Efficaciousness of Low Affinity Compared to High Affinity TSPO Ligands in the Inhibition of Hypoxic Mitochondrial Cellular Damage Induced by Cobalt Chloride in Human Lung H1299 Cells


 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. CoCl2 Exposure and Its Treatment with TSPO Ligands
2.3. ADP/ATP Ratio
2.4. Reactive Oxygen Species (ROS) Levels
2.5. Depolarization of the Mitochondrial Membrane Potential (ΔΨm)
2.6. Cellular Cytotoxicity Assay (LDH)
2.7. XTT Based Colorimetric Assay
2.8. Cell Density
2.9. Statistical Analysis and Data Presentation
3. Results
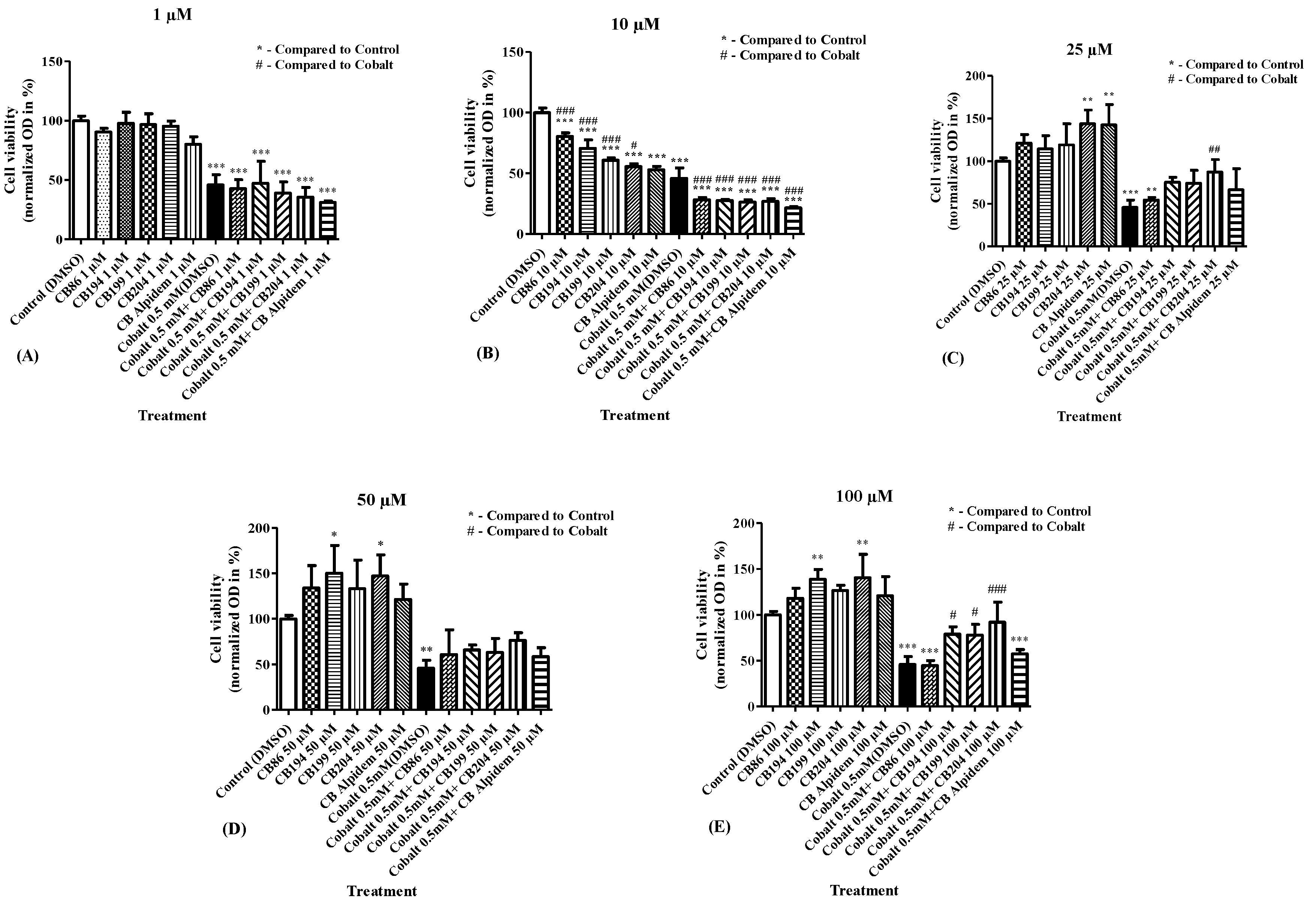
3.1. Screening of TSPO Ligand Efficacy in the Protection of Cellular Viability
3.2. Dose–Response Analyses for the Efficacy of CB86 and CB204 Ligands in the Prevention of CoCl2-Induced Decrease in Cellular Viability
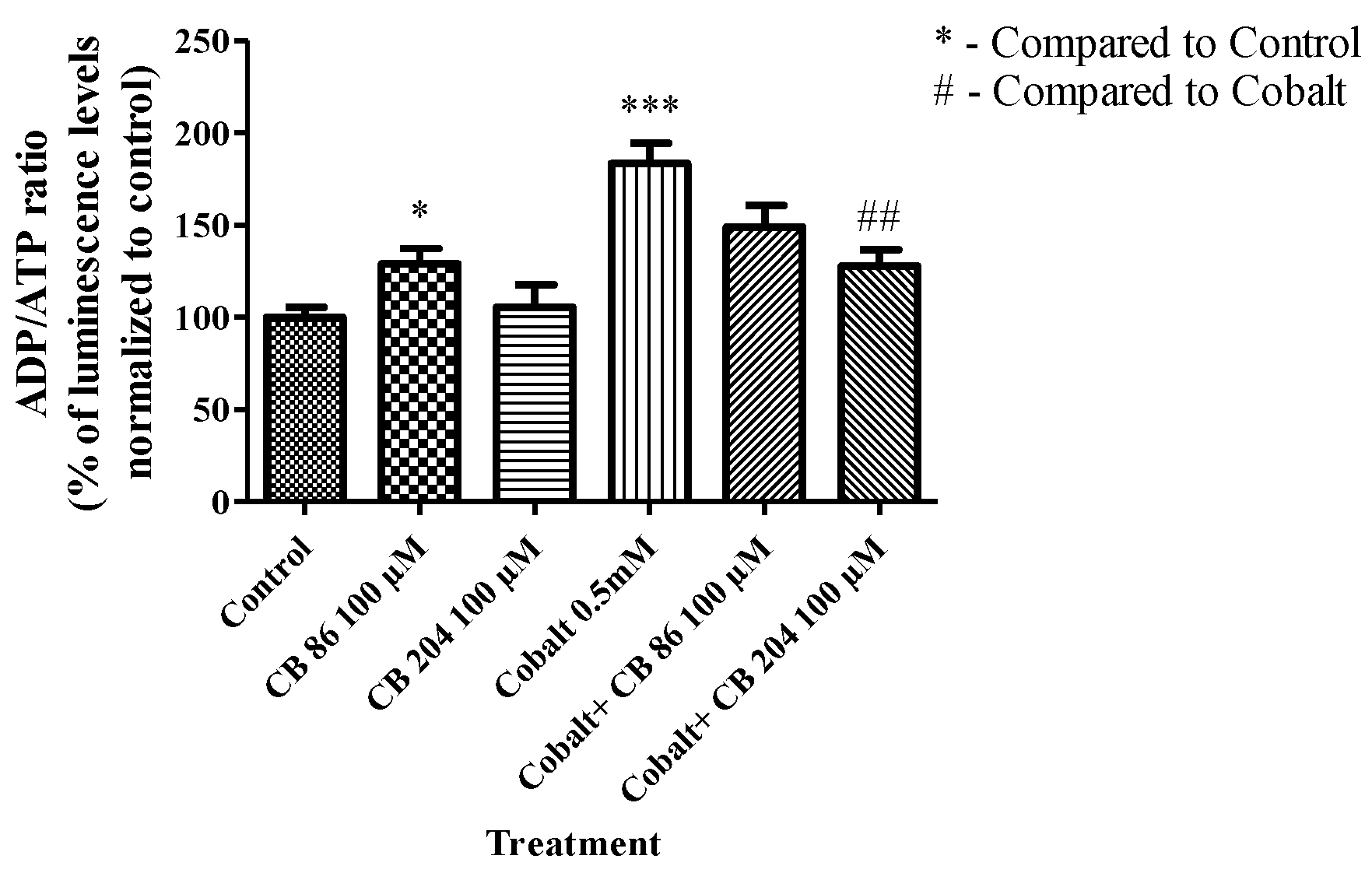
3.3. ADP/ATP Ratio
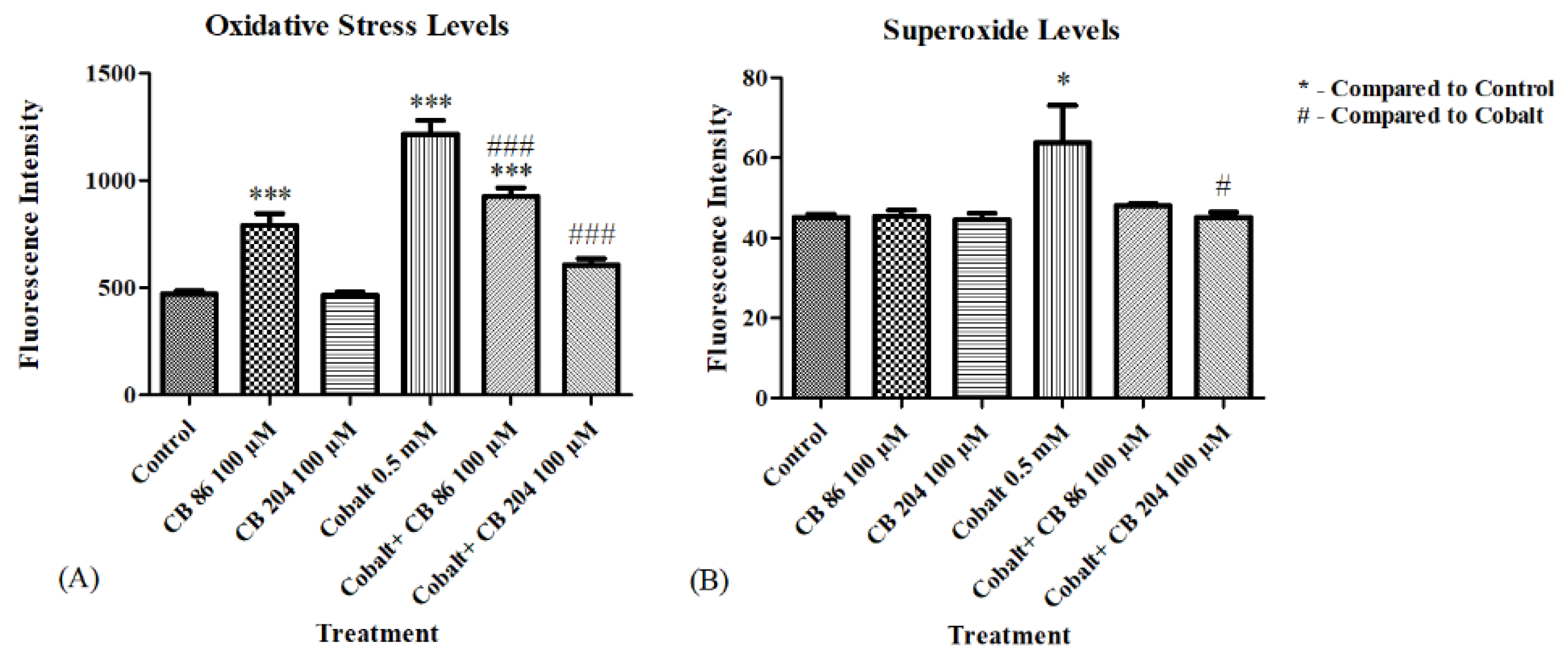
3.4. Oxidative Stress and Superoxide Levels
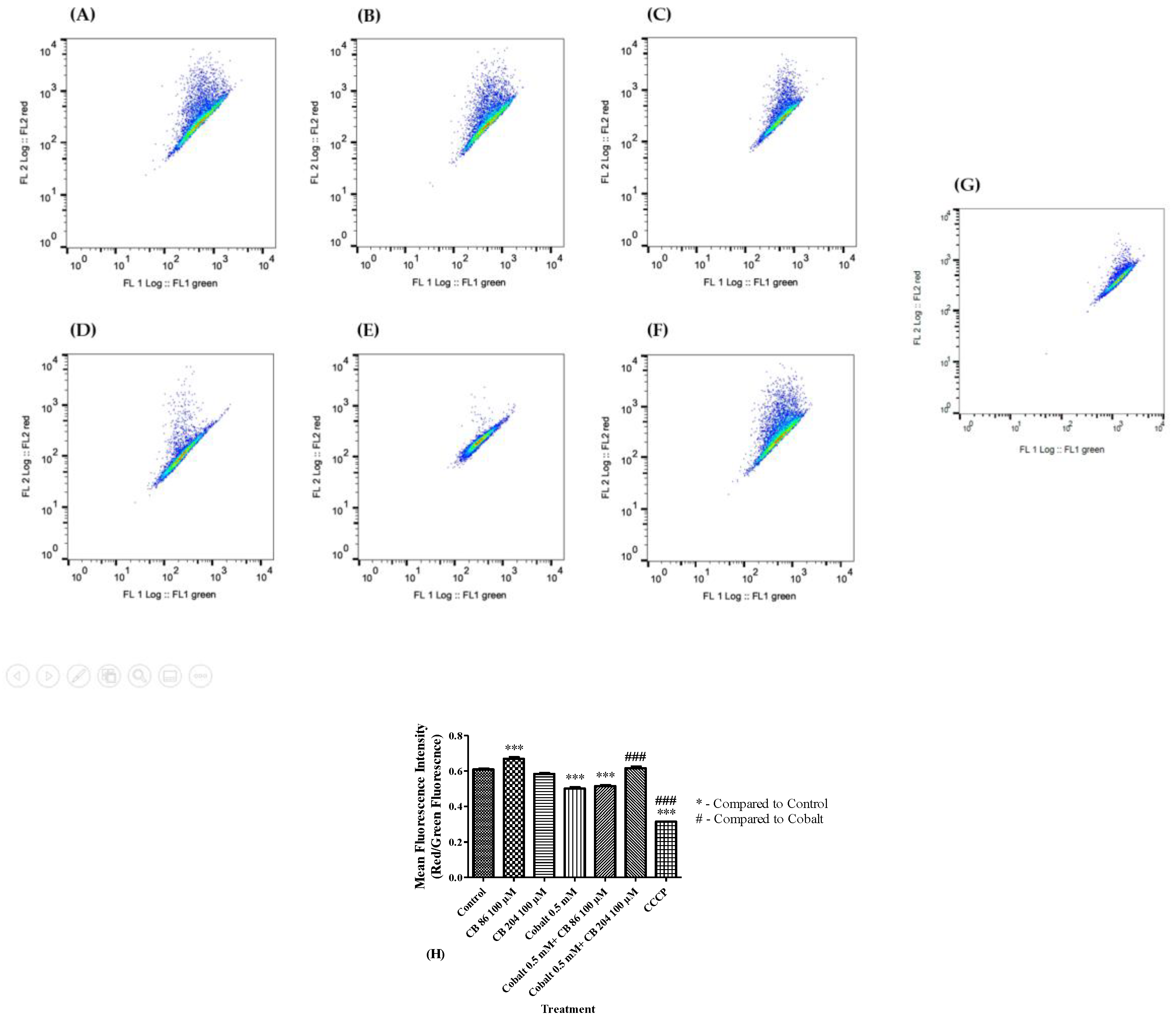
3.5. Depolarization of the Mitochondrial Membrane Potential (ΔΨm)
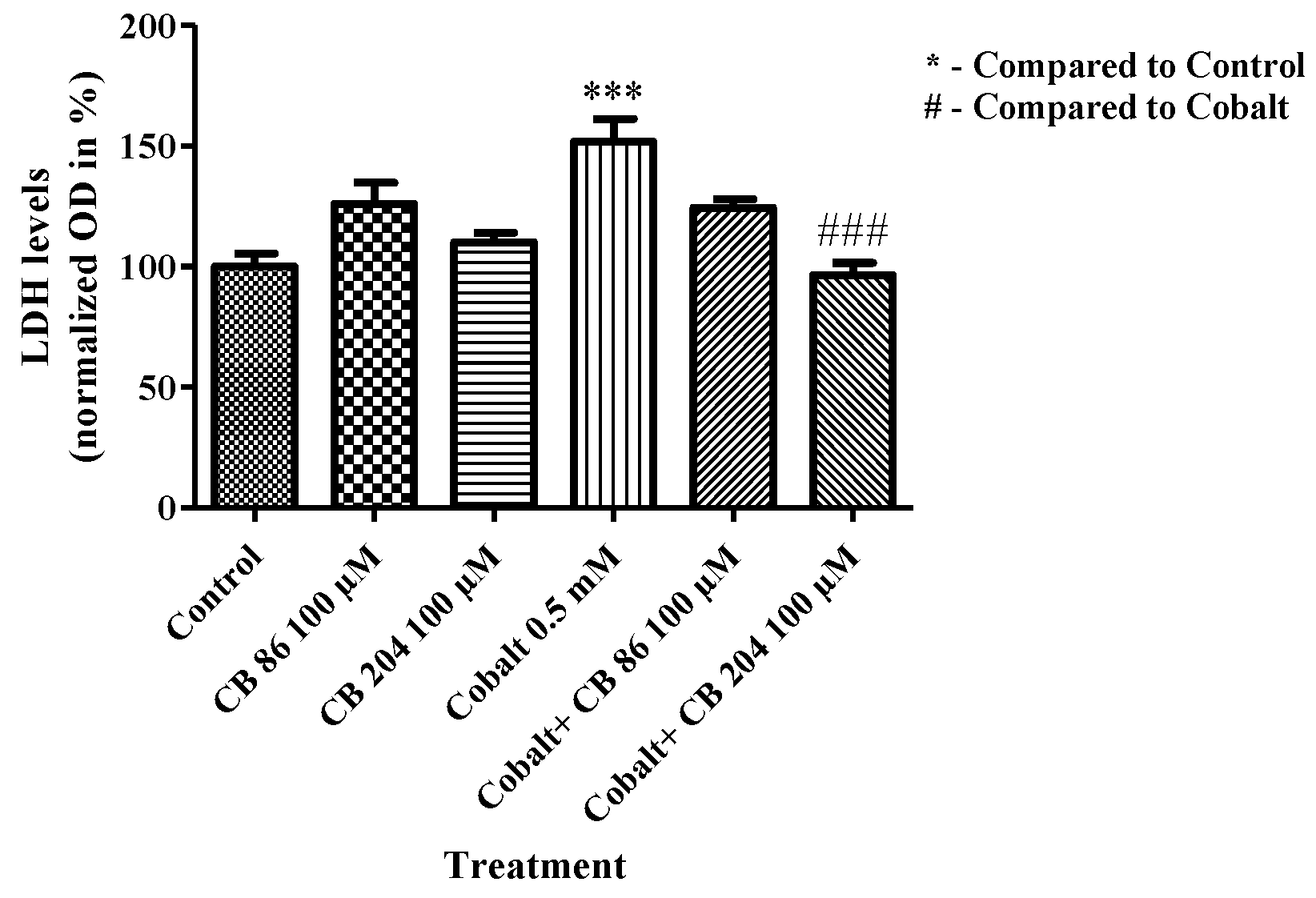
3.6. LDH Assay of Cell Death
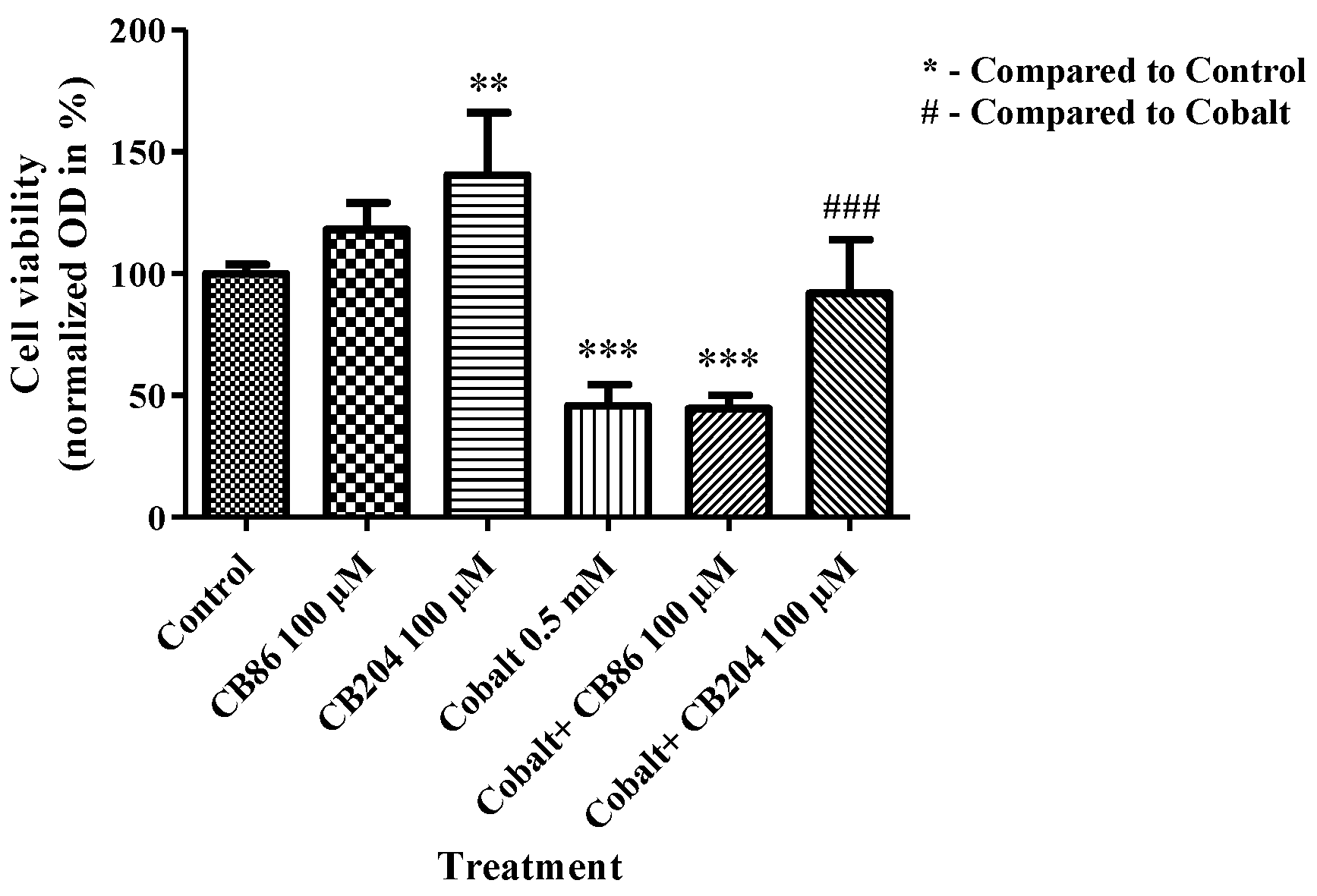
3.7. XTT Assay of Cell Viability
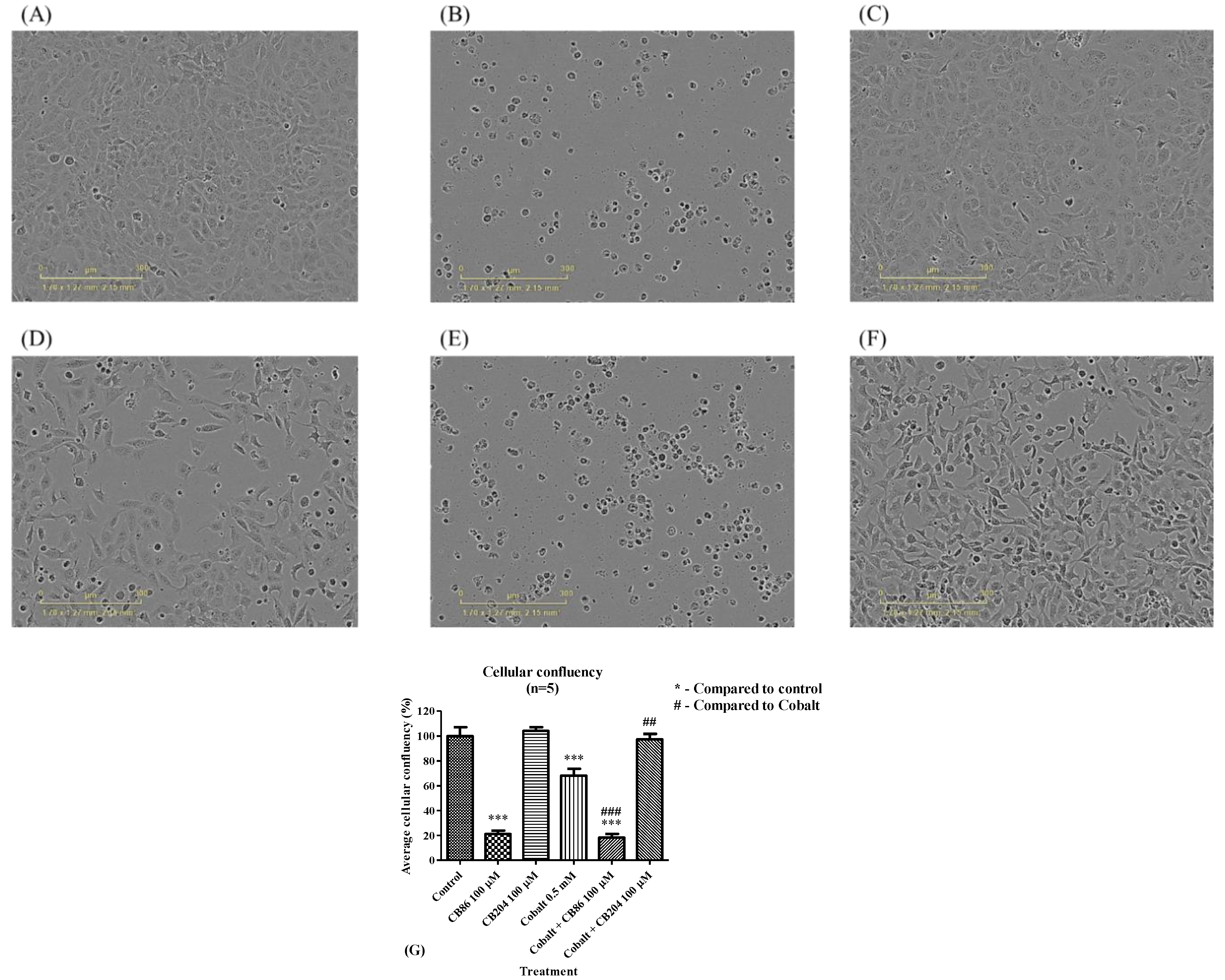
3.8. Cellular Density
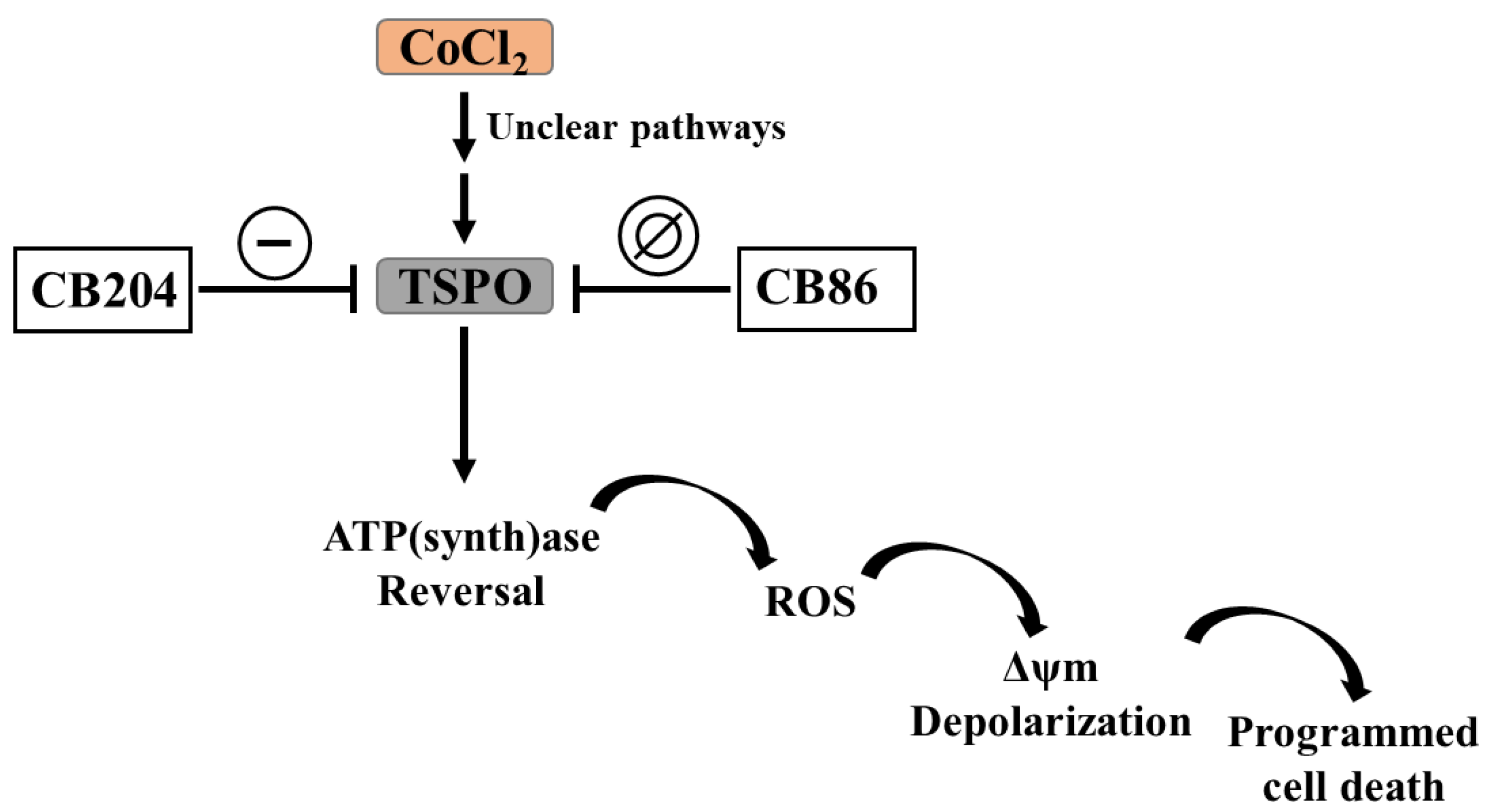
3.9. Summary of the Collected Data
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Tuder, R.M.; Yun, J.H.; Bhunia, A.; Fijalkowska, I. Hypoxia and chronic lung disease. J. Mol. Med. 2007, 85, 1317–1324. [Google Scholar] [CrossRef]
- Bensaid, S.; Fabre, C.; Fourneau, J.; Cieniewski-Bernard, C. Impact of different methods of induction of cellular hypoxia: Focus on protein homeostasis signaling pathways and morphology of C2C12 skeletal muscle cells differentiated into myotubes. J. Physiol. Biochem. 2019, 75, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fang, F.; Yang, Y.; Chen, J.; Xu, G.; Xu, Y.; Gao, Y. Brahma-related gene 1 (Brg1) epigenetically regulates CAM activation during hypoxic pulmonary hypertension. Cardiovasc. Res. 2013, 100, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yang, Y.; Cheng, X.; Fang, F.; Xu, G.; Yuan, Z.; Fang, M. Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 2015, 65, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Serocki, M.; Dobrucki, L.W.; Collawn, J.F.; Bartoszewski, R. eNOS expression and NO release during hypoxia is inhibited by miR-200b in human endothelial cells. Angiogenesis 2018, 21, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Sunamura, S. Protective Roles of Endothelial AMP-Activated Protein Kinase Against Hypoxia-Induced Pulmonary Hypertension in Mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372. [Google Scholar]
- Ferrero, E.; Fulgenzi, A.; Belloni, D.; Foglieni, C.; Ferrero, M.E. Cellfood improves respiratory metabolism of endothelial cells and inhibits hypoxia-induced reactive oxygen species (ros) generation. J. Physiol. Pharmacol. 2011, 62, 287–293. [Google Scholar]
- Debigaré, R.; Marquis, K.; Côté, C.H.; Tremblay, R.R.; Michaud, A.; LeBlanc, P.; Maltais, F. Catabolic/anabolic balance and muscle wasting in patients with COPD. Chest 2003, 124, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langen, R.C.J.; Gosker, H.R.; Remels, A.H.V.; Schols, A.M.W.J. Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int. J. Biochem. Cell Biol. 2013, 45, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.A.; Schneider, T.J. Similarities between the Oxygen-Sensing Mechanisms Regulating the Expression of Vascular Endothelial Growth-Factor and Erythropoietin. J. Biol. Chem. 1994, 269, 4355–4359. [Google Scholar] [PubMed]
- Griguer, C.E.; Oliva, C.R.; Kelley, E.E.; Giles, G.I.; Lancaster, J.R.; Gillespie, G.Y. Xanthine oxidase-dependent regulation of hypoxia-inducible factor in cancer cells. Cancer Res. 2006, 66, 2257–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, G.K.; Shi, X. Signaling by carcinogenic metals and metal-induced reactive oxygen species. Mutat. Res. 2003, 533, 183–200. [Google Scholar] [CrossRef]
