1. Introduction
Poxviruses are large, enveloped, double-stranded DNA viruses capable of causing disease in mammals, birds and insects. Binding and entry of poxviruses into vertebrate cells is an efficient process for a wide range of cell types, irrespective of the host species, with any host range restriction occurring after viral entry [
1]. The complex replication cycles of poxviruses take place exclusively in the cytoplasm, although it has long been suggested that poxviruses must interact with host nuclei for productive infection [
2,
3,
4,
5,
6]. Perhaps the best-studied antiviral host restriction mechanism is interferon (IFN)-mediated, against which almost all viruses have evolved defence mechanisms [
7,
8]. Some of the first viral anti-IFN defence mechanisms were elucidated using vaccinia virus (VACV), which expresses multiple, often redundant inhibitors of IFN induction, JAK/STAT signalling and IFN-stimulated genes (ISGs), as well as IFN-receptor antagonists and mimics of IFN ligands [
7,
8,
9,
10,
11,
12].
These potent immunomodulators are produced mainly during the early phase of VACV gene expression. However, poxviruses have strategies in place to prevent or evade immediate–early host innate responses induced as a consequence of the virus binding and fusing with the cell membrane. Poxviruses have proteinaceous substructures, termed lateral bodies (LBs), outside the core but within the mature virion’s membrane. These are analogous to herpesvirus tegument proteins, some of which perform immunomodulatory functions early during infection [
13,
14,
15,
16].
Schmidt et al. [
17] reported that VACV packages the conserved H1 phosphatase (also known as VH1) within LBs. When VH1 is released from LBs into the cytoplasm of the host cell following membrane fusion, it acts to block IFN-
γ-mediated STAT-dependent signalling prior to gene expression [
18]. Whether additional LB-resident viral immune modulators, capable of blocking other parts or the IFN system, are packaged in the LB of VACV or other poxviruses remain to be determined.
Relative to our understanding of the immunomodulation mediated by mammalian poxviruses, our knowledge of the strategies deployed by avipoxviruses to disarm the interferon response remains rudimentary. A prototypic member of the avipoxviruses, the fowlpox virus (FWPV) is the causal agent of a widespread, enzootic disease of domestic chickens and other gallinaceous birds [
19]. Like VACV, it has been developed for use as a recombinant vector for the expression of antigens from several avian and human pathogens in both poultry and humans [
19,
20]. The commercial FWPV recombinant vaccine (TROVAC-H5) expressing the hemagglutinin gene of H5N8 isolate A/turkey/Ireland/1378/83 has become the most extensively used live recombinant virus used, with almost 2 billion doses used against highly pathogenic influenza H5N2 [
20].
In common with the other poxviruses, FWPV has developed strategies to disarm the host IFN-response and has been found to efficiently block the pI:C-mediated induction of the IFN-β promoter and the IFN-stimulated induction of ISGs in chicken cells [
20]. Studies of FWPV immunomodulators have been complicated by the fact that only 110 (42%) of FWPV genes share significant similarity to those in other poxviruses [
21]. To identify the innate immunomodulatory factors encoded by FWPV, we previously conducted two broad-scale pan-genome analyses of FP9, a highly attenuated strain used as a vaccine vector in both poultry and mammals [
21,
22]. In the first study, we identified
FPV012 as a modulator of IFN induction by screening a knockout library of 65 nonessential FP9 genes [
20,
23]. In the second study, using a gain-of-function approach, in which 4–8 kbp fragments of FP9 were introduced into modified vaccinia Ankara (MVA), we found that FPV014 contributes to increased resistance to exogenous recombinant chicken IFN-
α [
24].
In this report, we use our existing FP9 knockout library [
20] to screen infected primary chicken embryo fibroblasts (CEFs) for FWPV genes that modulate the induction of interferon-regulated genes (IRG)s. Using this approach, we identified FPV184 as a third FWPV immunomodulatory protein blocking the induction of innate immune responses. Intriguingly, unlike the FWPV immunomodulators FPV012 and FPV014 (which are both early viral proteins), FPV184 was found to be a late, structural protein with a functional nuclear localisation signal. Consistent with its ability to modulate ISG responses soon after infection and long before de novo production, we show that FPV184 is packaged into FWPV particles where it resides in LBs. These results suggest that the packaging of late immunomodulatory proteins and their subsequent delivery into the nucleus of newly infected cells serve as an immediate–early innate immune evasion strategy.
2. Experimental Section
2.1. Cells and Viruses
Freshly isolated chicken embryo fibroblasts (CEFs) were provided by the former Institute for Animal Health, Compton, Berks, UK (now the Pirbright Institute, UK). Primary CEFs derived from special SPF chicken lines are normally used for propagation and titration of FWPV [
19]. CEFs were cultured in 199 media (Gibco
®, Invitrogen, Carlsbad, CA, USA), supplemented with 8% heat-inactivated newborn calf serum (NBCS; Gibco
®, Invitrogen), 10% tryptose phosphate broth (Sigma-Aldrich, St. Louis, MO, USA), 2% nystatin (Sigma-Aldrich) and 0.1% penicillin–streptomycin (Gibco
®, Invitrogen). DF-1 is a CEF-derived, spontaneously immortalised cell line, which, unlike CEFs, exhibits high transfection efficiency and, at the same time, supports satisfactory propagation of FWPV [
25]. HEK293T cells are immortalised human embryonic kidney cells that are commonly used in protein expression studies. DF-1 and HEK293T cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco
®, Invitrogen), and 0.1% penicillin–streptomycin. All cells were grown at 37 °C. The origin, propagation and titration (plaque assays) of the FP9 virus have been described previously [
21,
26,
27]. All transient transfections in the study were conducted with Fugene HD transfection reagent (Promega, Southampton, UK) unless otherwise specified. To collect the purified virus, confluent CEFs were infected with FP9 at 0.1 PFU per cell. At 5 days p.i., the supernatant was harvested, along with remaining cells. Virus purification has been described previously [
27].
2.2. Generation of Viable FPV184-Knockout Viruses (Deletion Mutant Viruses)
Generation of
FPV184 deletion mutant viruses was done using two different approaches—a PCR-mediated knockout with a guanine–phosphoribosyltransferase (GPT) insertion in the middle of the gene (FWPV∆184; used for the microarray study) and a transient dominant deletion mutant virus (TDdel184) [
28]—to produce recombinant viruses. The resulting knockout viruses were sequenced in the region of
FPV184, paying attention to the overlap with
FPV183, to check that no adventitious mutations had been inadvertently introduced during the knockout procedure. The nearest flanking genes to
FPV184,
FPV183 and
FPV186 are oriented such that the termini of the genes are towards
FPV184, therefore making it unlikely that manipulation of
FPV184 would disrupt their promoters. Generation of dually tagged
FPV184 was not possible as the sequences encoding the C-termini of
FPV183 and
FPV184 overlap by (about) 30 bp.
