Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article

Abstract
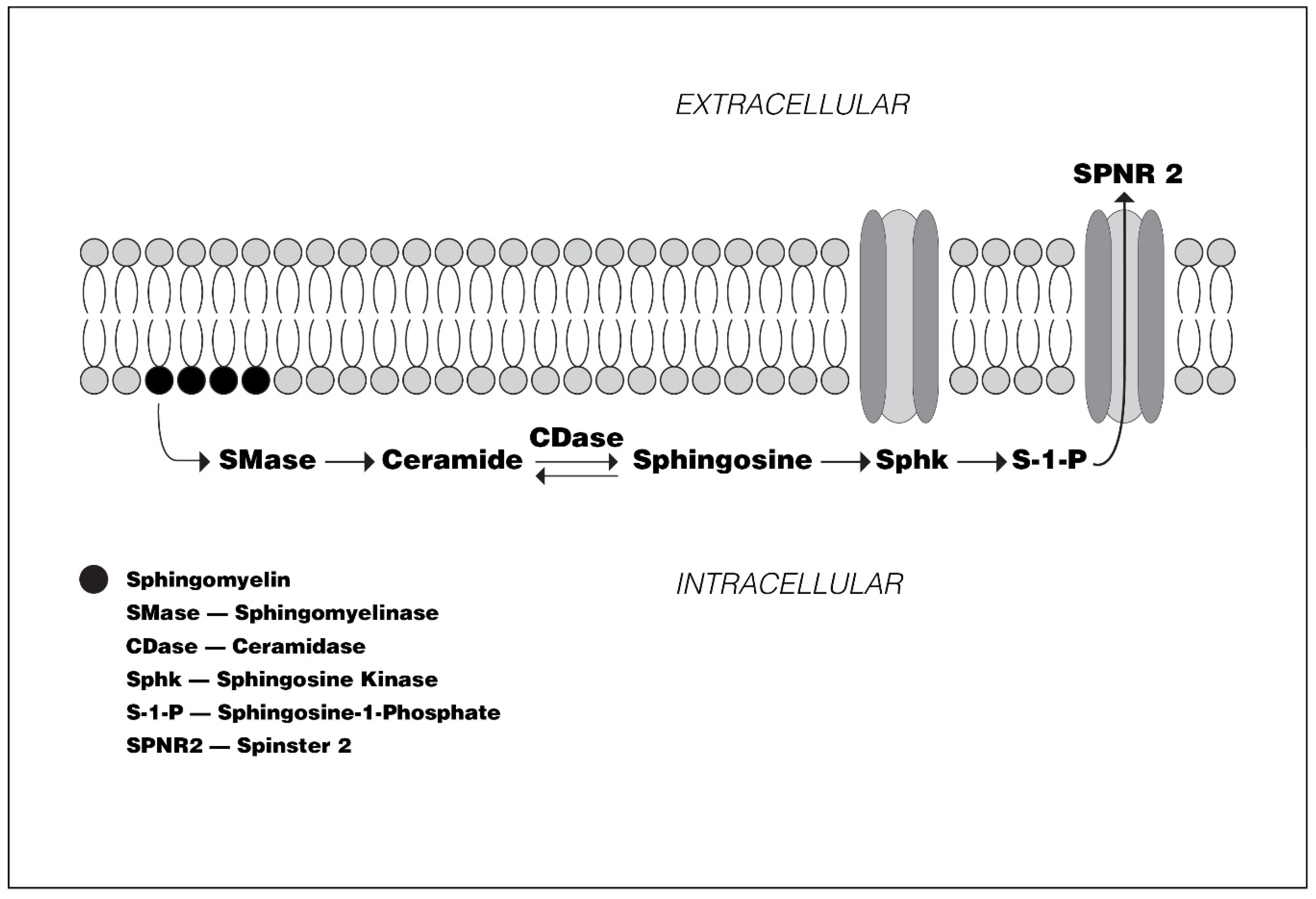
