Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects
2.2. Isolation and Culture of Fibroblasts from Keloid Scar and Control Samples
2.3. DNA Extraction and Bisulfate Conversion
2.4. DNA Methylation Assay
2.5. Data Processing for the 450k Methylation Array
2.6. Differential Methylation Statistical Analysis
2.7. Enrichment and Pathway Analysis
2.8. Interaction Network Analysis
3. Results
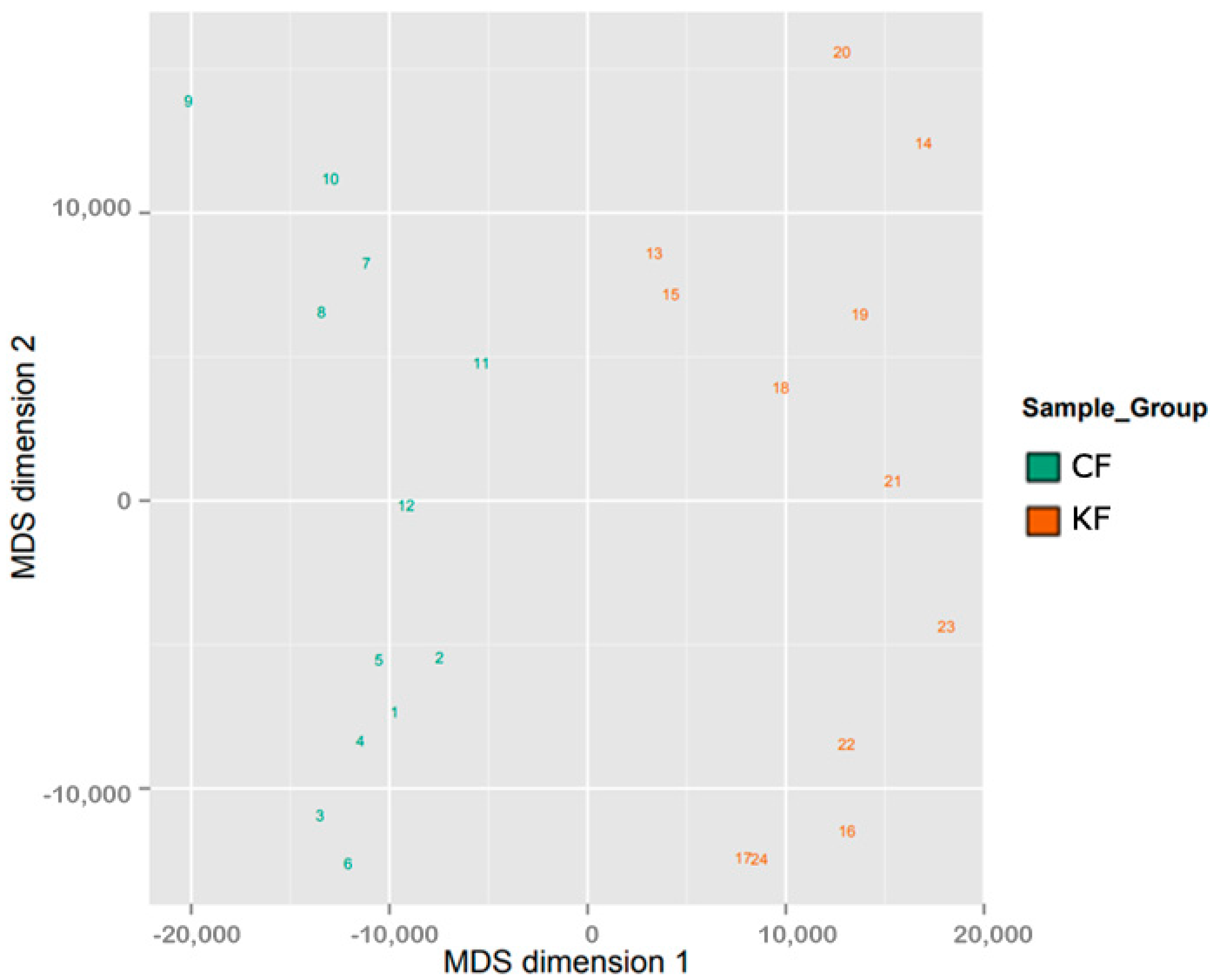
3.1. Samples Clustering
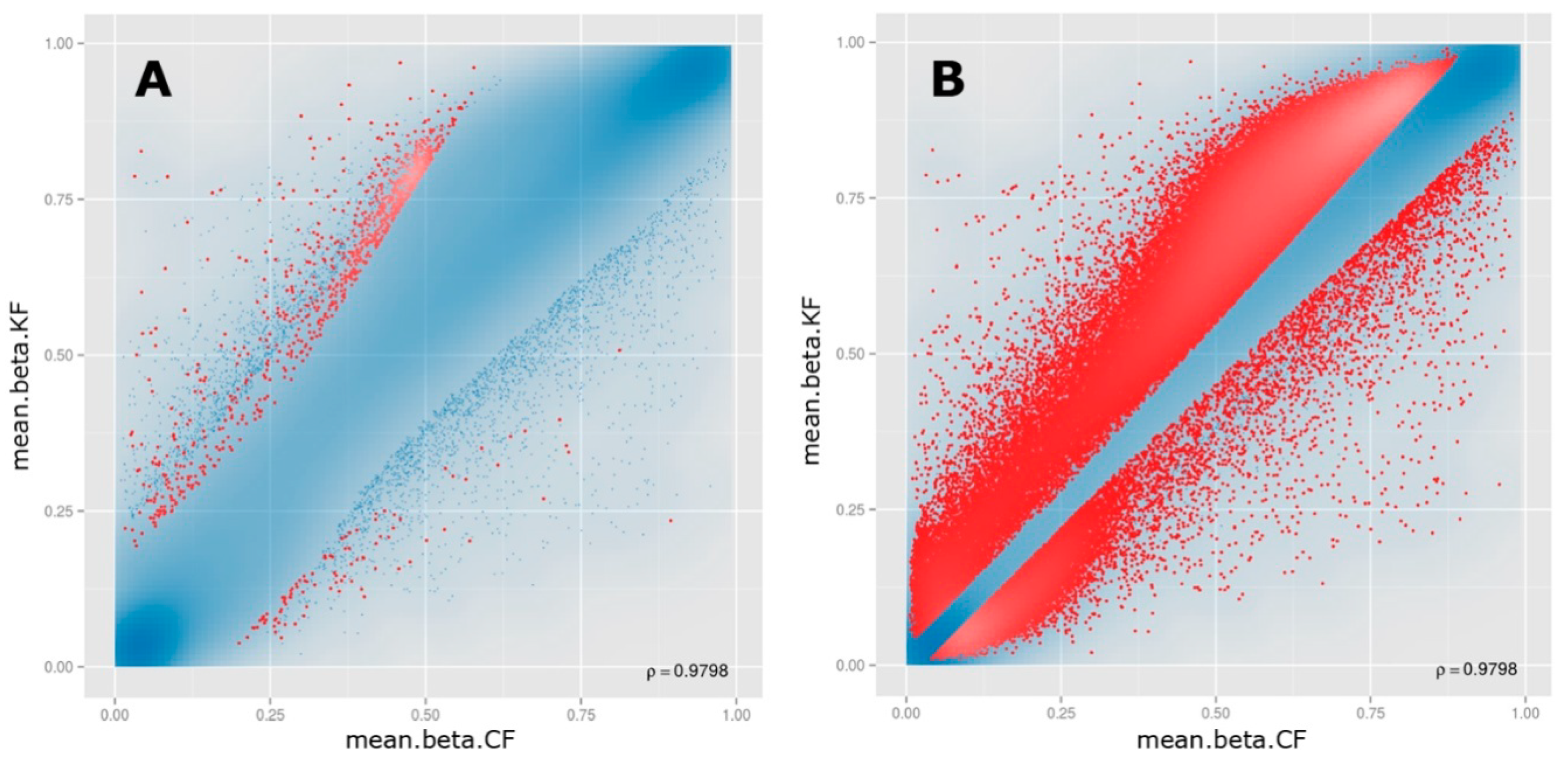
3.2. Differential Methylation of CpG Sites
3.3. Functional Enrichment Analysis
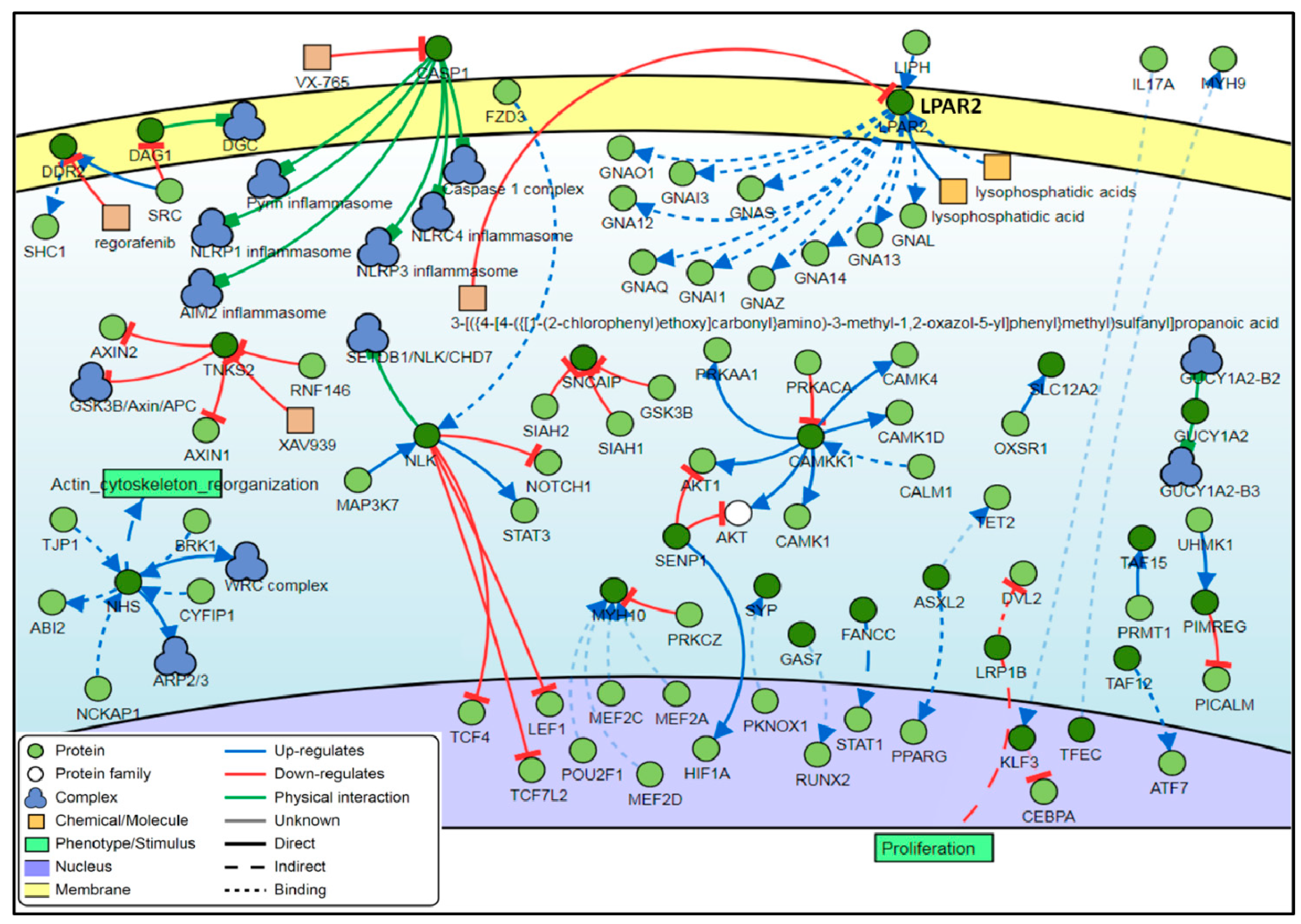
3.4. Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Gonzalez, A.C.D.O.; Andrade, Z.D.A.; Costa, T.F.; Medrado, A.R.A.P. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Potter, D.A.; Veitch, D.; Johnston, G.A. Scarring and wound healing. Br. J. Hosp. Med. 2019, 80, C166–C171. [Google Scholar] [CrossRef]
- Trace, A.P.; Enos, C.W.; Mantel, A.; Harvey, V.M. Keloids and hypertrophic scars: A Spectrum of clinical challenges. Am. J. Clin. Dermatol. 2016, 17, 201–223. [Google Scholar] [CrossRef]
- Safonov, I. Normotrophic scars. In Atlas of Scar Treatment and Correction; Safonov, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 161–188. [Google Scholar] [CrossRef]
- Shih, B.; Garside, E.; McGrouther, D.A.; Bayat, A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010, 18, 139–153. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [Green Version]
- Limandjaja, G.C.; van den Broek, L.J.; Waaijman, T.; van Veen, H.A.; Everts, V.; Monstrey, S.; Scheper, R.J.; Niessen, F.B.; Gibbs, S. Increased epidermal thickness and abnormal epidermal differentiation in keloid scars. Br. J. Dermatol. 2017, 176, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Lemonas, P.; Ahmad, I. Keloid scars: The hidden burden of disease. J. Pigment. Disord. 2015, 2, 1–5. [Google Scholar] [CrossRef]
- Zhang, G.; Guan, Q.Y.; Chen, G.; Qian, F.; Liang, J. DNA methylation of the CDC2L1 gene promoter region decreases the expression of the CDK11p58 protein and reduces apoptosis in keloid fibroblasts. Arch. Dermatol. Res. 2018, 310, 107–115. [Google Scholar] [CrossRef]
- Halim, A.S.; Emami, A.; Salahshourifar, I.; Kannan, T.P. Keloid scarring: Understanding the genetic basis, advances, and prospects. Arch. Plast. Surg. 2012, 39, 184–189. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Deng, Z.; Alghamdi, M.; Lu, L.; Fear, M.W.; He, L. From genetics to epigenetics: New insights into keloid scarring. Cell Prolif. 2017, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and hypertrophic scars: Pathophysiology, classification, and treatment. Dermatol. Surg. 2017, 43, S3–S18. [Google Scholar] [CrossRef]
- Mann, J.; Mann, D.A. Epigenetic regulation of wound healing and fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Neary, R.; Watson, C.J.; Baugh, J.A. Epigenetics and the overhealing wound: The role of DNA methylation in fibrosis. Fibrogenes Tissue Repair 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kader, F.; Ghai, M. DNA methylation-based variation between human populations. Mol. Genet. Genom. 2017, 292, 5–35. [Google Scholar] [CrossRef]