2.2.1. Isolation of FPV184 Deletion Mutant Virus (FWPV∆184) by PCR-Mediated Knockout
All primers used in the study are listed in
Table S1. Insertional mutagenesis has been described previously [
20,
23]. PCR-mediated knockout of
FPV184 was carried out using three sets of primers; FWPV DNA was used as the template for PCR with primers M2840 to M2841 and M2844 to M2845, whilst the previously described vector pGPTNEB193rev [
27] was used as the template for primers M2842 and M2843. The resultant PCR products were combined in equimolar amounts, and a further round of PCR was carried out using the flanking primer pair M2840 and M2845. The full-length PCR construct was then used to transfect FP9 infected-CEFs, and the recombinant viruses were selected using media containing mycophenolic acid (25 μg mL
−1), xanthine (250 μg mL
−1), and hypoxanthine (15 μg mL
−1; MXH). The virus was passaged three times in T25 flasks in the presence of MXH before plaquing. The passaged virus was then plaque-purified once in the absence of MXH and once more in the presence of MXH.
2.2.2. Isolation of FPV184 Deletion Mutant Virus (TDdel184) by Transient Dominant Selection
The
FPV184 deletion plasmid was constructed using the previously described vector pGNR [
26]. The 5′ end of the
FPV184 gene and 200 bp of the upstream sequence were amplified by PCR using primer pair M2854 and M2856; the 3′ end of the
FPV184 gene and 200 bp of the downstream sequence were amplified using primer pair M2857 and M2855. Following this first round of PCR, the products were combined in equimolar amounts and a second round of PCR was carried out using the flanking primer pair M2854 and M2855. Utilising
BamHI and
HindIII sites within M2854 and M2855, respectively, the second round PCR product was digested and cloned into pGNR/
BamHI/
HindIII to produce pGNRFPVΔ184. Deletion mutant virus (TDdel184) was isolated by the transient dominant selection method [
28], as described previously [
26].
2.2.3. Confirmation of FPV184 Knockout in Deletion Mutant Viruses (FWPV∆184 and TDdel184)
Each deletion mutant virus was screened by PCR with flanking primers (giving PCR products of specific sizes for wild-type and knocked-out genes), one flanking primer and one primer internal to the deleted sequences (detecting only the wild-type gene) or one flanking primer and one primer specific for the GPT gene (detecting the insertion of GPT into the wild-type gene). The primers used are as follows: flanking primers M530 TO M1257; internal primer M2919 to M1257; PCR-mediated deletion GPT primers M192 to M2854; transient dominant deletion GPT primers M192 to M1257.
2.2.4. Generation of Recombinant EGFP-Expressing Viruses (EGFP wt, NLS− and A19 NLS)
The
FPV184 gene was amplified by PCR with the primers M2952 and M2951. The product was digested with
XmaI and
SacII (within M2952 and M2951, respectively) and cloned into the expression/transfer vector pEF
gpt12S-CvectorEGFPmyc, which was derived from the vector pEF
gpt12S [
29,
30,
31] that was previously cloned with the coding sequence of EGFP (enhanced green fluorescent protein) from the pEGFP-C1 vector (Molecular Probes, Eugene, OR, USA) [
29,
31]. The cloning of the
FPV184 gene into pEF
gpt12S-CvectorEGFPmyc produced pCVecEGFP184 (EGFP at the N-terminus). In addition, mutations were introduced into the
FPV184 gene by PCR using the following primer pairs: M2953 and M2951 for NLS
−; M2956 and M2951 for VACV A19 NLS and two more controls to assess the effect of a putative phosphorylation site (PT; results not shown); primers M2954 and M2951 for NLS
−/PT
−; primers M2955 and M2951 for PT
−. All of the products were digested with
XmaI and
SacII and cloned into pEF
gpt12S-CvectorEGFPmyc/
XmaI/
SacII. Following transfection of constructs into FP9-infected CEF, recombinant EGFP-expressing viruses (EGFP wild-type (wt), NLS
− and A19 NLS) were selected using mycophenolic acid and plaque-purified twice.
2.3. Multistep Growth Curve
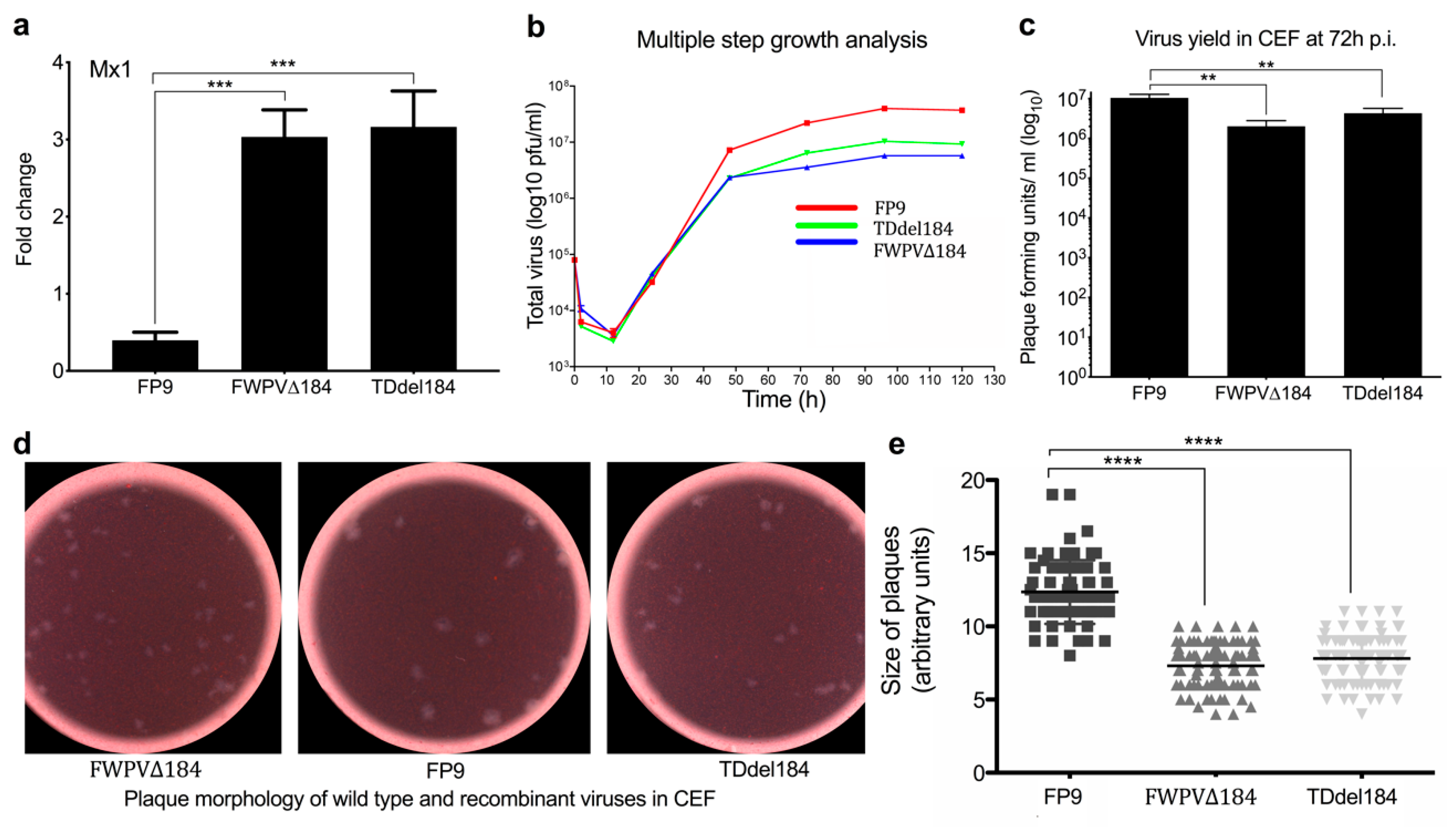
Confluent CEFs were infected with the PCR-mediated (FWPV∆184) and transient dominant (Tddel184) knockout viruses of FPV184 or with the wild-type virus (FP9) at 0.01 PFU per cell. The inoculum was removed 1 h later and replaced by fresh medium. At different times p.i., the extracellular medium was collected, and the cells were overlaid with 1 mL of fresh medium and stored at −70 °C. Intracellular and extracellular viruses were subjected to titer determination by plaque assay [
32]. Plaque sizes between wild-type and knockout viruses were evaluated as before. Briefly, plaques areas were digitally enlarged and calculated in arbitrary units using ImageJ v.32 image analysis software. Scatterplots were created with GraphPad prism v.6.0 (GraphPad software. San Diego, CA, USA).