- Hervouet, E.; Pecina, P.; Demont, J.; Vojtíšková, A.; Simonnet, H.; Houštěk, J.; Godinot, C. Inhibition of cytochrome c oxidase subunit 4 precursor processing by the hypoxia mimic cobalt chloride. Biochem. Biophys. Res. Commun. 2006, 344, 1086–1093. [Google Scholar] [CrossRef]
- Leonard, S.S.; Harris, G.K.; Shi, X. Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med. 2004, 37, 1921–1942. [Google Scholar] [CrossRef]
- Santore, M.T.; McClintock, D.S.; Lee, V.Y.; Budinger, G.S.; Chandel, N.S. Anoxia-induced apoptosis occurs through a mitochondria-dependent pathway in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L727–L734. [Google Scholar] [CrossRef] [Green Version]
- Vijayasarathy, C.; Damle, S.; Lenka, N.; Avadhani, N.G. Tissue variant effects of heme inhibitors on the mouse cytochrome c oxidase gene expression and catalytic activity of the enzyme complex. Eur. J. Biochem. 1999, 266, 191–200. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Chen, S.; Shen, B.; Yu, F.; Zhang, Y.; Shen, R. Apocynin Attenuates Cobalt Chloride-Induced Pheochromocytoma Cell Apoptosis by Inhibiting P38-MAPK/Caspase-3 Pathway. Cell. Physiol. Biochem. 2018, 48, 208–214. [Google Scholar] [CrossRef]
- Padmanaban, G.; Sarma, P.S. Cobalt toxicity and iron metabolism in Neurospora crassa. Biochem. J. 1966, 98, 330–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeno, S.; Zaaroor, M.; Leschiner, S.; Veenman, L.; Gavish, M. CoCl(2) induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry 2009, 48, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Zeno, S.; Veenman, L.; Katz, Y.; Bode, J.; Gavish, M.; Zaaroor, M. The 18 kDa mitochondrial translocator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygen species (ROS). Curr. Mol. Med. 2012, 12, 494–501. [Google Scholar] [PubMed]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Kugler, W.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Lakomek, M.; Gavish, M. Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell. Oncol. 2008, 30, 435–450. [Google Scholar] [PubMed]