- Jones, L.R.; Young, W.; Divine, G.; Datta, I.; Chen, K.M.; Ozog, D.; Worsham, M.J. Genome-wide scan for methylation profiles in keloids. Dis. Markers 2015, 2015. [Google Scholar] [CrossRef]
- Jones, L.R.; Greene, J.; Chen, K.M.; Divine, G.; Chitale, D.; Shah, V.; Datta, I.; Worsham, M.J. Biological significance of genome-wide DNA methylation profiles in keloids. Laryngoscope 2017, 127, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Mardaryev, A.N. Epigenetic regulation of skin wound healing. In Epigenetic Regulation of Skin Development and Regeneration; Stem Cell Biology and Regenerative Medicine; Botchkarev, V., Millar, S., Eds.; Humana Press: Cham, Switzerland, 2018; pp. 293–314. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Russell, S.B.; Russell, J.D.; Trupin, K.M.; Gayden, A.E.; Opalenik, S.R.; Nanney, L.B.; Broquist, A.H.; Raju, L.; Williams, S.M. Epigenetically altered wound healing in keloid fibroblasts. J. Investig. Dermatol. 2010, 130, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Keira, S.M.; Ferreira, L.M.; Gragnani, A.; Duarte, I.d.S.; Santos, I.A.N. Experimental model for fibroblast culture. Acta Cir. Bras. 2004, 19, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Tucci-Viegas, V.M.; Hochman, B.; Frana, J.P.; Ferreira, L.M. Keloid explant culture: A model for keloid fibroblasts isolation and cultivation based on the biological differences of its specific regions. Int. Wound J. 2010, 7, 339–348. [Google Scholar] [CrossRef]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Licata, L.; Lo Surdo, P.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling network open resource 2.0: 2019 Update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef]
- Deng, C.C.; Zhu, D.H.; Chen, Y.J.; Huang, T.Y.; Peng, Y.; Liu, S.Y.; Lu, P.; Xue, Y.H.; Xu, Y.P.; Yang, B.; et al. TRAF4 promotes fibroblast proliferation in keloids by destabilizing p53 via interacting with the deubiquitinase USP10. J. Investig. Dermatol. 2019, 139, 1925–1935.e5. [Google Scholar] [CrossRef]
- Arno, A.I.; Gauglitz, G.G.; Barret, J.P.; Jeschke, M.G. Up-to-date approach to manage keloids and hypertrophic scars: A useful guide. Burns 2014, 40, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Coppola, M.M.; Salzillo, R.; Segreto, F.; Persichetti, P. Triamcinolone acetonide intralesional injection for the treatment of keloid scars: Patient selection and perspectives. Clin. Cosmet. Investig. Dermatol. 2018, 11, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, P.L.; Rea, S.M.; Wood, F.M.; Fear, M.W.; Viola, H.M.; Hool, L.C.; Gankande, T.U.; Alghamdi, M.; Stevenson, A.W.; Manzur, M.; et al. Verapamil is less effective than triamcinolone for prevention of keloid scar recurrence after excision in a randomized controlled trial. Acta Derm. Venereol. 2016, 96, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Robles, D.T.; Moore, E.; Draznin, M.; Berg, D. Keloids: Pathophysiology and management. Dermatol. Online J. 2007, 13, 9. [Google Scholar]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.N. Gene expression, epigenetic regulation, and cancer. In Epigenetic Advancements in Cancer; Mishra, M., Bishnupuri, K., Eds.; Springer: Cham, Switzerland, 2016; pp. 79–96. [Google Scholar] [CrossRef]