2.4. Generation of Expression Constructs (pcDNA6FPV184V5His, pFPV184, pVACV-A19)
The FPV184 gene was amplified by PCR with M2892 and M2893. The product was digested with HindIII and XhoI and cloned into pcDNA6V5His/HindIII/XhoI (Invitrogen) to give pcDNA6FPV184V5His or amplified with 4279 and 4280 and digested with XhoI and SalI into pCIFLAG(N-terminus)/XhoI/SalI to give pFPV184. VACV A19 was amplified with 4344 and 4345 and digested with XhoI and SalI into pCIFLAG/XhoI/SalI to give pVACV-A19.
2.5. Infection of CEFs for Microarray and qPCR Analyses
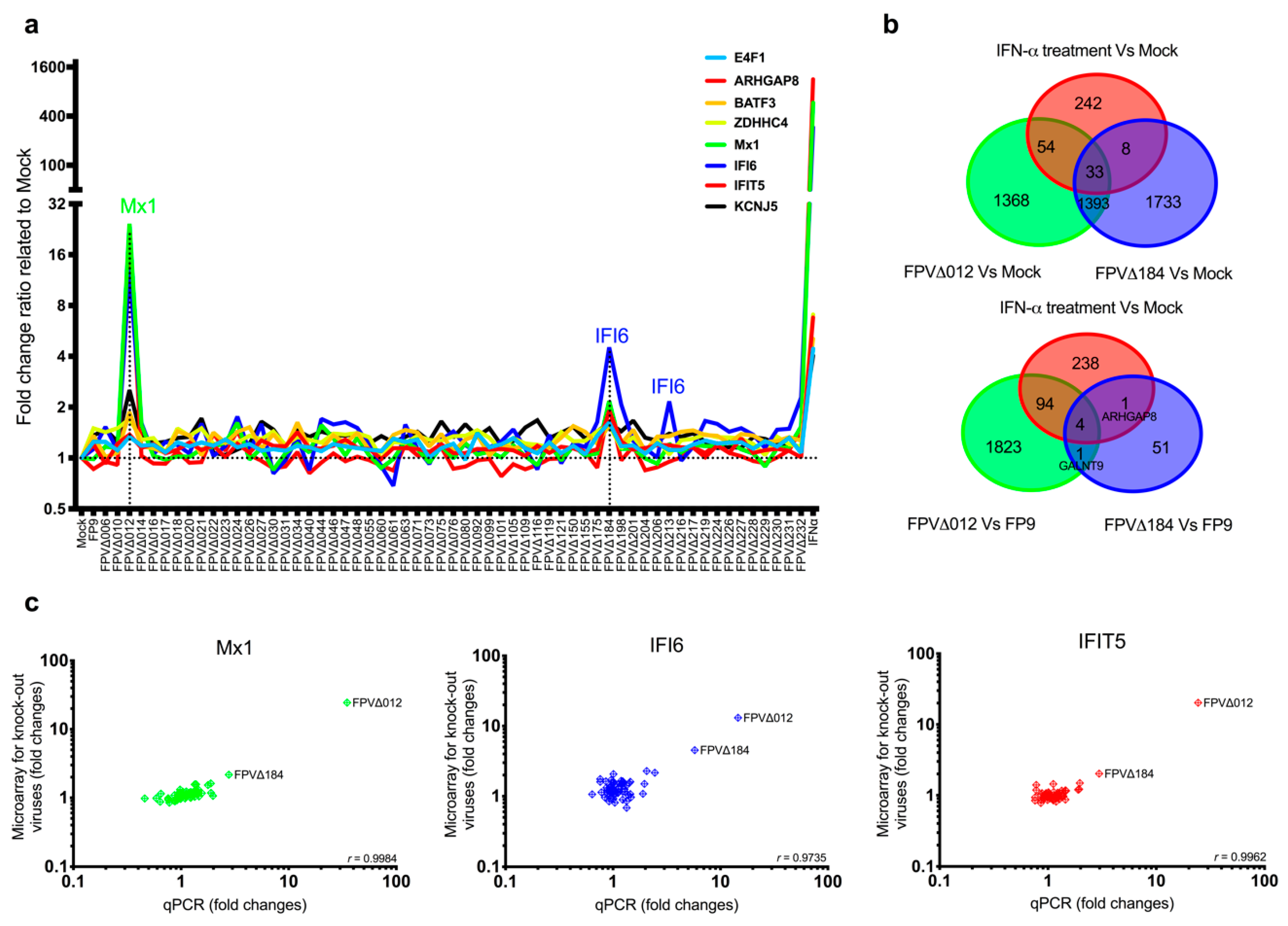
Media was removed from fully confluent CEFs (in T25 flasks; Greiner Bio-one (Alphen a/d Rijn, The Netherlands); 5.6 × 106 cells/flask) and replaced with 8 mL DMEM containing DMEM (mock-infected), FP9 (MOI; multiplicity of infection: 5) or one of the knockout viruses (MOI: 5). After 2 h, DMEM or virus-containing DMEM was replaced with culture media (199 media supplemented with 2% NBCS, 10% TPB, 2% nystatin and 0.1% penicillin/streptomycin) and cells were then incubated for a further 14 h before harvesting. Mock- and virus-infected cells were harvested at 16 h postinfection and stored at −80 °C in RNALater (Sigma-Aldrich) until RNA extraction. The experiment was repeated in triplicate for each knockout virus (for FWPVΔ228, virus duplicates were used) using three different batches of CEFs.