- Levin, E.; Premkumar, A.; Veenman, L.; Kugler, W.; Leschiner, S.; Spanier, I.; Pasternak, G.W. The peripheral-type benzodiazepine receptor and tumorigenicity: Isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry 2005, 44, 9924–9935. [Google Scholar] [CrossRef]
- Shoukrun, R.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Karry, R.; Gavish, M. The 18-kDa translocator protein, formerly known as the peripheral-type benzodiazepine receptor, confers proapoptotic and antineoplastic effects in a human colorectal cancer cell line. Pharm. Genom. 2008, 18, 977–988. [Google Scholar] [CrossRef]
- Veenman, L.; Levin, E.; Weisinger, G.; Leschiner, S.; Spanier, I.; Snyder, S.H.; Gavish, M. Peripheral-type benzodiazepine receptor density and in vitro tumorigenicity of glioma cell lines. Biochem. Pharmacol. 2004, 68, 689–698. [Google Scholar] [CrossRef]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef]
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.J.; Lindemann, P.; Gavish, M. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Gavish, M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther. 2006, 110, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Denora, N.; Laquintana, V.; Pisu, M.G.; Dore, R.; Murru, L.; Latrofa, A.; Sanna, E. 2-Phenyl-imidazo[1,2-a]pyridine Compounds Containing Hydrophilic Groups as Potent and Selective Ligands for Peripheral Benzodiazepine Receptors: Synthesis, Binding Affinity and Electrophysiological Studies. J. Med. Chem. 2008, 51, 6876–6888. [Google Scholar] [CrossRef] [PubMed]
- Azrad, M.; Zeineh, N.; Weizman, A.; Veenman, L.; Gavish, M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int. J. Mol. Sci. 2019, 20, 594. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Veenman, L.; Shterenberg, A.; Singh, S.; Masarwa, A.; Dutta, B.; Maniv, I. Quinazoline-based tricyclic compounds that regulate programmed cell death, induce neuronal differentiation, and are curative in animal models for excitotoxicity and hereditary brain disease. Cell Death Discov. 2015, 1, 1–17. [Google Scholar] [CrossRef]