- Robinson, C.M.; Watson, C.J.; Baugh, J.A. Epigenetics within the matrix, a neo-regulator of fibrotic disease. Epigenetics 2012, 7, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Dowson, C.; O’Reilly, S. DNA methylation in fibrosis. Eur. J. Cell Biol. 2016, 95, 323–330. [Google Scholar] [CrossRef]
- Al-Eitan, L.N.; Alghamdi, M.A.; Tarkhan, A.H.; Al-Qarqaz, F.A. Genome-wide tiling array analysis of HPV-induced warts reveals aberrant methylation of protein-coding and non-coding regions. Genes 2020, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Al-Eitan, L.N.; Alghamdi, M.A.; Tarkhan, A.H.; Al-Qarqaz, F.A. Epigenome-wide analysis of common warts reveals aberrant promoter methylation. Int. J. Med. Sci. 2020, 17, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Al-Eitan, L.N.; Alghamdi, M.A.; Tarkhan, A.H.; Al-Qarqaz, F.A. Genome-wide CpG island methylation profiles of cutaneous skin with and without HPV infection. Int. J. Mol. Sci. 2019, 20, 4822. [Google Scholar] [CrossRef] [Green Version]
- Ti, D.; Li, M.; Fu, X.; Han, W. Causes and consequences of epigenetic regulation in wound healing. Wound Repair Regen. 2014, 22, 305–312. [Google Scholar] [CrossRef]
- Antequera, F.; Bird, A. CpG islands: A historical perspective. In CpG Islands. Methods in Molecular Biology; Vavouri, T., Peinado, M., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1766, pp. 3–13. [Google Scholar] [CrossRef]
- Lea, A.J.; Vockley, C.M.; Johnston, R.A.; Del Carpio, C.A.; Barreiro, L.B.; Reddy, T.E.; Tung, J. Genome-wide quantification of the effects of DNA methylation on human gene regulation. Elife 2018, 7, 1–27. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA hypermethylation associated with upregulated gene expression in prostate cancer demonstrates the diversity of epigenetic regulation. BMC Med. Genom. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Dedeurwaerder, S.; Fumagalli, D.; Fuks, F. Unravelling the epigenomic dimension of breast cancers. Curr. Opin. Oncol. 2011, 23, 559–565. [Google Scholar] [CrossRef]
- Magalhães, M.; Tost, J.; Pineau, F.; Rivals, I.; Busato, F.; Alary, N.; Mely, L.; Leroy, S.; Murris, M.; Caimmi, D.; et al. Dynamic changes of DNA methylation and lung disease in cystic fibrosis: Lessons from a monogenic disease. Epigenomics 2018, 10, 1131–1145. [Google Scholar] [CrossRef]
- Moore, K.; McKnight, A.J.; Craig, D.; O’Neill, F. Epigenome-wide association study for Parkinson’s disease. NeuroMolecular Med. 2014, 16, 845–855. [Google Scholar] [CrossRef]
- Farkas, S.A.; Milutin-Gašperov, N.; Grce, M.; Nilsson, T.K. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 2013, 8, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Ashktorab, H.; Daremipouran, M.; Goel, A.; Varma, S.; Leavitt, R.; Sun, X.; Brim, H. DNA methylome profiling identifies novel methylated genes in African American patients with colorectal neoplasia. Epigenetics 2014, 9, 503–512. [Google Scholar] [CrossRef]