2.6. RNA Extraction and Processing of Samples for Microarray
RNA isolation and processing of samples and microarrays was done as previously described [
25]. Total RNA was extracted from mock-, infected-, and IFN-stimulated DF-1 and CEFs using the RNeasy kit (Qiagen, West Sussex, UK) according to the manufacturer’s instructions, as previously described [
25]. On-column DNA digestion was performed using RNase-free DNase (Qiagen) to remove contaminating genomic DNA. RNA samples were quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and their quality was evaluated using a 2100 bioanalyser (Agilent Technologies, Santa Clara, CA, USA). All RNA samples had an RNA integrity number ≥9.6. RNA samples were processed for microarray using the GeneChip
® 3′ IVT Express Kit (Affymetrix, Emeryville, CA, USA) according to the manufacturer’s instructions, as previously described [
25]. Total RNA (100 ng) was used as input, and quality checks were performed using the bioanalyser at all stages. RNA samples were processed in batches of 12, and batch mixing was used at every stage to avoid creating experimental bias. Hybridisation of RNA to chips and scanning of arrays was performed by the Medical Research Council’s Clinical Sciences Centre (CSC) Genomics Laboratory, Hammersmith Hospital, London, UK, as previously described. The RNA was hybridised to GeneChip Chicken Genome Array chips (Affymetrix; containing comprehensive coverage of 32,773 transcripts corresponding to over 28,000 chicken genes) in a GeneChip hybridisation oven (Affymetrix), the chips were stained and washed on a GeneChip Fluidics Station 450 (Affymetrix), and the arrays were scanned in a GeneChip Scanner 3000 7G with an autoloader (Affymetrix).
2.7. Microarray Data Analysis
A one-way ANOVA adjusted with the Benjamini–Hochberg multiple-testing correction (false discovery rate (FDR) of
p < 0.05) was performed with Partek Genomics Suite (v6.6, Partek Inc, St Louis, MO, USA) across all samples, as previously described [
25,
33]. Comparisons were conducted between infected cells versus mock-treated cells and between infected cells with the KO viruses versus CEFs infected with the parental FP9 strain. The analysis cut-off criteria were fold change ≥ ±1.5 and
p-value ≤0.05. The Affymetrix chicken genome arrays contain probe sets for detecting transcripts from 17 avian viruses, including FWPV, allowing confirmation of viral infection. Visualisation of gene expression data was conducted with GeneSpring GX (v.13.1, Agilent Technologies) and GraphPad Prism (v.6.0). Original microarray data produced in this study have been deposited according to the MIAME guidelines in the public database ArrayExpress (
http://www.ebi.ac.uk/microarray-as/ae/; Acc. No: E-MTAB-7276). A catalogue of 337 ISGs was created by applying a fold change >3 compared to mock and false discovery rates (FDRs) <0.05 on previously published microarray data ([
34]; ArrayExpress accession: E-MTAB-3711 and
Table S2).
2.8. Quantitative Real-Time RT PCR
Quantitative real-time RT PCR was performed on RNA samples using a two-step procedure, as previously described [
35]. RNA was first reverse-transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) according to manufacturer’s instructions. qPCR was then conducted on the cDNA in a 384-well plate with an ABI-7900HT Fast qPCR system (Applied Biosystems, Foster City, CA, USA). Mesa Green qPCR MasterMix (Eurogentec, Seraing, Belgium) was added to the cDNA (5 μL for every 2 μL of cDNA). The following amplification conditions were used: 95 °C for 5 min; 40 cycles of 95 °C for 15 s, 57 °C for 20 s, and 72 °C for 20 s; 95 °C for 15 s; 60 °C for 15 s; 95 °C for 15 s. Primer sequences for genes that were used in the study are shown in
Table S1. The output Ct values and dissociation curves were analysed using SDS v2.3 and RQ Manager v1.2 (Applied Biosystems). Gene expression data were normalised against the housekeeping gene GAPDH and compared with the mock controls using the comparative C
T method (also referred to as the 2
−ΔΔCT method [
36]). All samples were loaded in triplicate.
2.9. Confocal and Widefield Fluorescence Microscopy
Immunofluorescence labelling was carried out using CEFs seeded at 2.5 × 105 cells/well on coverslips, incubated in 6-well plates at 37 °C, 5% CO2 and infected with 0.5–1 pfu of virus for 24 h. The medium was aspirated; cells were washed 3 times (3×) with PBS and fixed with 4% paraformaldehyde in PBS at room temperature (RT). Coverslips were washed 3× in PBS, and cells were permeabilised (0.5% Triton X-100 in PBS) at RT for 10 min with shaking. Following further 3× PBS washing, cells were blocked (0.5% bovine serum albumin in PBS) for 1 h at RT with shaking. Primary antibodies were applied at 1:200 in blocking solution for 1 h at RT with shaking, followed by 5× 5 min PBS washing at RT. Secondary Alexa dye-conjugated antibodies (Molecular Probes) were applied at 1:200 in blocking solution for 1 h at RT in the dark, followed by 5× 5 min PBS washing. For double-labelling experiments, a second round of incubation with primary antibody, washing and secondary antibody was carried out. To label DNA within cells, coverslips were incubated with 1:5000 TOPRO3 (Molecular Probes) for 10 min at RT, followed by 3× PBS washing. Coverslips were dipped in SuperQ water briefly, drained and mounted on Vectorshield mounting media (Vector Labs, Burlingame, CA, USA). Coverslips mounted with hard-set mounting media were allowed to set at 4 °C overnight; all other coverslips were sealed with nail varnish. Confocal microscopy was performed using a Leica TCS NT confocal microscope (Leica, Heidelberg, Germany).
For widefield fluorescence microscopy, CEFs were washed 2× with PBS at RT and fixed with 10% buffered formaldehyde. The images were acquired on Evos fluorescence microscope (Evos FL Imaging System, Thermo Fisher Scientific).
