Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy

 ,
, 
Abstract
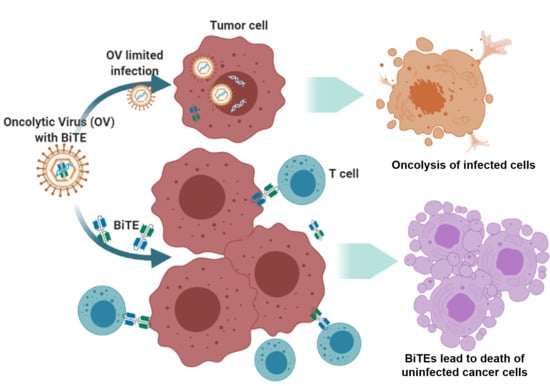
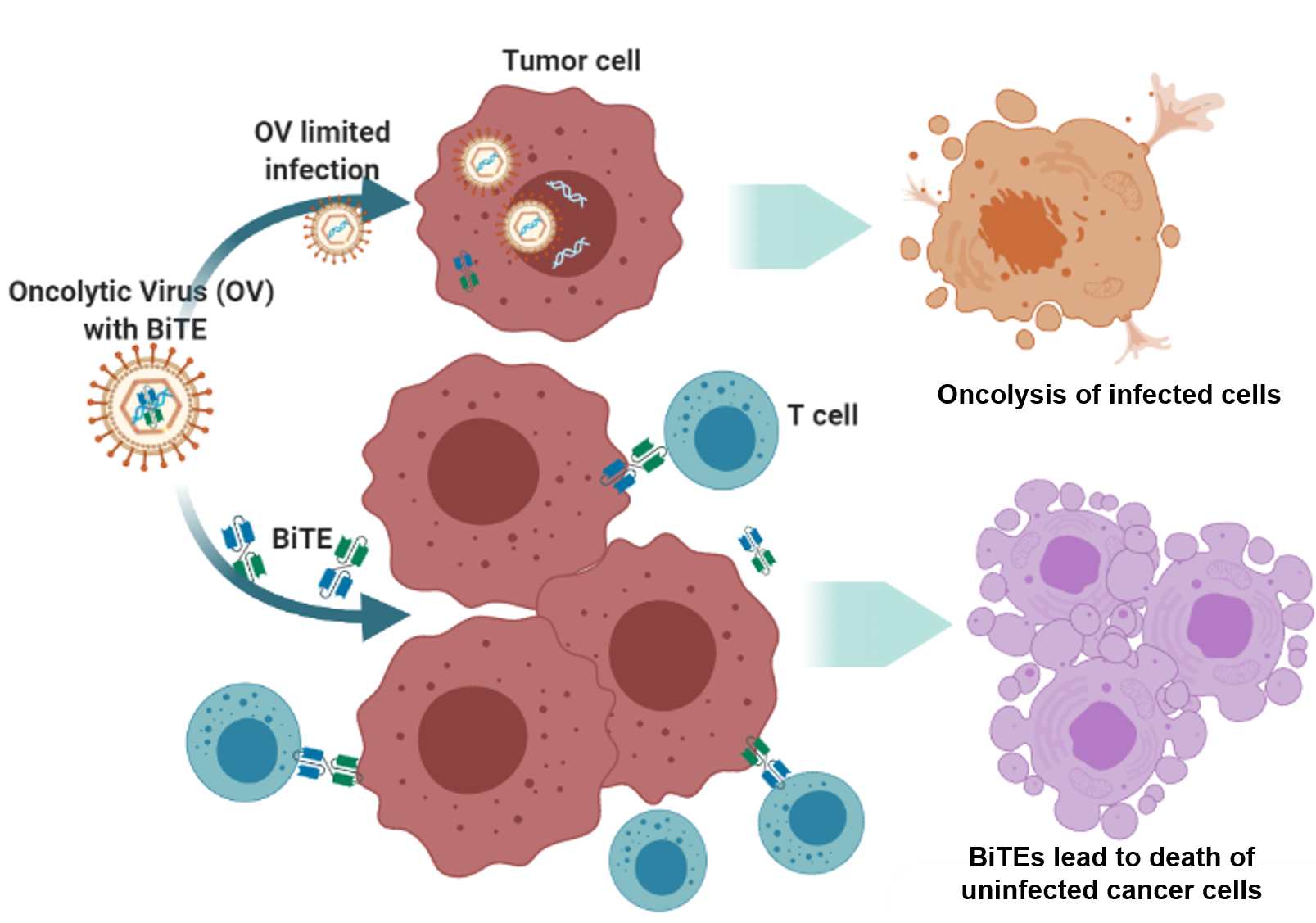
:
1. Introduction
2. Oncolytic Virus-Mediated Immunotherapy
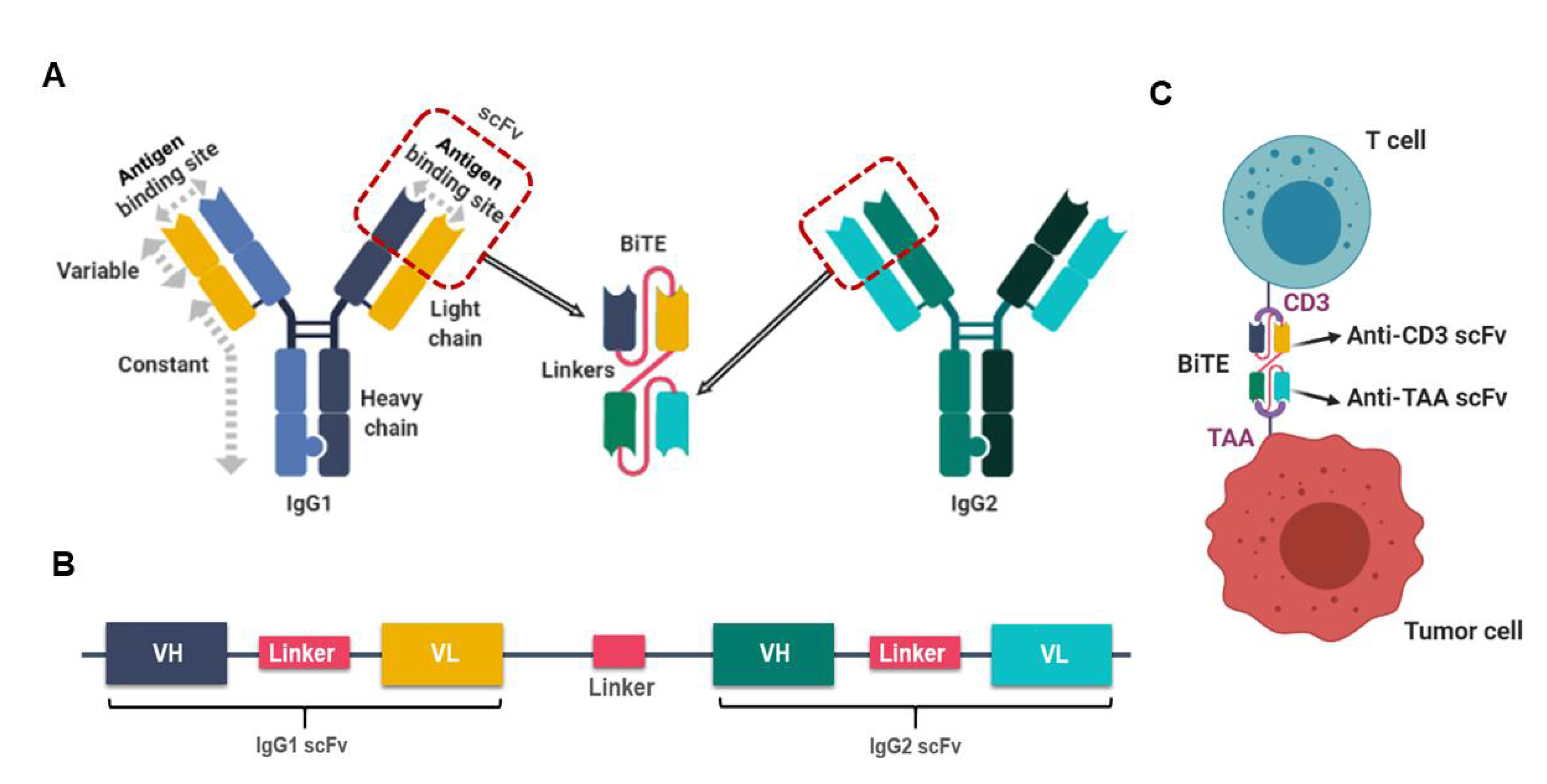
3. Bi- and Tri-Specific Antibodies, Bi- and Tri-specific T Cell Engagers (BiTEs and TriTE) for Integration into OV-Based Cancer Therapies
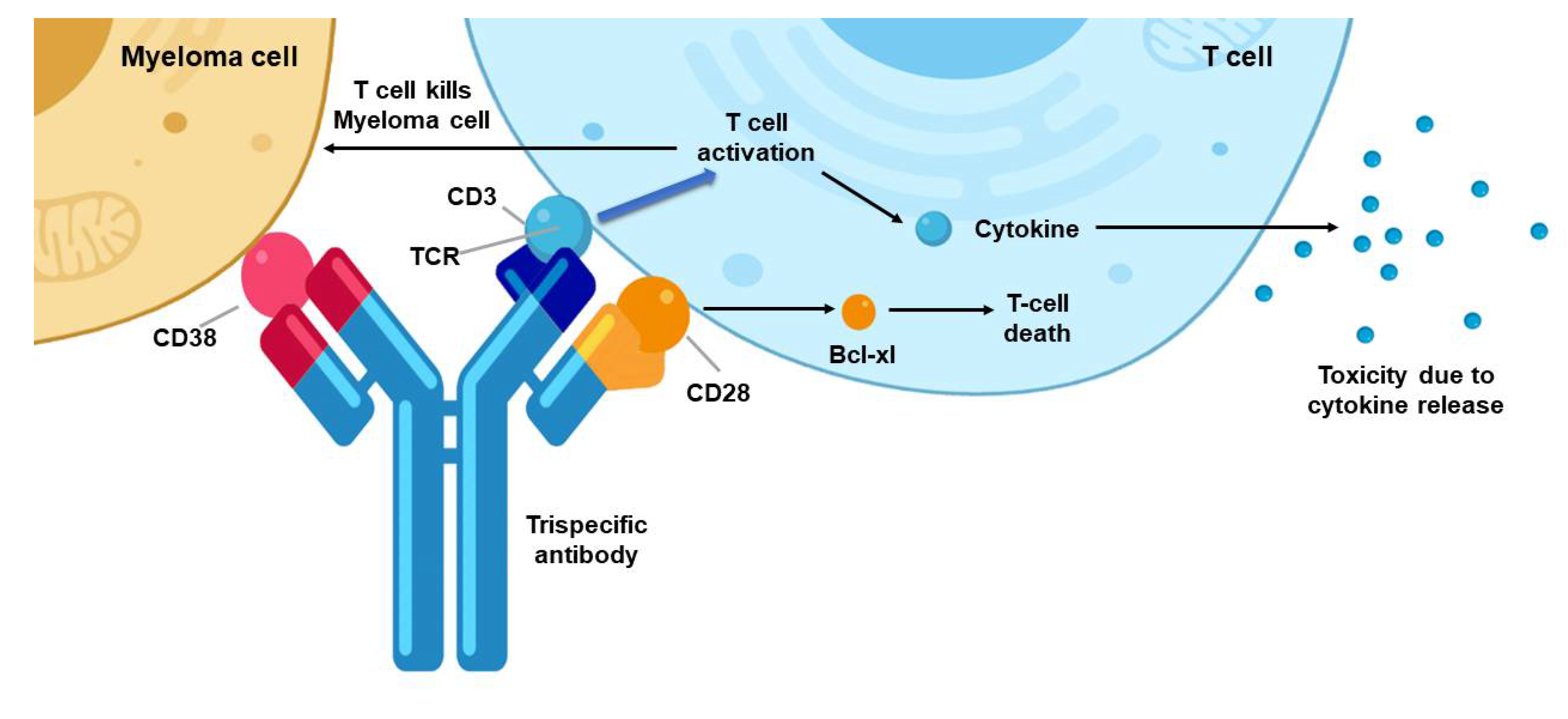
4. BiTE- and TriTE-Armed OVs Mediate Superior Therapeutic Efficacy
5. Despite Many Strengths, There Remain Significant Hurdles for This Approach as an Effective Immunotherapy
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Kowalsky, S.; Feist, M.; Kalinski, P.; Lu, B.; Storkus, W.J.; Bartlett, D.L. Oncolytic immunotherapy: Conceptual evolution, current strategies, and future perspectives. Front. Immunol. 2017, 8, 555. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, S.; Bell, J.; Diallo, J.S. SnapShot: Cancer immunotherapy with oncolytic viruses. Cell 2019, 176, 1240. [Google Scholar] [CrossRef]
- Jing, Y.; Tong, C.; Zhang, J.; Nakamura, T.; Iankov, I.; Russell, S.J.; Merchan, J.R. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009, 69, 1459–1468. [Google Scholar] [CrossRef] [Green Version]
- Ilkow, C.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Ben Neriah, D.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic immunity is required for effective cancer immunotherapy. Cell 2017, 168, 487–502. [Google Scholar] [CrossRef]
- Guo, Z.S.; Bartlett, D.L. Oncolytic viruses as platform for multimodal cancer therapeutics: A promising land. Cancer Gene Ther. 2014, 21, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Martin, N.T.; Bell, J.C. Oncolytic virus combination therapy: Killing one bird with two stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [Green Version]