- Ashktorab, H.; Rahi, H.; Daremiporan, M.; Lee, E.L.; Frederick, W.A.; Laiyemo, A.O.; Nouraie, M.; Brim, H. Sa1989 novel genes mutation and methylation targets in colon cancer using whole exome sequencing. Gastroenterology 2013, 144, S-353. [Google Scholar] [CrossRef]
- Morán, A.; Fernández-Marcelo, T.; Carro, J.; De Juan, C.; Pascua, I.; Head, J.; Gómez, A.; Hernando, F.; Torres, A.J.; Benito, M.; et al. Methylation profiling in non-small cell lung cancer: Clinical implications. Int. J. Oncol. 2012, 40, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, M.P.; Desai, A.; Palakal, M.J. Systems biology approach to stage-wise characterization of epigenetic genes in lung adenocarcinoma. BMC Syst. Biol. 2013, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Angulo, J.C.; Andrés, G.; Ashour, N.; Sánchez-Chapado, M.; López, J.I.; Ropero, S. Development of castration resistant prostate cancer can be predicted by a DNA hypermethylation profile. J. Urol. 2016, 195, 619–626. [Google Scholar] [CrossRef]
- Kim, Y.H.; Lee, H.C.; Kim, S.Y.; Yeom, Y.I.; Ryu, K.J.; Min, B.H.; Kim, D.H.; Son, H.J.; Rhee, P.L.; Kim, J.J.; et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann. Surg. Oncol. 2011, 18, 2338–2347. [Google Scholar] [CrossRef]
- How-Kit, A.; Dejeux, E.; Dousset, B.; Renault, V.; Baudry, M.; Terris, B.; Tost, J. DNA methylation profiles distinguish different subtypes of gastroenteropancreatic neuroendocrine tumors. Epigenomics 2015, 7, 1245–1258. [Google Scholar] [CrossRef]
- Almén, M.S.; Jacobsson, J.A.; Moschonis, G.; Benedict, C.; Chrousos, G.P.; Fredriksson, R.; Schiöth, H.B. Genome wide analysis reveals association of a FTO gene variant with epigenetic changes. Genomics 2012, 99, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Lacunza, E.; Canzoneri, R.; Rabassa, M.E.; Zwenger, A.; Segal-Eiras, A.; Croce, M.V.; Abba, M.C. RHBDD2: A 5-fluorouracil responsive gene overexpressed in the advanced stages of colorectal cancer. Tumor Biol. 2012, 33, 2393–2399. [Google Scholar] [CrossRef]
- Abba, M.C.; Lacunza, E.; Nunez, M.I.; Colussi, A.; Isla-Larrain, M.; Segal-Eiras, A.; Croce, M.V.; Aldaz, C.M. Rhomboid domain containing 2 (RHBDD2): A novel cancer-related gene over-expressed in breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Canzoneri, R.; Lacunza, E.; Isla Larrain, M.; Croce, M.V.; Abba, M.C. Rhomboid family gene expression profiling in breast normal tissue and tumor samples. Tumor Biol. 2014, 35, 1451–1458. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, K.; Lin, Z.; Bai, S.; Wu, J.; Li, P.; Xue, H.; Du, J.; Shen, B.; Wang, H.; et al. Identification of susceptibility gene mutations associated with the pathogenesis of familial nonmedullary thyroid cancer. Mol. Genet. Genom. Med. 2019, 7, 1–7. [Google Scholar] [CrossRef]