2.10. Virion Fractionation
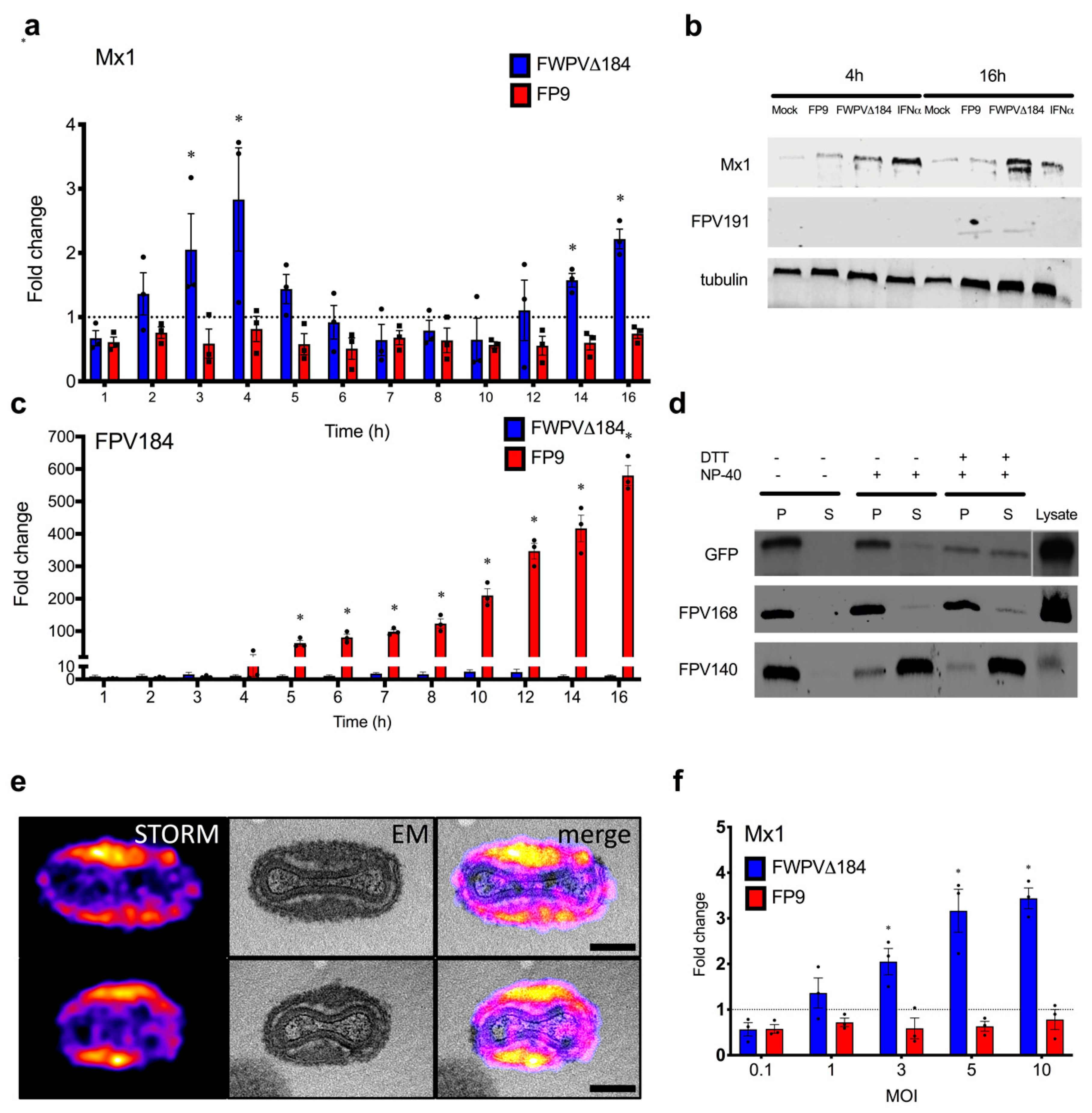
Purified particles of the recombinant virus, which has EGFP fused with FPV184, were incubated in a reaction mixture containing 50 mM Tris-HCl, pH 7.5 and 1% (vol/vol) NP-40, with or without 50 mM dithiothreitol (DTT) for 1 h at 37 °C. The insoluble and soluble materials were separated by centrifugation at 20,000× g for 30 min at 4 °C.
2.11. Western Blotting
Proteins for Western blots were harvested from CEFs and DF-1 cells. Cell pellets were lysed with CelLytic-M solution (Sigma-Aldrich) and the supernatant collected by centrifugation at 15,000×
g, 15 min, prior to protease inhibitor (Roche, Welwyn Garden City, UK) addition. To every 20 μL of sample, 5 μL of 4× loading buffer (Bio-Rad, Hemel Hempstead, UK) was added, and the samples were heated to 60 °C for 5 min. They were then separated on a 12% sodium dodecylsulfate polyacrylamide gel, alongside a protein ladder (Chameleon Dual Colour Standards, LICOR, Cambridge, UK). Samples (20 μg) were loaded for each well, and the gel was run at 150 V for 2 h. Protein samples fractionated by SDS-PAGE were electroblotted onto a nitrocellulose membrane (Hybond-ECL; Amersham Biosciences, Amersham, UK) by following standard protocols. After transfer, the membranes, blocked overnight in 5% nonfat dry milk in PBS buffer, were incubated for 2 h at room temperature with one of the following antibodies: mouse monoclonal anti-GFP (Sigma-Aldrich), mouse monoclonal anti-FLAG (M2) (Sigma-Aldrich), rabbit monoclonal anti-tubulin (Cell Signalling Technology; Ipswich, MA, USA), mouse polyclonal anti-chicken Mx1 (AbMart, Shanghai, China), and mouse monoclonal anti-FPV168 (GB9), anti-FPV140 (DF6) and anti-FPV191 (DE9; in PBS + 0.1% Tween-20 (Sigma-Aldrich) + 2% nonfat dried skimmed milk) [
27,
37] at a dilution of 1:(1000–5000), followed by washing with PBS five times for 5 min. Secondary antibody (goat anti-rabbit horseradish peroxidase-conjugated or goat anti-rabbit or donkey anti-mouse secondary antibodies (LICOR)) was diluted in PBS + 0.1% Tween-20 and added to the blot. Incubation was allowed to proceed for 1 h, followed by washing with PBS five times for 5 min each. Labelled proteins were detected by incubation with either the ECL detection reagent (Amersham Biosciences) and exposure to Hyperfilm ECL chemiluminescence film (Amersham Biosciences) or scanned with the Odyssey Imaging System (LICOR).