- Sova, P.; Ren, X.W.; Ni, S.; Bernt, K.M.; Mi, J.; Kiviat, N.; Lieber, A. A tumor-targeted and conditionally replicating oncolytic adenovirus vector expressing TRAIL for treatment of liver metastases. Mol. Ther. 2004, 9, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Ziauddin, M.F.; Guo, Z.S.; O’Malley, M.E.; Austin, F.; Popovic, P.J.; Kavanagh, M.A.; Li, J.; Sathaiah, M.; Thirunavukarasu, P.; Fang, B.; et al. TRAIL gene-armed oncolytic poxvirus and oxaliplatin can work synergistically against colorectal cancer. Gene Ther. 2010, 17, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Sham, J.; Yang, J.; Su, C.; Xue, H.; Chua, D.; Sun, L.; Zhang, Q.; Cui, Z.; Wu, M.; et al. Potent antitumor efficacy of an E1B 55kDa-deficient adenovirus carrying murine endostatin in hepatocellular carcinoma. Int. J. Cancer 2005, 113, 640–648. [Google Scholar] [CrossRef]
- Wong, R.J.; Chan, M.-K.; Yu, Z.; Ghossein, R.A.; Ngai, I.; Adusumilli, P.S.; Stiles, B.M.; Shah, J.P.; Singh, B.; Fong, Y. Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 2004, 10, 4509–4516. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ge, Y.; Wang, H.; Ma, C.; Feist, M.; Ju, S.; Guo, Z.S.; Bartlett, D.L. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat. Commun. 2018, 9, 4682. [Google Scholar] [CrossRef] [Green Version]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Dai, E.; Liu, Z.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Situ therapeutic cancer vaccination with an oncolytic virus expressing Membrane-Tethered IL-2. Mol. Ther. Oncolytics 2020, 17, 350–360. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Heiniö, C.; Cervera-Carrascon, V.; A Quixabeira, D.C.; Siurala, M.; Havunen, R.; Butzow, R.; Zafar, S.; De Gruijl, T.; Lassus, H.; et al. Oncolytic adenovirus shapes the ovarian tumor microenvironment for potent tumor-infiltrating lymphocyte tumor reactivity. J. Immunother. Cancer 2020, 8, e000188. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.W.; Wagner, M.; Fankhauser, M.; Steinskog, E.S.; Broggi, M.A.; Spranger, S.; Gajewski, T.F.; Alitalo, K.; Eikesdal, H.P.; Wiig, H.; et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Investig. 2016, 126, 3389–3402. [Google Scholar] [CrossRef]