- Veenman, L.; Vainshtein, A.; Yasin, N.; Azrad, M.; Gavish, M. Tetrapyrroles as Endogenous TSPO Ligands in Eukaryotes and Prokaryotes: Comparisons with Synthetic Ligands. Int. J. Mol. Sci. 2016, 17, 880. [Google Scholar] [CrossRef]
- Chen, Y.; Veenman, L.; Singh, S.; Ouyang, F.; Liang, J.; Huang, W.; Gavish, M. 2-Cl-MGV-1 Ameliorates Apoptosis in the Thalamus and Hippocampus and Cognitive Deficits After Cortical Infarct in Rats. Stroke 2017, 48, 3366–3374. [Google Scholar] [CrossRef]
- Shehadeh, M.; Palzur, E.; Apel, L.; Soustiel, J.F. Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. Int. J. Mol. Sci. 2019, 20, 2639. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef]
- Caballero, B.; Veenman, L.; Bode, J.; Leschiner, S.; Gavish, M. Concentration-dependent bimodal effect of specific 18 kDa translocator protein (TSPO) ligands on cell death processes induced by ammonium chloride: Potential implications for neuropathological effects due to hyperammonemia. CNS Neurol. Disord. Drug Targets 2014, 13, 574–592. [Google Scholar] [CrossRef]
- Zeineh, N.; Nagler, R.; Gabay, M.; Weizman, A.; Gavish, M. Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes. Cells 2019, 8, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcar, A.; Menze, M.A.; Toner, M.; Hand, S.C. Metabolic preconditioning of mammalian cells: Mimetic agents for hypoxia lack fidelity in promoting phosphorylation of pyruvate dehydrogenase. Cell Tissue Res. 2013, 351, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Rajagopal, V.; Gonsalves, C.; Johnson, C.; Kalra, V.K. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. 2006, 177, 7211–7224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.M.; Kardashyan, L.; Callaway, J.K.; Lee, E.M.; Beart, P.M. Long-term functional and protective actions of preconditioning with hypoxia, cobalt chloride, and desferrioxamine against hypoxic-ischemic injury in neonatal rats. Pediatr. Res. 2008, 63, 620–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.Y.; Roh, K.H.; Jeong, Y.J.; Kim, S.H.; Lee, E.J.; Kim, M.S.; Kim, W.J. Estradiol protects PC12 cells against CoCl2-induced apoptosis. Brain Res. Bull. 2008, 76, 579–585. [Google Scholar] [CrossRef]