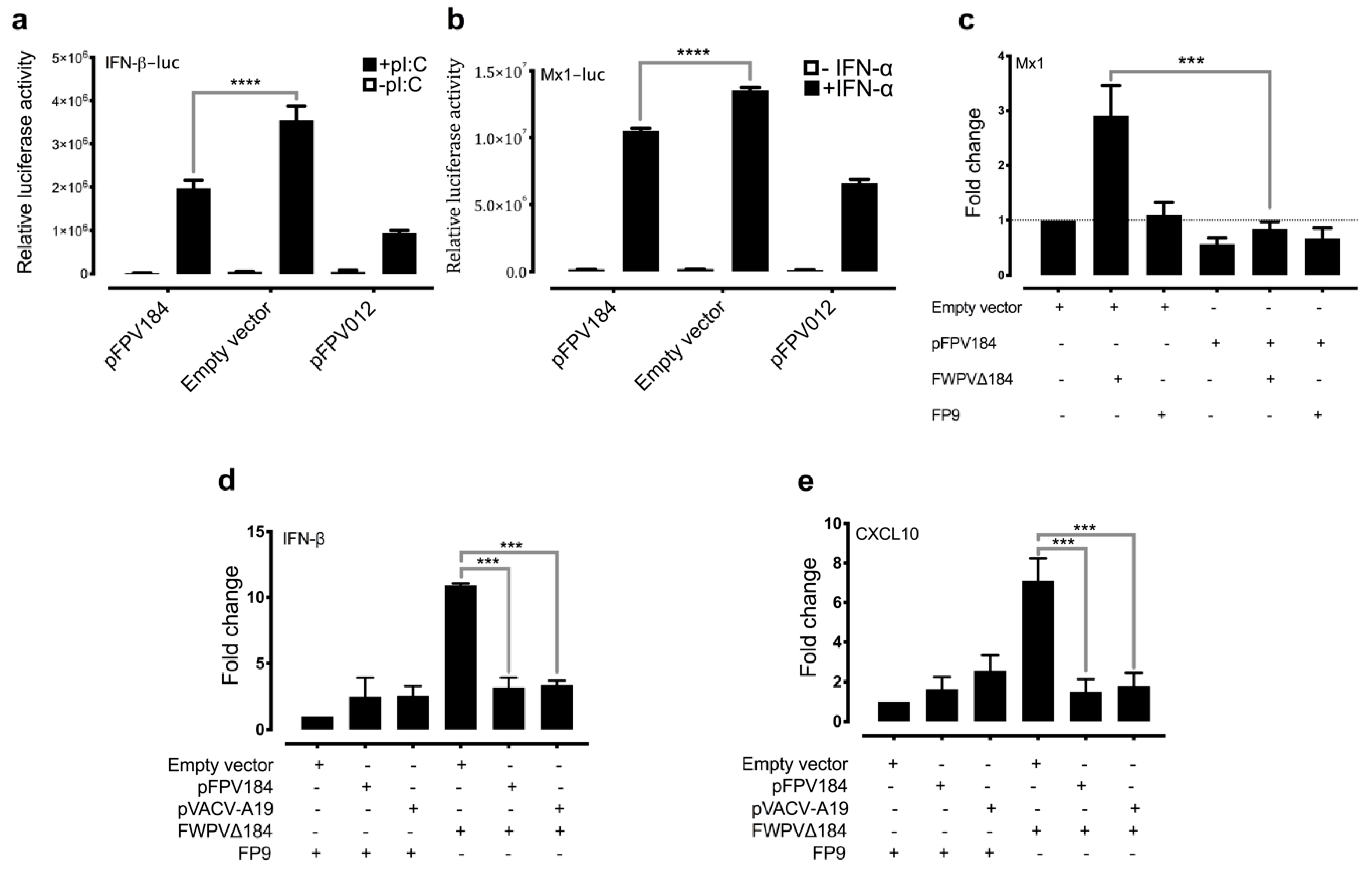
2.12. Transfection of Cells with POLYI:C and Assay of Luciferase Reporters
Chicken fibroblast DF-1 cells were transfected in 12-well plates with either chicken Mx1 or chicken IFN-β promoter reporters (100 ng) [
38], the constitutive reporter plasmid pJATlacZ (100 ng) [
39] or cotransfected with plasmids driving the overexpression of FPV184 or FPV012 or VACV A19 or the empty control vector. Following recovery for 24 h, cells were either left untreated or treated with 1000 IU/mL recombinant IFN-α and incubated for 6 h or transfected overnight with high molecular weight polyI:C (10 μg/mL) purchased from InvivoGen (San Diego, CA, USA) using the Polyfect transfection reagent (Qiagen). Luciferase assays were carried out, and data were normalised using β-galactosidase measurements and expressed as relative luciferase activity. Cell lysate β-galactosidase concentrations were measured by incubation of 10 μL of cell lysate with ortho-nitrophenyl-β-galactoside (50 μL of 0.5 mg mL
−1 diluted in 60 mM Na
2HPO
4·7H
2O, 40 mM Na
2H
2PO
4·H
2O, 10 mM KCl, 1 mM MgSO
4·7H
2O, 2.7 mL litre
−1 β-mercaptoethanol). The reaction mixture was incubated at 37 °C until a yellow colouration appeared; then, the
A420 was measured using a spectrophotometer.
2.13. Correlative Super-Resolution Light and Electron Microscopy (CSRLEM)
Viruses expressing FPV184 with EGFP fused to its N-terminus were pelleted through 36% sucrose and then band-purified on a 25% to 40% sucrose gradient, as described previously [
40]. Virions were diluted in 20 µl 1 mM Tris pH 9, placed in the centre of clean coverslips for 30 min and bound virus was fixed with 4% EM-grade formaldehyde (TAAB). A small asymmetric scratch was made in the middle of the coverslip using a diamond scorer to enable localisation of the super-resolution imaging region of interest within the resin block for trimming, targeting and, subsequently, in sections during electron imaging. The samples were permeabilised for 30 min with 1% Triton X-100 in PBS and blocked with PBS containing 5% BSA (Sigma-Aldrich), 1% FCS for 30 min. Then, the samples were immunostained overnight with anti-GFP nanobody (Chromotek, Martinsried, Germany), conjugated in-house to AlexaFluor647-NHS (Invitrogen) in PBS containing 5% BSA at 4 °C. Coverslips were washed 3 times with PBS and mounted on a microscope slide with parafilm gasket in 1% (
v/
v)
β-mercaptoethanol (Sigma-Aldrich), 150 mM Tris, 1% glucose, 1% glycerol, 10 mM NaCl, pH 8 with 0.25 mg/mL glucose-oxidase and 20 µg/mL catalase.
Super-resolution microscopy was performed on an Elyra PS.1 inverted microscope (Zeiss, Dublin, CA, USA) using an alpha Plan-Apochromat 100×/1.46 NA oil DIC M27 objective. STORM [
41] images were acquired with a 1.6× tube lens on an iXon 897 EMCCD camera (Andor) with 20 ms exposure time, 642 nm excitation at 100% laser power and a 655 nm LP filter. Fluorophore activation was dynamically controlled with a 405 nm laser at 0–2% laser power. Images were processed in Fiji [
42] using ThunderSTORM [
43]. Localisations were fitted with a maximum-likelihood estimator, and lateral drift was corrected by cross-correlation; localisations <20 nm apart within ≤1 frame were merged, and images were rendered using a Gaussian profile.
After super-resolution imaging of the region of interest, a series of phase-contrast images using objectives with 10×, 20×, 40× magnification and fluorescence images using an objective with 100× magnification were taken to map the region of interest and its localisation in relation to the scratch. Coverslips were washed twice in PBS and fixed with 1.5% glutaraldehyde and 2% EM-grade paraformaldehyde (TAAB) in 0.1 M sodium cacodylate for 45 min at room temperature. The samples were treated with reduced osmium and tannic acid, dehydrated through an ethanol series, and embedded in epon resin, as previously described [
44]. After resin polymerisation, the coverslip was removed using liquid nitrogen, revealing the positive pattern of the coverslip-scratch on the surface of the resin block. Using the scratch mark as a reference and viral clusters from phase images as fiducials, the region of interest that had been imaged for super-resolution imaging was identified and targeted for serial sectioning. Sections were collected on formvar-coated slot grids, stained with lead citrate and imaged using a transmission electron microscope (Tecnai T12, Thermo Fisher Scientific, Waltham, MA, USA) equipped with a charge-coupled device camera (SIS Morada; Olympus, Tokyo, Japan). EM and STORM images were registered using NanoJ [
45]. It should be noted that preparation of samples for EM after super-resolution microscopy may move or remove individual virus particles, which mandates careful registration and manual overlay of the images. However, the orientation and structure of the fluorescence signal are clearly reminiscent of the lateral body structure observed in EM.