- Bedognetti, D.; Ceccarelli, M.; Galluzzi, L.; Lu, R.; Palucka, K.; Samayoa, J.; Spranger, S.; Warren, S.; Wong, K.-K.; Ziv, E.; et al. Toward a comprehensive view of cancer immune responsiveness: A synopsis from the SITC workshop. J. Immunother. Cancer 2019, 7, 131. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Thorne, S.H.; Bartlett, D.L. Oncolytic virotherapy: Molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim. Biophys. Acta 2008, 1785, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Howells, A.; Marelli, G.; Lemoine, N.R.; Wang, Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front. Oncol. 2017, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.X.; Liu, G.; Wong-Staal, F. Oncolytic virotherapy as a personalized cancer vaccine. Int. J. Cancer 2008, 123, 493–499. [Google Scholar] [CrossRef]
- Aitken, A.S.; Roy, D.G.; Bourgeois-Daigneault, M.C. Taking a stab at cancer; oncolytic virus-mediated anti-cancer vaccination strategies. Biomedicines 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gurlevik, E.; Ureche, C.I.; Schumacher, A.; Kuhnel, F. Oncolytic viruses as anticancer vaccines. Front. Oncol. 2014, 4, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.J.; Barber, G.N. Oncolytic viruses as Antigen-Agnostic cancer vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ’hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Ylosmaki, E.; Cerullo, V. Design and application of oncolytic viruses for cancer immunotherapy. Curr. Opin. Biotechnol. 2019, 65, 25–36. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Nisonoff, A.; Wissler, F.C.; Lipman, L.N. Properties of the major component of a peptic digest of rabbit antibody. Science 1960, 132, 1770–1771. [Google Scholar] [CrossRef]
