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J. Thorac. Oncol. 2010, 5, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhang, W.; Zhong, W.Q.; Liu, Z.J.; Li, H.M.; Yu, Z.L.; Zhao, Y.F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Lederle, W.; Depner, S.; van Rooijen, N.; Gutschalk, C.M.; Mueller, M.M. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 2012, 227, 17–28. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Fu, X.T.; Dai, Z.; Song, K.; Zhang, Z.J.; Zhou, Z.J.; Zhou, S.L.; Zhao, Y.M.; Xiao, Y.S.; Sun, Q.M.; Ding, Z.B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Lu, G.; Yao, Y.; Gu, W. An Autocrine IL-6/IGF-1R Loop Mediates EMT and Promotes Tumor Growth in Non-small Cell Lung Cancer. Int. J. Biol. Sci. 2019, 15, 1882–1891. [Google Scholar] [CrossRef]
- Masola, V.; Carraro, A.; Granata, S.; Signorini, L.; Bellin, G.; Violi, P.; Lupo, A.; Tedeschi, U.; Onisto, M.; Gambaro, G.; et al. In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J. Transl. Med. 2019, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Liang, F.; Cheng, W.; Zhou, R.; Wu, X.; Feng, Y.; Wang, Y. The mechanisms for lung cancer risk of PM2.5: Induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 2017, 32, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- da Silva, I.L.; Montero-Montero, L.; Martin-Villar, E.; Martin-Perez, J.; Sainz, B.; Renart, J.; Toscano Simoes, R.; Soares Veloso, E.; Salviano Teixeira, C.; de Oliveira, M.C.; et al. Reduced expression of the murine HLA-G homolog Qa-2 is associated with malignancy, epithelial-mesenchymal transition and stemness in breast cancer cells. Sci. Rep. 2017, 7, 6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Express 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Storci, G.; Sansone, P.; Mari, S.; D’Uva, G.; Tavolari, S.; Guarnieri, T.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Chieco, P.; et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010, 225, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Segui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar]
- Manso, B.A.; Zhang, H.; Mikkelson, M.G.; Gwin, K.A.; Secreto, C.R.; Ding, W.; Parikh, S.A.; Kay, N.E.; Medina, K.L. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia 2019, 33, 638–652. [Google Scholar] [CrossRef]
- Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Casciaro, M.; Gangemi, S. Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role. Cancers (Basel) 2020, 12, 524. [Google Scholar] [CrossRef] [Green Version]
- Dayer, J.M. From supernatants to cytokines: A personal view on the early history of IL-1, IL-1Ra, TNF and its inhibitor in rheumatology. Arthritis Res. Ther. 2018, 20, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. An expanding role for interleukin-1 blockade from gout to cancer. Mol. Med. 2014, 20 (Suppl. 1), S43–S58. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Chen, R.; Zhou, J. E2F1 and NF-kappaB: Key Mediators of Inflammation-associated Cancers and Potential Therapeutic Targets. Curr. Cancer Drug Targets 2016, 16, 765–772. [Google Scholar] [CrossRef]