2.14. Phylogenetic Analysis
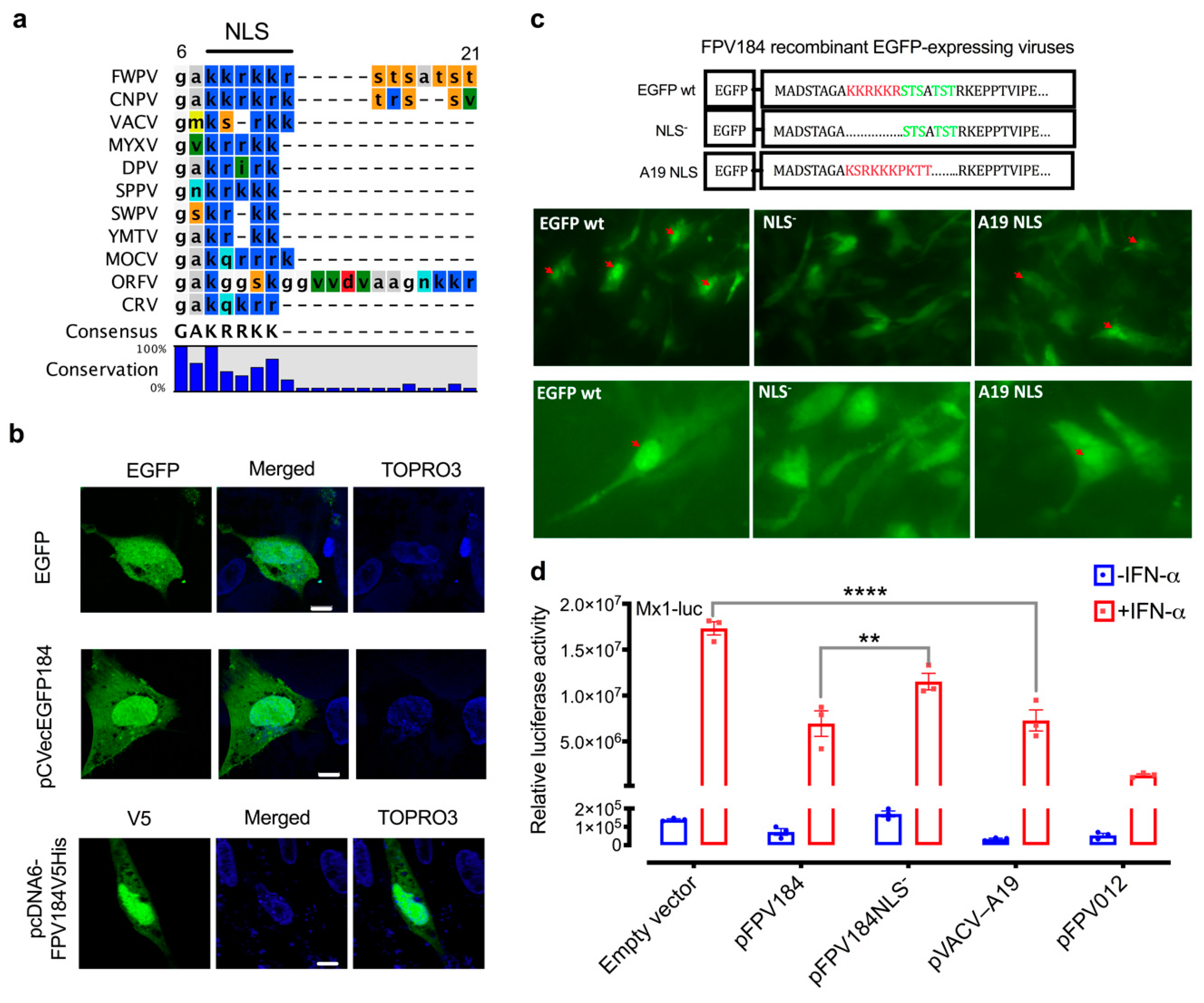
The amino acid sequences of FPV184 orthologues from each genus of chordopoxvirus were subjected to multiple alignments using CLC Workbench 7 (CLC Bio, Qiagen, Aarhus, Denmark). Protein sequence accession numbers for the indicated viruses are as follows: fowlpox virus (FPV184, NP_039147), canarypox virus (CNPV258, NP_955281), VACV (A19, P68714), myxoma Virus (m109 L, AQT34599), deerpox virus (DpV83gp120, YP_227495), sheeppox virus (SPPV_106, NP_659683), swinepox virus (SPV108, NP_570268), Yaba monkey tumor virus (111L, NP_938366), molluscum contagiosum virus (MC124, AQY16697), orf virus (ORF096, NP_957873), and crocodilepox virus (P4b, YP_784314).
2.15. Statistical Analysis
To determine the significance of differences between experimental groups, a Shapiro–Wilk normality test was initially used to confirm whether the data followed a normal distribution for parametric or nonparametric testing. Subsequently, one- or two-way ANOVA parametric analyses were performed using the fold change scores with a Tukey’s or Dunnett’s multiple-comparisons test depending on the application [
46].
p-values were set at 0.05 (
p ≤ 0.05) unless indicated otherwise. Error bars represent the standard error of the mean (SEM). The correlation of expression values between microarray analysis and qRT-PCR was statistically assessed by calculation of Pearson’s correlation coefficient using the built-in function of GraphPad Prism (v.6.0).
4. Discussion
A common strategy to generate new or improved live recombinant poxvirus vaccines is to target and delete immunomodulatory genes in the vector. There has been considerable study of multiple immunomodulatory proteins of VACV [
8,
9,
10,
12] but, in contrast, few immunomodulators have been reported in other poxviruses. In FWPV, two immunomodulatory proteins have been reported so far: FPV012 and FPV014 [
20,
24]. Both are members of the ankyrin repeat protein superfamily (Pfam clan CLO465), are expressed early during viral replication and are not essential for virus replication in culture.
In a systematic, large-scale study, we interrogated the innate immune function of nonessential FWPV genes by transcriptomic analysis of a library of knockout viruses derived from the highly attenuated FWPV vaccine strain FP9. We identified a third immunomodulatory protein (FPV184), which, unlike the other two immunomodulators, is not an ankyrin protein, is expressed late during FP9 replication and encodes a very small structural protein packaged in the virions. There is a good degree of conservation between FPV184 and its VACV orthologue A19. A19 is also expressed at the intermediate/late stages of replication, but, unlike FPV184, it has been reported as essential for viral replication [
50], being involved in the maturation of VACV virions and in early viral transcription in newly-infected cells, where it interacts directly with the viral RNA polymerase and other members of the early transcription complex [
49,
50]. Since A19 is a late protein and has an essential role in virion morphogenesis, A19 knockout viruses were not constructed and, thus, the potential role of A19 as an immunomodulator was not evaluated. Whilst conservation of A19 and FPV184 suggests an important function for the latter, we were able to generate FWPV lacking
FPV184 using two different methodologies, showing that the gene is nonessential for replication in vitro, albeit with a reduced replication rate.