- Perez, P.; A Titus, J.; Lotze, M.T.; Cuttitta, F.; Longo, D.L.; Groves, E.S.; Rabin, H.; Durda, P.J.; Segal, D.M. Specific lysis of human tumor cells by T cells coated with anti-T3 cross-linked to anti-tumor antibody. J. Immunol. 1986, 137, 2069–2072. [Google Scholar]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies-BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-Specific and tri-specific antibodies-the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Yu, S.; Li, A.; Liu, Q.; Yuan, X.; Xu, H.; Han, X.; Pestell, R.G.; Han, X.; Wu, K. Recent advances of bispecific antibodies in solid tumors. J. Hematol. Oncol. 2017, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A comparison between T Cell-Redirection Strategies for cancer treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef] [Green Version]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R. IL15 thepecific Killer Engagers (TriKE) make natural killer cells specific to CD33+ Targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [Green Version]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Garfall, A.L.; June, C.H. Trispecific antibodies offer a third way forward for anticancer immunotherapy. Nature 2019, 575, 450–451. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, J. T cell-redirecting bispecific antibodies in cancer immunotherapy: Recent advances. J. Cancer Res. Clin. Oncol. 2019, 145, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-hodgkin lymphoma: Final results from a phase i study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Gore, L.; et al. A randomized phase 3 trial of blinatumomab vs. chemotherapy as post-reinduction therapy in high and Intermediate Risk (HR/IR) first relapse of B-Acute Lymphoblastic Leukemia (B-ALL) in children and Adolescents/Young Adults (AYAs) demonstrates superior efficacy and tolerability of blinatumomab: A report from children’s oncology group study AALL1331. Blood 2019, 134, LBA-1. [Google Scholar]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, N.