- Wu, K.C.; Cheng, K.S.; Wang, Y.W.; Chen, Y.F.; Wong, K.L.; Su, T.H.; Leung, Y.M. Perturbation of Akt Signaling, Mitochondrial Potential, and ADP/ATP Ratio in Acidosis-Challenged Rat Cortical Astrocytes. J. Cell Biochem. 2017, 118, 1108–1117. [Google Scholar] [CrossRef]
- Weiner, D.; Levy, Y.; Khankin, E.V.; Reznick, A.Z. Inhibition of salivary amylase activity by cigarette smoke aldehydes. J. Physiol. Pharmacol. 2008, 59 (Suppl. S6), 727–737. [Google Scholar]
- Le Fur, G.; Vaucher, N.; Perrier, M.L.; Flamier, A.; Benavides, J.; Renault, C.; Uzan, A. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983, 33, 449–457. [Google Scholar] [CrossRef]
- Marginedas-Freixa, I.; Alvarez, C.L.; Moras, M.; Hattab, C.; Bouyer, G.; Chene, A.; Ostuni, M.A. Induction of ATP Release, PPIX Transport, and Cholesterol Uptake by Human Red Blood Cells Using a New Family of TSPO Ligands. Int. J. Mol. Sci. 2018, 19, 3098. [Google Scholar] [CrossRef] [Green Version]
- Canat, X.; Carayon, P.; Bouaboula, M.; Cahard, D.; Shire, D.; Roque, C.; Casellas, P. Distribution profile and properties of peripheral-type benzodiazepine receptors on human hemopoietic cells. Life Sci. 1993, 52, 107–118. [Google Scholar] [CrossRef]
- Olson, J.M.; Ciliax, B.J.; Mancini, W.R.; Young, A.B. Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur. J. Pharmacol. 1988, 152, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Ostuni, M.A.; Ducroc, R.; Peranzi, G.; Tonon, M.C.; Papadopoulos, V.; Lacapere, J.J. Translocator protein (18 kDa) ligand PK 11195 induces transient mitochondrial Ca2+ release leading to transepithelial Cl- secretion in HT-29 human colon cancer cells. Biol. Cell 2007, 99, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.; Gavish, M. Species differences and heterogeneity of solubilized peripheral-type benzodiazepine binding sites. Biochem. Pharmacol. 1989, 38, 3843–3849. [Google Scholar] [CrossRef]