- Kaler, P.; Godasi, B.N.; Augenlicht, L.; Klampfer, L. The NF-kappaB/AKT-dependent Induction of Wnt Signaling in Colon Cancer Cells by Macrophages and IL-1beta. Cancer Microenviron. 2009, 2, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Nussinov, R.; Keskin, O. The structural pathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms of oncogenic mutations and SNPs in inflammation and cancer. PLoS Comput. Biol. 2014, 10, e1003470. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 2006, 42, 751–759. [Google Scholar] [CrossRef]
- Takeuchi, H.; Katayama, I. Interleukin 1 (IL-1 alpha and IL-1 beta) induces differentiation/activation of B cell chronic lymphoid leukemia cells. Cytokine 1994, 6, 243–246. [Google Scholar] [CrossRef]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1beta as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef]
- Zhao, G.; Zhu, G.; Huang, Y.; Zheng, W.; Hua, J.; Yang, S.; Zhuang, J.; Ye, J. IL-6 mediates the signal pathway of JAK-STAT3-VEGF-C promoting growth, invasion and lymphangiogenesis in gastric cancer. Oncol. Rep. 2016, 35, 1787–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzaye, O.; Hu, F.; Derkow, K.; Haage, V.; Euskirchen, P.; Harms, C.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma Stem Cells but Not Bulk Glioma Cells Upregulate IL-6 Secretion in Microglia/Brain Macrophages via Toll-like Receptor 4 Signaling. J. Neuropathol. Exp. Neurol. 2016, 75, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.M.; Mariano, V.S.; Pastrez, P.R.A.; Pinto, M.C.; Castro, A.G.; Syrjanen, K.J.; Longatto-Filho, A. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS ONE 2017, 12, e0181125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, L.; Yu, J.; Ghia, E.M.; Choi, M.Y.; Zhang, L.; Zhang, S.; Sanchez-Lopez, E.; Widhopf, G.F., 2nd; Messer, K.; et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF-kappaB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood 2019, 134, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Buggins, A.G.; Patten, P.E.; Richards, J.; Thomas, N.S.; Mufti, G.J.; Devereux, S. Tumor-derived IL-6 may contribute to the immunological defect in CLL. Leukemia 2008, 22, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Dozmorov, I.; Li, W.; Yancopoulos, S.; Sison, C.; Centola, M.; Jain, P.; Allen, S.L.; Kolitz, J.E.; Rai, K.R.; et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood 2011, 118, 5201–5210. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Chen, L.; Shi, Y.; Zhu, X.; Guo, W.; Zhang, M.; Che, Y.; Tang, L.; Yang, X.; You, Q.; Liu, Z. IL10 secreted by cancerassociated macrophages regulates proliferation and invasion in gastric cancer cells via cMet/STAT3 signaling. Oncol. Rep. 2019, 42, 595–604. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. OncoTargets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; Gao, Q.; Ji, S.; Chen, X.; Lei, Q.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-kappaB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 2019, 145, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, C.; Li, W.; Gao, Z.; Guo, K.; Song, S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life 2014, 66, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Fiorcari, S.; Maffei, R.; Audrito, V.; Martinelli, S.; Ten Hacken, E.; Zucchini, P.; Grisendi, G.; Potenza, L.; Luppi, M.; Burger, J.A.; et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget 2016, 7, 65968–65981. [Google Scholar] [CrossRef] [PubMed]