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA approval: Blinatumomab for patients with B-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell maturation antigen BiTE Molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018, 10, eaat5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative target affinities of T-Cell-Dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebenko, M.; Goebeler, M.-E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef] [PubMed]
- Benonisson, H.; Altıntaş, I.; Sluijter, M.; Verploegen, S.; Labrijn, A.F.; Schuurhuis, D.H.; Houtkamp, M.A.; Verbeek, J.S.; Schuurman, J.; Van Hall, T. CD3-Bispecific antibody therapy turns solid tumors into inflammatory sites but does not install protective memory. Mol. Cancer Ther. 2019, 18, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmacher, V. New insights into mechanisms of long-term protective anti-tumor immunity induced by cancer vaccines modified by virus infection. Biomedicines 2020, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for combining bispecific T cell activating antibodies with checkpoint blockade for cancer therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.A.; Lutterbuese, R.; Roff, S.; Lutterbuese, P.; Schlereth, B.; Bruckheimer, E.; Kinch, M.S.; Coats, S.; Baeuerle, P.A.; Kufer, P.; et al. Selective targeting and potent control of tumor growth using an EphA2/CD3-Bispecific single-chain antibody construct. Cancer Res. 2007, 67, 3927–3935. [Google Scholar] [CrossRef] [Green Version]
- Song, X.T. Combination of virotherapy and T-cell therapy: Arming oncolytic virus with T-cell engagers. Discov. Med. 2013, 16, 261–266. [Google Scholar] [PubMed]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; De Sostoa, J.; June, C.H.; Alemany, R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, J.D.; Hagel, J.P.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D.; et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087. [Google Scholar] [CrossRef] [PubMed]