- Kanegawa, N.; Collste, K.; Forsberg, A.; Schain, M.; Arakawa, R.; Jucaite, A.; Halldin, C. In vivo evidence of a functional association between immune cells in blood and brain in healthy human subjects. Brain Behav. Immun. 2016, 54, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, M.A.R.T.I.N.E.; Dimchev, A.B.; Boujrad, N.O.U.R.E.D.D.I.N.E.; Price, J.M.; Musto, N.A.; Papadopoulos, V.A.S.S.I.L.I.O.S. In vitro reconstitution of a functional peripheral-type benzodiazepine receptor from mouse Leydig tumor cells. Mol. Pharmacol. 1994, 45, 201–211. [Google Scholar]
- Veenman, L.; Leschiner, S.; Spanier, I.; Weisinger, G.; Weizman, A.; Gavish, M. PK 11195 attenuates kainic acid-induced seizures and alterations in peripheral-type benzodiazepine receptor (PBR) protein components in the rat brain. J. Neurochem. 2002, 80, 917–927. [Google Scholar] [CrossRef]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Lacapere, J.J. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Caballero, B.; Veenman, L.; Gavish, M. Role of mitochondrial translocator protein (18 kDa) on mitochondrial- related cell death processes. Recent Pat. Endocr. Metab. Immune Drug Discov. 2013, 7, 86–101. [Google Scholar] [CrossRef]
- Veenman, L.; Alten, J.; Linnemannstöns, K.; Shandalov, Y.; Zeno, S.; Lakomek, M.; Kugler, W. Potential involvement of F0F1-ATP(synth)ase and reactive oxygen species in apoptosis induction by the antineoplastic agent erucylphosphohomocholine in glioblastoma cell lines: A mechanism for induction of apoptosis via the 18 kDa mitochondrial translocator protein. Apoptosis 2010, 15, 753–768. [Google Scholar]
- Veenman, L.; Gavish, M.; Kugler, W. Apoptosis induction by erucylphosphohomocholine via the 18 kDa mitochondrial translocator protein: Implications for cancer treatment. Anticancer Agents Med. Chem. 2014, 14, 559–577. [Google Scholar] [CrossRef]