- Alhakeem, S.S.; McKenna, M.K.; Oben, K.Z.; Noothi, S.K.; Rivas, J.R.; Hildebrandt, G.C.; Fleischman, R.A.; Rangnekar, V.M.; Muthusamy, N.; Bondada, S. Chronic Lymphocytic Leukemia-Derived IL-10 Suppresses Antitumor Immunity. J. Immunol. 2018, 200, 4180–4189. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, T.; Tang, W.; Deng, B.; Chen, Y.; Zhu, J.; Shen, X. Hepatocellular Carcinoma Cells Induce Regulatory T Cells and Lead to Poor Prognosis via Production of Transforming Growth Factor-beta1. Cell. Physiol. Biochem. 2016, 38, 306–318. [Google Scholar] [CrossRef]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Haque, S.; Morris, J.C. Transforming growth factor-beta: A therapeutic target for cancer. Hum. Vaccines Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-beta in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: A foe or ally? Cell. Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, Y.D.; Zhan, Y.T.; Zhu, Y.H.; Li, Y.; Xie, D.; Guan, X.Y. High levels of CCL2 or CCL4 in the tumor microenvironment predict unfavorable survival in lung adenocarcinoma. Thorac. Cancer 2018, 9, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; He, S.J.; Yang, J. MCP-1/CCL2 Mediated by Autocrine Loop of PDGF-BB Promotes Invasion of Lung Cancer Cell by Recruitment of Macrophages Via CCL2-CCR2 Axis. J. Interferon Cytokine Res. 2019, 39, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Nelson, D.; Tian, S.; Mulvey, E.; Patel, B.; Conti, I.; Jaen, J.; Rollins, B.J. CCL2/CCR2 Regulates the Tumor Microenvironment in HER-2/neu-Driven Mammary Carcinomas in Mice. PLoS ONE 2016, 11, e0165595. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, V.; Balakrishnan, K. CCL2 in chronic lymphocytic leukemia: A macro in microenvironment? Leuk. Lymphoma 2012, 53, 1849–1850. [Google Scholar] [CrossRef]
- Dal-Bo, M.; Bertoni, F.; Forconi, F.; Zucchetto, A.; Bomben, R.; Marasca, R.; Deaglio, S.; Laurenti, L.; Efremov, D.G.; Gaidano, G.; et al. Intrinsic and extrinsic factors influencing the clinical course of B-cell chronic lymphocytic leukemia: Prognostic markers with pathogenetic relevance. J. Transl. Med. 2009, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Gine, E.; Martinez, A.; Villamor, N.; Lopez-Guillermo, A.; Camos, M.; Martinez, D.; Esteve, J.; Calvo, X.; Muntanola, A.; Abrisqueta, P.; et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica 2010, 95, 1526–1533. [Google Scholar] [CrossRef]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef]
- Filip, A.A.; Cisel, B.; Koczkodaj, D.; Wasik-Szczepanek, E.; Piersiak, T.; Dmoszynska, A. Circulating microenvironment of CLL: Are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol. Dis. 2013, 50, 263–270. [Google Scholar] [CrossRef]
- Boissard, F.; Fournie, J.J.; Laurent, C.; Poupot, M.; Ysebaert, L. Nurse like cells: Chronic lymphocytic leukemia associated macrophages. Leuk. Lymphoma 2015, 56, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Caligaris-Cappio, F. Role of the microenvironment in chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 123, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Boissard, F.; Fournie, J.J.; Quillet-Mary, A.; Ysebaert, L.; Poupot, M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J. 2015, 5, e355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.Y.; Kashyap, M.K.; Kumar, D. The chronic lymphocytic leukemia microenvironment: Beyond the B-cell receptor. Best Pract. Res. Clin. Haematol. 2016, 29, 40–53. [Google Scholar] [CrossRef] [PubMed]