- Roos, L.; Sandling, J.K.; Bell, C.G.; Glass, D.; Mangino, M.; Spector, T.D.; Deloukas, P.; Bataille, V.; Bell, J.T. Higher nevus count exhibits a distinct DNA methylation signature in healthy human skin: Implications for melanoma. J. Investig. Dermatol. 2017, 137, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y.; Li, S.-Z.; Zhang, H.-H.; Wu, Q.-R.; Gong, J.; Liang, T.; Gao, L.; Xing, N.-N.; Liu, W.-B.; Du, R.-L.; et al. Stabilization of ATF5 by TAK1–nemo-like kinase critically regulates the interleukin-1β-stimulated C/EBP signaling pathway. Mol. Cell. Biol. 2015, 35, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Ishitani, T.; Hirao, T.; Suzuki, M.; Isoda, M.; Ishitani, S.; Harigaya, K.; Kitagawa, M.; Matsumoto, K.; Itoh, M. Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 2010, 12, 278–285. [Google Scholar] [CrossRef]
- Zhang, B.; Li, K.Y.; Chen, H.Y.; Pan, S.D.; Chen, S.F.; Zhang, W.F.; Xia, C.P.; Jiang, L.C.; Liu, X.B.; Zhao, F.J.; et al. Lentivirus-based RNA silencing of nemo-like kinase (NLK) inhibits the CAL 27 human adenosquamos carcinoma cells proliferation and blocks G0/G1 phase to S phase. Int. J. Med. Sci. 2013, 10, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.R.; Guo, N.; Zhao, J.P.; Liu, P.D.; Feng, H.H.; Li, Y. Inhibition of nemo-like kinase increases taxol sensitivity in laryngeal cancer. Asian Pac. J. Cancer Prev. 2013, 14, 7137–7141. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Xu, L.; Tang, Z.; Zhang, W.; Wei, Y.; Ni, J.; Zhang, S.; Feng, J. Knockdown of Nemo-like kinase promotes metastasis in non-small-cell lung cancer. Oncol. Rep. 2019, 42, 1090–1100. [Google Scholar] [CrossRef]
- Suwei, D.; Liang, Z.; Zhimin, L.; Ruilei, L.; Yingying, Z.; Zhen, L.; Chunlei, G.; Zhangchao, L.; Yuanbo, X.; Jinyan, Y.; et al. NLK functions to maintain proliferation and stemness of NSCLC and is a target of metformin. J. Hematol. Oncol. 2015, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; He, J.; Du, Y.; Gao, X.H.; Liu, Y.; Liu, Q.Z.; Chang, W.J.; Cao, G.W.; Fu, C.G. Upregulation of nemo-like kinase is an independent prognostic factor in colorectal cancer. World J. Gastroenterol. 2015, 21, 8935–8942. [Google Scholar] [CrossRef]
- Kodiha, M.; Ho-Wo-Cheong, D.; Stochaj, U. Pharmacological AMP-kinase activators have compartment-specific effects on cell physiology. Am. J. Physiol. Cell Physiol. 2011, 301, 1307–1315. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M.; Schlattner, U. Bypassing AMPK phosphorylation. Chem. Biol. 2014, 21, 567–569. [Google Scholar] [CrossRef]
- Kitayama, J.; Shida, D.; Sako, A.; Ishikawa, M.; Hama, K.; Aoki, J.; Arai, H.; Nagawa, H. Over-expression of lysophosphatidic acid receptor-2 in human invasive ductal carcinoma. Breast Cancer Res. 2004, 6, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Radhika, V.; Ha, J.H.; Jayaraman, M.; Tsim, S.T.; Dhanasekaran, N. Mitogenic signaling by lysophosphatidic acid (LPA) involves Gα12. Oncogene 2005, 24, 4597–4603. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Fukushima, K.; Tanaka, K.; Minami, K.; Ishimoto, K.; Otagaki, S.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Involvement of LPA signaling via LPA receptor-2 in the promotion of malignant properties in osteosarcoma cells. Exp. Cell Res. 2018, 369, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lee, S.J.; Shim, H.; Chun, J.; Yun, C.C. The absence of LPA receptor 2 reduces the tumorigenesis by Apc Min mutation in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wang, D.; Iyer, S.; Ghaleb, A.M.; Shim, H.; Yang, V.W.; Chun, J.; Yun, C.C. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology 2009, 136, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; He, P.; Yun, C.C. Transgenic expression of human lysophosphatidic acid receptor LPA2 in mouse intestinal epithelial cells induces intestinal dysplasia. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.Y.J.; Zamudio, R.; Frost, J.H.; Almuedo, A.; Steinberg, H.; Clipman, S.J.; Duran, G.; Marcus, R.; Crawford, T.; Alyesh, D.; et al. Association of caspase-1 polymorphisms with chagas cardiomyopathy among individuals in Santa Cruz, Bolivia. Rev. Soc. Bras. Med. Trop. 2017, 50, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.N.; Brighton, L.E.; Mueller, L.; Xiang, Z.; Rager, J.E.; Fry, R.C.; Peden, D.B.; Jaspers, I. Influenza enhances caspase-1 in bronchial epithelial cells from asthmatic volunteers and is associated with pathogenesis. J. Allergy Clin. Immunol. 2012, 130, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Gong, Z.; Xing, S.; Xing, Q. Overexpression of caspase-1 in aorta of patients with coronary atherosclerosis. Heart Lung Circ. 2014, 23, 1070–1074. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef] [Green Version]
- Burdon, K.P.; McKay, J.D.; Sale, M.M.; Russell-Eggitt, I.M.; Mackey, D.A.; Wirth, M.G.; Elder, J.E.; Nicoll, A.; Clarke, M.P.; FitzGerald, L.M.; et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 2003, 73, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.P.; Coccia, M.; Tang, H.R.; Kanuga, N.; Machesky, L.M.; Bailly, M.; Cheetham, M.E.; Hardcastle, A.J. The Nance-Horan syndrome protein encodes a functional WAVE homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology. Hum. Mol. Genet. 2010, 19, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Shoshany, N.; Avni, I.; Morad, Y.; Weiner, C.; Einan-Lifshitz, A.; Pras, E. NHS gene mutations in Ashkenazi Jewish families with Nance–Horan syndrome. Curr. Eye Res. 2017, 42, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome sequencing of 18 Chinese families with congenital cataracts: A new sight of the NHS gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, L.; Sun, Z.; Yuan, Z.; Wu, S.; Sui, R. A novel small deletion in the NHS gene associated with Nance-Horan syndrome. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Sui, R.; Yao, F.; Wu, Z.; Zhang, X.; Zhang, S. Whole exome sequencing identified a novel truncation mutation in the NHS gene associated with Nance-Horan syndrome. BMC Med. Genet. 2019, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Felkl, M.; Leube, R.E. Interaction assays in yeast and cultured cells confirm known and identify novel partners of the synaptic vesicle protein synaptophysin. Neuroscience 2008, 156, 344–352. [Google Scholar] [CrossRef]
- Pawson, T.; Gish, G.D.; Nash, P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001, 11, 504–511. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.; Li, H.; Qian, Q.; Yang, L.; Glatt, S.J.; Faraone, S.V.; Wang, Y. Association between SYP with attention-deficit/hyperactivity disorder in Chinese Han subjects: Differences among subtypes and genders. Psychiatry Res. 2013, 210, 308–314. [Google Scholar] [CrossRef]