Like VACV A19, FPV184 may act as an early viral transcription factor or a regulator. The two CxxC motifs involved in the binding of the VACV orthologue to viral RNA polymerase [
49,
50] are well conserved in FPV184. Although not investigated in this study, the presence of CxxC motifs is indicative of a zinc finger motif [
51,
52]. The zinc-binding domains, initially identified as DNA binding motifs in transcription factors, are now grouped into superfamilies based on their amino-acid composition. A number of transcription factors have been found to bind the DNA sequence (A/T)GATA(A/G) in the regulatory region of genes. These have been termed GATA-binding transcription factors and are able to bind DNA via a conserved zinc-finger domain in which the zinc ion is coordinated by four cysteine residues [
53]. In VACV, early transcription factor null mutants have displayed defects in morphogenesis [
50,
54,
55]. We have not conducted morphological studies of the FWPV∆184 virions, e.g., using electron microscopy; consequently, a role for FPV184 in morphogenesis cannot be excluded.
It is well established that FWPV blocks the launch of the avian type I IFN response and the induction of ISGs entirely. We recently showed that FWPV DNA is sensed by the chicken cGAS/STING DNA-sensing pathway, but the downstream signalling response leading to type I IFN production is effectively blocked by the wild-type virus [
56]. Infection of chicken macrophages with FWPV∆184 resulted in IFN and ISG transcription, which was lost in cGAS and STING CRISPR knockout lines [
56]. In this study, the relatively subtle induction of a subset of ISGs by FWPV∆184 could be explained by the presence of a multigenic, redundant system in FP9 to control host interferon response, as observed in VACV and other poxviruses or by relatively mild stimuli. It is possible that the complementary effects of other FWPV immunomodulatory proteins alleviate the effects caused by the absence of FPV184. For example, FPV138 is a homologue of VACV H1, a protein-tyrosine phosphatase that resides in the LBs, which, upon release into the cytoplasm, dampens type I IFN-induced, STAT1-mediated signalling. ISG expression was higher in cells infected with FWPV∆012 than with FPV∆184, in terms of both numbers and expression levels; there was, however, a significant overlap between the ISGs induced by both viruses (
Figure 1b). Furthermore, when FPV012 and FPV184 were transiently coexpressed in DF-1 cells, they were not synergistic in inhibiting the pI:C-mediated induction of IFN-
β (
Figure S3), suggesting they may target the same host immune pathways but at different time points during infection. FPV184′s role may be to shut down immediate–early host immune responses and its function may be gradually superseded by that of the early immunomodulatory proteins expressed de novo upon infection.
The putative role of FPV184 in blocking immediate–early host immune response is also supported by our demonstration, with super-resolution microscopy and CSRLEM, that the protein is packaged within the LB and outside the confines of the viral cores, where early viral transcription is executed. LBs have been described as a poxvirus mechanism for the delivery of viral proteins to the cytoplasm of cells soon after fusion of the MV membrane with cellular plasma or endosomal membranes; their release and disaggregation are believed to depend upon reduction and proteasomal activity [
17]. Parallels exist with the herpes virus tegument, which is also located inside the virion (under the envelope but outside of the capsid) and is known to deliver virion host effector proteins into cells [
17,
57]. Our hypothesis is that FWPV LBs contain additional packaged immunomodulatory factors that can act before core activation and early gene expression to establish a favourable environment in the cytoplasm. Other proteins found in LBs in VACV [
17] include phosphoprotein F17 and oxidoreductase G4, both involved in morphogenesis, as well as the dual-specificity phosphatase H1 discussed above (FWPV orthologues, FPV103, FPV077 and FPV138, respectively). H1 has an immunomodulatory function by virtue of its ability to target STAT-mediated signalling, required for IFN-mediated induction of ISG expression. Our demonstration that FPV184 and VACV A19 are also packaged in LBs, with functional evidence that they both have an immunomodulatory function, shows that they could complement H1, primarily by blocking the induction of IFN but, to a lesser extent, also by blocking IFN-mediated induction of ISGs.
Another unexpected observation was that FPV184 is preferentially localised in the nuclei of host cells. Few host nuclear proteins have been shown to play a part in the poxvirus lifecycle, and fewer poxviral proteins (e.g., VACV C6, VACV F16) have been found to enter the nucleus during an infection [
58,
59]. There is no evidence that host proteins are required either for DNA replication or early gene transcription [
60], but it is known that host proteins are necessary for VACV postreplication transcription; intermediate transcription requires host protein VITF-2, which resides within the nucleus of uninfected cells [
61]. Although some poxviral proteins have been reported to enter the nucleus, to date, there has been no identification of a poxvirus-encoded protein containing an identifiable and functional NLS. Furthermore, the nuclear localisation of FPV184 was found to influence its immunomodulatory ability. Using a luciferase assay, we showed that a construct expressing FPV184 without the NLS only partially abrogated the ability of the protein to inhibit the IFN-mediated induction of the Mx1 promoter (
Figure 5d). Whether nuclear localisation is partially or fully required for the immunomodulatory ability of FPV184 was unclear as, due to its small size (9.5 kDa), low levels of the protein can enter the nucleus without an NLS via passive diffusion. Myxoma virus encodes a protein termed myxoma nuclear factor (MNF) [
62], an ankyrin repeat-containing protein that localises to the nucleus in the absence of an NLS and sequesters NF-κB. The cowpox virus protein CrmA, even though it is small enough to shuttle between the nucleus and the cytoplasm by passive diffusion, requires a leucine-rich nuclear export signal (NES) for its nuclear export [
63]. It is possible that in the presence of the active NLS, the accumulation of FPV184 in the nucleus is dependent on its lack of an NES.
Although the conserved block of lysines and arginines found at the N terminus of FPV184 is characteristic of an NLS, it could also constitute a DNA/RNA binding domain. The possibility that FPV184 might be a dsRNA-binding protein could explain the inhibition of polyI:C mediated-induction of the IFN-β promoter but would not explain the inhibition of IFN-mediated induction of the Mx1 promoter we observed.
Collectively, the findings of this study indicate a late-expressed poxviral protein, packaged outside of the viral cores, with a nonessential phenotype, a functional NLS and an immediate–early effect on the innate immune response, properties that make FPV184 resemble a herpesvirus tegument protein rather than a typical early, immunomodulatory poxviral protein. The precise mechanism(s) and function(s) of FPV184 are obscure. Nevertheless, these findings extend the paradigm by which poxvirus structural proteins can block the induction of innate immune responses, immediately after infection, which might be useful in vaccine development.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