- Speck, T.; Veinalde, R.; Jaeger, D.; Von Kalle, C.; Ungerechts, G.; Heidbuechel, J.P.; Ball, C.R.; Engeland, C.E. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Hong, B.; Song, X.-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl. Med. 2017, 3, 122–132. [Google Scholar]
- Freedman, J.D.; Duffy, M.R.; Lei, J.; Muntzer, A.; Scott, E.M.; Hagel, J.P.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [Green Version]
- de Sostoa, J.; Fajardo, C.A.; Moreno, R.; Ramos, M.D.; Farrera-Sal, M.; Alemany, R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef]
- Barlabe, P.; Sostoa, J.; Fajardo, C.A.; Alemany, R.; Moreno, R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020, 27, 383–388. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.A.; Schendel, D.J.; Butterfield, L.H.; Aamdal, S.; Allison, J.P.; Ascierto, P.A.; Atkins, M.B.; Bartunkova, J.; Bergmann, L.; Berinstein, N.L.; et al. Defining the critical hurdles in cancer immunotherapy. J. Transl. Med. 2011, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic adenovirus armed with BiTE, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Ilyas, S.; Yang, J.C. Landscape of tumor antigens in T cell immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Braun, S.; Hepp, F.; Sommer, H.L.; Pantel, K. Tumor-antigen heterogeneity of disseminated breast cancer cells: Implications for immunotherapy of minimal residual disease. Int. J. Cancer 1999, 84, 1–5. [Google Scholar] [CrossRef]
- Wurz, G.T.; Kao, C.J.; DeGregorio, M.W. Novel cancer antigens for personalized immunotherapies: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2016, 8, 4–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, S.; Galon, J.; Fuchs, C.S.; Dranoff, G. Cancer immunology—Analysis of host and tumor factors for personalized medicine. Nat. Rev. Clin. Oncol. 2011, 8, 711–719. [Google Scholar] [CrossRef]