- Shen, Y.C.; Tsai, H.M.; Ruan, J.W.; Liao, Y.C.; Chen, S.F.; Chen, C.H. Genetic and functional analyses of the gene encoding synaptophysin in schizophrenia. Schizophr. Res. 2012, 137, 14–19. [Google Scholar] [CrossRef]
- Kuwano, N.; Kato, T.A.; Mitsuhashi, M.; Sato-Kasai, M.; Shimokawa, N.; Hayakawa, K.; Ohgidani, M.; Sagata, N.; Kubo, H.; Sakurai, T.; et al. Neuron-related blood inflammatory markers as an objective evaluation tool for major depressive disorder: An exploratory pilot case-control study. J. Affect. Disord. 2018, 240, 88–98. [Google Scholar] [CrossRef]
- Reilly, P.T.; Yu, Y.; Hamiche, A.; Wang, L. Cracking the ANP32 whips: Important functions, unequal requirement, and hints at disease implications. BioEssays 2014, 36, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Gursoy-Yuzugullu, O.; Ayrapetov, M.K.; Price, B.D. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 7507–7512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obri, A.; Ouararhni, K.; Papin, C.; Diebold, M.L.; Padmanabhan, K.; Marek, M.; Stoll, I.; Roy, L.; Reilly, P.T.; Mak, T.W.; et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 2014, 505, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Kadkol, S.S.; Brody, J.R.; Pevsner, J.; Bai, J.; Pasternack, G.R. Modulation of oncogenic potential by alternative gene use in human prostate cancer. Nat. Med. 1999, 5, 275–279. [Google Scholar] [CrossRef]
- Kadkol, S.S.; Naga, G.A.E.; Brody, J.R.; Bai, J.; Gusev, Y.; Dooley, W.C.; Pasternack, G.R. Expression of pp32 gene family members in breast cancer. Breast Cancer Res. Treat. 2001, 68, 65–73. [Google Scholar] [CrossRef]
- Xiong, Z.; Ye, L.; Zhenyu, H.; Li, F.; Xiong, Y.; Lin, C.; Wu, X.; Deng, G.; Shi, W.; Song, L.; et al. ANP32E induces tumorigenesis of triple-negative breast cancer cells by upregulating E2F1. Mol. Oncol. 2018, 12, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; Pena, L.D.; Kim, K.; Burton, B.; Hempel, M.; Schoch, K.; Walkiewicz, M.; McLaughlin, H.M.; Cho, M.; Stong, N.; et al. De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am. J. Hum. Genet. 2016, 99, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Izawa, T.; Rohatgi, N.; Fukunaga, T.; Wang, Q.T.; Silva, M.J.; Gardner, M.J.; McDaniel, M.L.; Abumrad, N.A.; Semenkovich, C.F.; Teitelbaum, S.L.; et al. ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep. 2015, 11, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Park, U.H.; Kang, M.R.; Kim, E.J.; Kwon, Y.S.; Hur, W.; Yoon, S.K.; Song, B.J.; Park, J.H.; Hwang, J.T.; Jeong, J.C.; et al. ASXL2 promotes proliferation of breast cancer cells by linking ERα to histone methylation. Oncogene 2016, 35, 3742–3752. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject ID 1 | Age | Gender 2 | Type of Tissue | Site of Tissue | Ethnicity |
|---|---|---|---|---|---|
| P 1 | 40 | F | keloid scar | shoulder | East Asian |