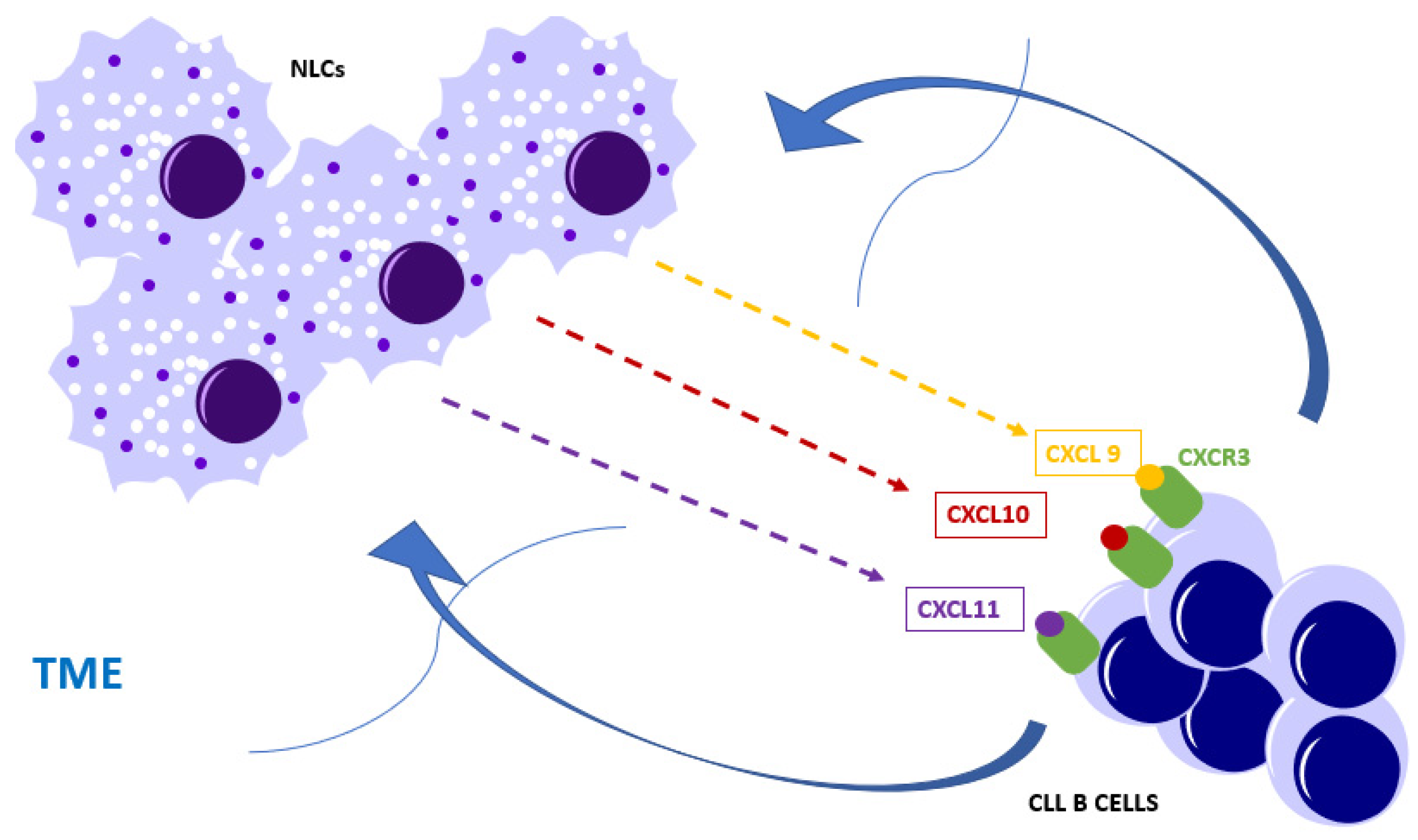
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The Role of CXC Chemokine Receptors 1-4 on Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Ocana, E.; Delgado-Perez, L.; Campos-Caro, A.; Munoz, J.; Paz, A.; Franco, R.; Brieva, J.A. The prognostic role of CXCR3 expression by chronic lymphocytic leukemia B cells. Haematologica 2007, 92, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Burger, M.; Kipps, T.J. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood 1999, 94, 3658–3667. [Google Scholar] [CrossRef]
- Burger, M.; Hartmann, T.; Krome, M.; Rawluk, J.; Tamamura, H.; Fujii, N.; Kipps, T.J.; Burger, J.A. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood 2005, 106, 1824–1830. [Google Scholar] [CrossRef] [Green Version]
- Ganghammer, S.; Gutjahr, J.; Hutterer, E.; Krenn, P.W.; Pucher, S.; Zelle-Rieser, C.; Johrer, K.; Wijtmans, M.; Leurs, R.; Smit, M.J.; et al. Combined CXCR3/CXCR4 measurements are of high prognostic value in chronic lymphocytic leukemia due to negative co-operativity of the receptors. Haematologica 2016, 101, e99–e102. [Google Scholar] [CrossRef]
- Muller, G.; Hopken, U.E.; Lipp, M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol. Rev. 2003, 195, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Burkle, A.; Niedermeier, M.; Schmitt-Graff, A.; Wierda, W.G.; Keating, M.J.; Burger, J.A. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood 2007, 110, 3316–3325. [Google Scholar] [CrossRef] [PubMed]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.W., Jr.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Quiroga, M.P.; Hartmann, E.; Burkle, A.; Wierda, W.G.; Keating, M.J.; Rosenwald, A. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood 2009, 113, 3050–3058. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Punt, J.; Stranford, S.A.; Jones, P.P.; Owen, J.A. Kuby Immunology, 8th ed.; Macmillan Learning: New York, NY, USA, 2019; Volume 1. [Google Scholar]
- Cols, M.; Barra, C.M.; He, B.; Puga, I.; Xu, W.; Chiu, A.; Tam, W.; Knowles, D.M.; Dillon, S.R.; Leonard, J.P.; et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL, and CD40L. J. Immunol. 2012, 188, 6071–6083. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Toedt, G.; Zenz, T.; Stilgenbauer, S.; Lichter, P.; Seiffert, M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: A dominant role of CCL2. Haematologica 2011, 96, 408–416. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Yang, J.; Bi, Y.; Wang, H. ZAP-70 in chronic lymphocytic leukemia: A meta-analysis. Clin. Chim. Acta 2018, 483, 82–88. [Google Scholar] [CrossRef]
- Lafarge, S.T.; Hou, S.; Pauls, S.D.; Johnston, J.B.; Gibson, S.B.; Marshall, A.J. Differential expression and function of CD27 in chronic lymphocytic leukemia cells expressing ZAP-70. Leuk. Res. 2015, 39, 773–778. [Google Scholar] [CrossRef]
- Fayad, L.; Keating, M.J.; Reuben, J.M.; O’Brien, S.; Lee, B.N.; Lerner, S.; Kurzrock, R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: Correlation with phenotypic characteristics and outcome. Blood 2001, 97, 256–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Shaim, H.; Estrov, Z.; Harris, D.; Hernandez Sanabria, M.; Liu, Z.; Ruvolo, P.; Thompson, P.A.; Ferrajoli, A.; Daher, M.; Burger, J.; et al. The CXCR4-STAT3-IL-10 Pathway Controls the Immunoregulatory Function of Chronic Lymphocytic Leukemia and Is Modulated by Lenalidomide. Front. Immunol. 2017, 8, 1773. [Google Scholar] [CrossRef] [PubMed]