- Wang, R.F.; Wang, H.Y. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017, 27, 11–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Nakamura, Y. Cancer Precision medicine: From Cancer screening to drug selection and personalized immunotherapy. Trends Pharmacol. Sci. 2017, 38, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; Guerreiro, M.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Trenevska, I.; Li, D.; Banham, A.H. Therapeutic antibodies against intracellular tumor antigens. Front. Immunol. 2017, 8, 1001. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Salazar GTa Zhang, N.; An, Z. T-cell receptor mimic (TCRm) antibody therapeutics against intracellular proteins. Antib. Ther. 2019, 2, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, N.; Wakata, Y.; Ida, K.; Midorikawa, A.; Isobe, M. High throughput development of TCR-mimic antibody that targets survivin-2B80–88/HLA-A*A24 and its application in a bispecific T-cell engager. Sci. Rep. 2019, 9, 9827. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol. Biosci. 2018, 18, 1700187. [Google Scholar] [CrossRef]
- Li, H.; Er Saw, P.; Song, E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 130, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for cancer therapy: Barriers and recent advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-Targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Vafa, O.; Trinklein, N.D. Perspective: Designing T-cell engagers with better therapeutic windows. Front. Oncol. 2020, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- In Lim, S. Fine-Tuning bispecific therapeutics. Pharmacol. Ther. 2020, 212, 107582. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
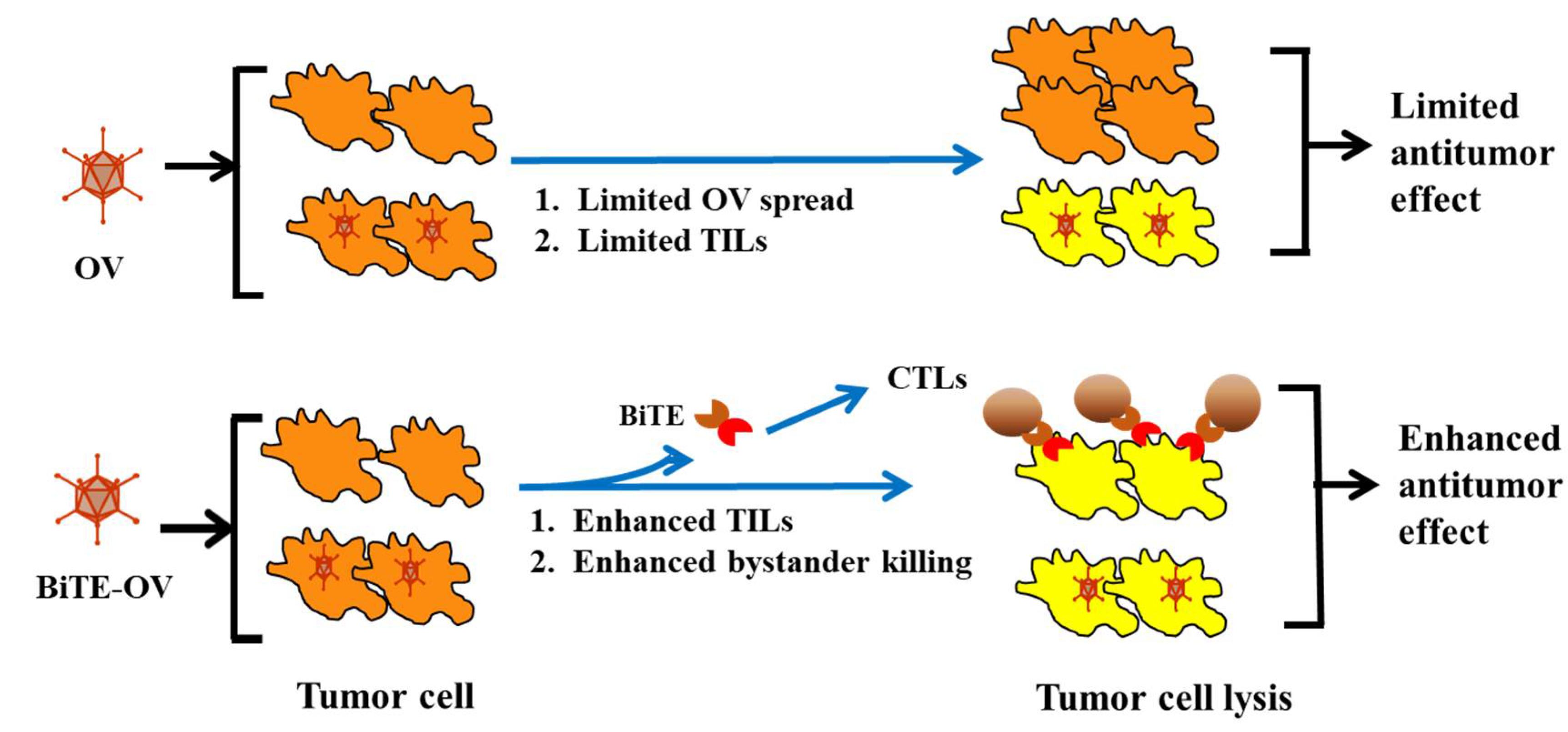{kind=link}
| OV | BiTE or TriTE | Tumor Model | Main Observations | Reference |
|---|---|---|---|---|
| VV | CD3/EphA2 | Human lung cancer in SCID/ beige mice | (1). Tumor cells infected with the OV-induced T cell activation. In coculture assays, the armed OV not only killed infected tumor cells, but in the presence of T cells, it also induced bystander killing of non-infected tumor cells. (2). EphA2-TEA-VV combined with human T cells had potent antitumor activity. | [69] |