- Yasin, N.; Veenman, L.; Singh, S.; Azrad, M.; Bode, J.; Vainshtein, A.; Gavish, M. Classical and Novel TSPO Ligands for the Mitochondrial TSPO Can Modulate Nuclear Gene Expression: Implications for Mitochondrial Retrograde Signaling. Int. J. Mol. Sci. 2017, 18, 786. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Yao, J.; Hiramatsu, N.; Hayakawa, K.; Araki, I.; Takeda, M.; Kitamura, M. Involvement of hypoxia-triggered endoplasmic reticulum stress in outlet obstruction-induced apoptosis in the urinary bladder. Lab. Invest. 2008, 88, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, B.; Yang, W.W.; Yang, H.T. Expression pattern of E2F6 in physical and chemical hypoxia-induced apoptosis. Sheng Li Xue Bao 2008, 60, 1–10. [Google Scholar] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Name | CB86 | CB194 | CB199 | CB204 | Alpidem |
|---|---|---|---|---|---|
| Molecular weight | 384 | 484 | 470 | 498 | 404 |
| TSPO Ki (nM) | 1.6 | 285.3 | 193.1 | 117.7 | 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeineh, N.; Denora, N.; Laquintana, V.; Franco, M.; Weizman, A.; Gavish, M. Efficaciousness of Low Affinity Compared to High Affinity TSPO Ligands in the Inhibition of Hypoxic Mitochondrial Cellular Damage Induced by Cobalt Chloride in Human Lung H1299 Cells. Biomedicines 2020, 8, 106. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8050106
Zeineh N, Denora N, Laquintana V, Franco M, Weizman A, Gavish M. Efficaciousness of Low Affinity Compared to High Affinity TSPO Ligands in the Inhibition of Hypoxic Mitochondrial Cellular Damage Induced by Cobalt Chloride in Human Lung H1299 Cells. Biomedicines. 2020; 8(5):106. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8050106
Chicago/Turabian StyleZeineh, Nidal, Nunzio Denora, Valentino Laquintana, Massimo Franco, Abraham Weizman, and Moshe Gavish. 2020. "Efficaciousness of Low Affinity Compared to High Affinity TSPO Ligands in the Inhibition of Hypoxic Mitochondrial Cellular Damage Induced by Cobalt Chloride in Human Lung H1299 Cells" Biomedicines 8, no. 5: 106. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8050106