- Abbaci, A.; Talbot, H.; Saada, S.; Gachard, N.; Abraham, J.; Jaccard, A.; Bordessoule, D.; Fauchais, A.L.; Naves, T.; Jauberteau, M.O. Neurotensin receptor type 2 protects B-cell chronic lymphocytic leukemia cells from apoptosis. Oncogene 2018, 37, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Talbot, H.; Saada, S.; Barthout, E.; Gallet, P.F.; Gachard, N.; Abraham, J.; Jaccard, A.; Troutaud, D.; Lalloue, F.; Naves, T.; et al. BDNF belongs to the nurse-like cell secretome and supports survival of B chronic lymphocytic leukemia cells. Sci. Rep. 2020, 10, 12572. [Google Scholar] [CrossRef]
- Boissard, F.; Tosolini, M.; Ligat, L.; Quillet-Mary, A.; Lopez, F.; Fournie, J.J.; Ysebaert, L.; Poupot, M. Nurse-like cells promote CLL survival through LFA-3/CD2 interactions. Oncotarget 2017, 8, 52225–52236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzoni, M.; Doglioni, C.; Caligaris-Cappio, F. Chronic lymphocytic leukemia: The pathologist’s view of lymph node microenvironment. Semin. Diagn. Pathol. 2011, 28, 161–166. [Google Scholar] [CrossRef]
- Damle, R.N.; Temburni, S.; Calissano, C.; Yancopoulos, S.; Banapour, T.; Sison, C.; Allen, S.L.; Rai, K.R.; Chiorazzi, N. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood 2007, 110, 3352–3359. [Google Scholar] [CrossRef] [Green Version]
- Calissano, C.; Damle, R.N.; Hayes, G.; Murphy, E.J.; Hellerstein, M.K.; Moreno, C.; Sison, C.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood 2009, 114, 4832–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaksic, O.; Paro, M.M.; Kardum Skelin, I.; Kusec, R.; Pejsa, V.; Jaksic, B. CD38 on B-cell chronic lymphocytic leukemia cells has higher expression in lymph nodes than in peripheral blood or bone marrow. Blood 2004, 103, 1968–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaglio, S.; Vaisitti, T.; Aydin, S.; Bergui, L.; D’Arena, G.; Bonello, L.; Omede, P.; Scatolini, M.; Jaksic, O.; Chiorino, G.; et al. CD38 and ZAP-70 are functionally linked and mark CLL cells with high migratory potential. Blood 2007, 110, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Aydin, S.; Grand, M.M.; Vaisitti, T.; Bergui, L.; D’Arena, G.; Chiorino, G.; Malavasi, F. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol. Med. 2010, 16, 87–91. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesaros, O.; Jimbu, L.; Neaga, A.; Popescu, C.; Berceanu, I.; Tomuleasa, C.; Fetica, B.; Zdrenghea, M. Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells. Biomedicines 2020, 8, 516. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110516
Mesaros O, Jimbu L, Neaga A, Popescu C, Berceanu I, Tomuleasa C, Fetica B, Zdrenghea M. Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells. Biomedicines. 2020; 8(11):516. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110516
Chicago/Turabian StyleMesaros, Oana, Laura Jimbu, Alexandra Neaga, Cristian Popescu, Iulia Berceanu, Ciprian Tomuleasa, Bogdan Fetica, and Mihnea Zdrenghea. 2020. "Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells" Biomedicines 8, no. 11: 516. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8110516