| Ad | CD3/EGFR | Human lung and colorectal cancers in SCID/beige mice | (1). ICOVIR-15K-cBiTE–mediated oncolysis resulted in robust T cell activation, proliferation, and bystander cell-mediated cytotoxicity. (2). Intratumoral injection increased the persistence and accumulation of TILs in vivo, and combined delivery with PBMCs or T cells enhanced antitumor efficacy. | [72] |
| EnAd | CD3/EpCAM | Primary pleural effusions and peritoneal malignant ascites | Infection of cancer cells leads to the activation of endogenous T cells to kill endogenous tumor cells. | [73] |
| Measles viruses | CD3/CEA or CD20 | (1). MC38-CEA model; (2). Human primary colorectal cancer-derived spheroids | (1). Therapeutic efficacy against established tumors in fully immunocompetent mice. (2). Therapeutic efficacy in xenograft models of patient-derived primary colorectal carcinoma spheroids with transfer of PBMCs. | [74] |
| VV or Ad | CD3/FAP | (1). B16 models; (2). Human colon and lung cancers | BiTE-armed OVs combine direct oncolysis of cancer cells with endogenous T cell activation to attack stromal fibroblasts. | [75,76,77] |
| EnAd | CD3, CD206 folate receptor β | Human cancer samples in vitro | T cells activated by BiTE- or TriTE-armed EnAd preferentially killed M2-like autologous macrophages. | [78] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.-T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070204
Guo ZS, Lotze MT, Zhu Z, Storkus WJ, Song X-T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines. 2020; 8(7):204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070204
Chicago/Turabian StyleGuo, Zong Sheng, Michael T. Lotze, Zhi Zhu, Walter J. Storkus, and Xiao-Tong Song. 2020. "Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy" Biomedicines 8, no. 7: 204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070204