| P 2 | 38 | F | keloid scar | neck (thyroid) | Southeast European |
| P 3 | 29 | M | keloid scar | forearm | Hispanic |
| P 4 | 30 | M | keloid scar | sternum | Northwest European |
| P 5 | 53 | M | keloid scar | sternum | Northwest European |
| P 6 | 28 | M | keloid scar | upper arm | Northwest European |
| P 7 | 18 | F | keloid scar | shoulder | East Asian |
| P 8 | 42 | M | keloid scar | ear | Northwest European |
| P 9 | 30 | M | keloid scar | sternum | East Asian |
| P 10 | 21 | M | keloid scar | sternum | Northwest European and East Asian |
| P 11 | 47 | F | keloid scar | sternum | Northwest European |
| P 12 | 29 | M | keloid scar | sternum | East Asian |
| C 1 | 29 | M | normotrophic scar | forearm | South East Asian |
| normal skin | contralateral forearm | ||||
| C 2 | 25 | M | normotrophic scar | forearm | Caucasian |
| normal skin | contralateral forearm | ||||
| C 3 | 19 | M | normotrophic scar | forearm | Caucasian |
| normal skin | contralateral forearm | ||||
| C 4 | 25 | M | normotrophic scar | forearm | Caucasian |
| normal skin | contralateral forearm | ||||
| C 5 | 30 | M | normotrophic scar | forearm | Caucasian |
| normal skin | contralateral forearm | ||||
| C 6 | 19 | M | normotrophic scar | forearm | Caucasian |
| normal skin | contralateral forearm |
| Category 1 | Term | p-Value 2 | Genes |
|---|---|---|---|
| MF | GO:0042169~SH2 domain binding | 0.002 | SYP, NLK, DAG1 |
| BP | GO:0006355~regulation of transcription, DNA-templated | 0.005 | ASXL2, PKNOX2, ZNF718, BPTF, TFEC, NLK, SCML1, GAS7, BRD8, KLF3 |
| CC | GO:0005634~nucleus | 0.011 | ANP32C, NLK, ANP32E, SCML1, TKT, CAMKK1, TNKS2, PKNOX2, PSMA1, SERPINB9, ZNF718, FAM64A, SENP1, TAF15, BPTF, C19ORF66, USP36, FANCC, BRD8, MYH10, KLF3 |
| BP | GO:1903955~positive regulation of protein targeting to mitochondrion | 0.016 | HSPA1L, NBPF3, USP36 |
| CC | GO:0005654~nucleoplasm | 0.020 | ASXL2, NLK, FANK1, DAG1, TKT, HSPA1L, PSMA1, SENP1, BPTF, TAF15, TFEC, BRD8, FANCC |
| CC | GO:0000812~Swr1 complex | 0.021 | ANP32E, BRD8 |
| BP | GO:0043486~histone exchange | 0.025 | ANP32C, ANP32E |
| BP | GO:0071310~cellular response to organic substance | 0.038 | SYP, CASP1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghamdi, M.A.; Wallace, H.J.; Melton, P.E.; Moses, E.K.; Stevenson, A.; Al-Eitan, L.N.; Rea, S.; Duke, J.M.; Danielsen, P.L.; Prêle, C.M.; et al. Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars. Biomedicines 2020, 8, 181. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070181
Alghamdi MA, Wallace HJ, Melton PE, Moses EK, Stevenson A, Al-Eitan LN, Rea S, Duke JM, Danielsen PL, Prêle CM, et al. Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars. Biomedicines. 2020; 8(7):181. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070181
Chicago/Turabian StyleAlghamdi, Mansour A., Hilary J. Wallace, Phillip E. Melton, Eric K. Moses, Andrew Stevenson, Laith N. Al-Eitan, Suzanne Rea, Janine M. Duke, Patricia L. Danielsen, Cecilia M. Prêle, and et al. 2020. "Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars" Biomedicines 8, no. 7: 181. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070181