:1. Introduction
2. Preclinical Studies
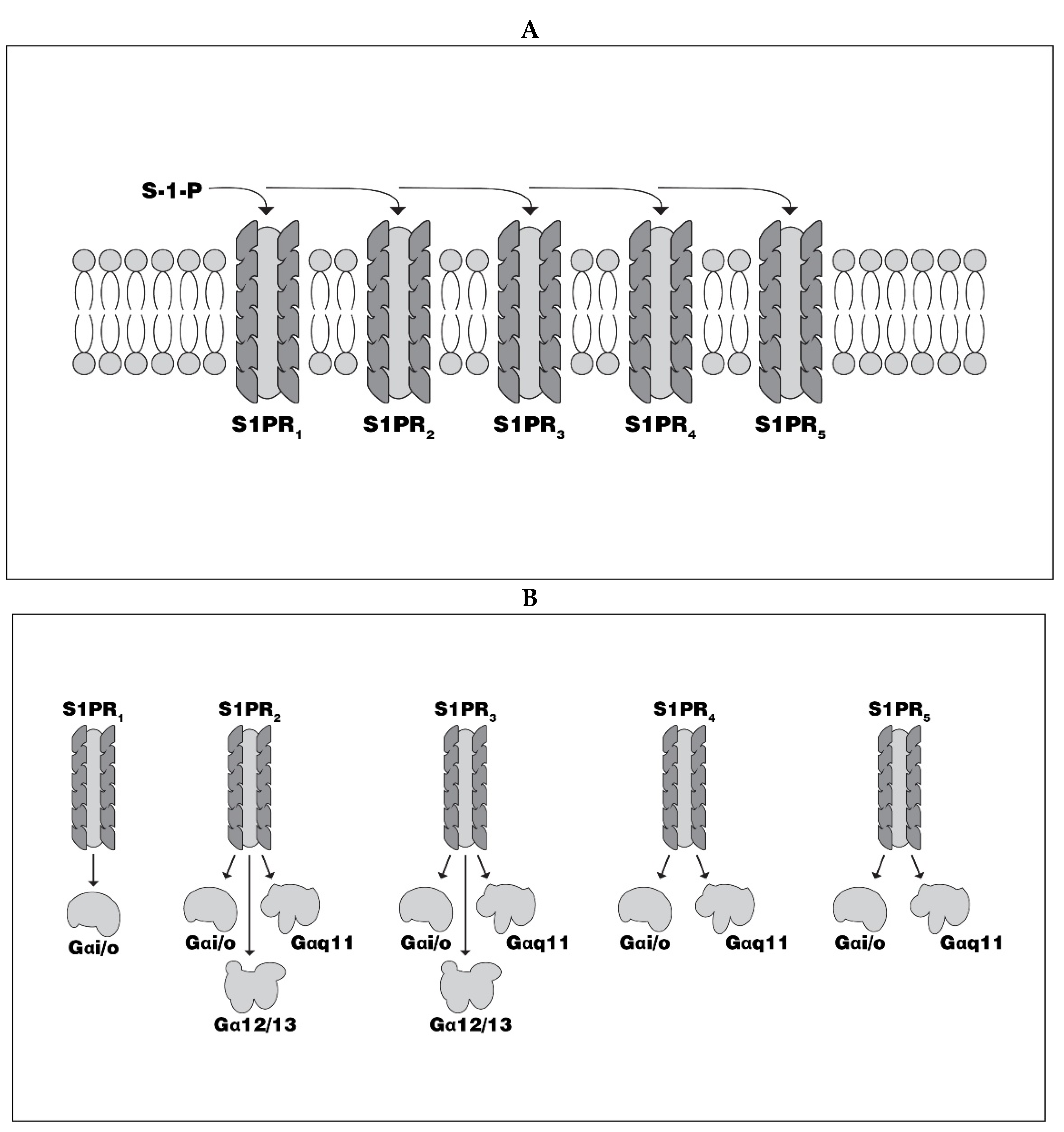
2.1. S1P Receptors
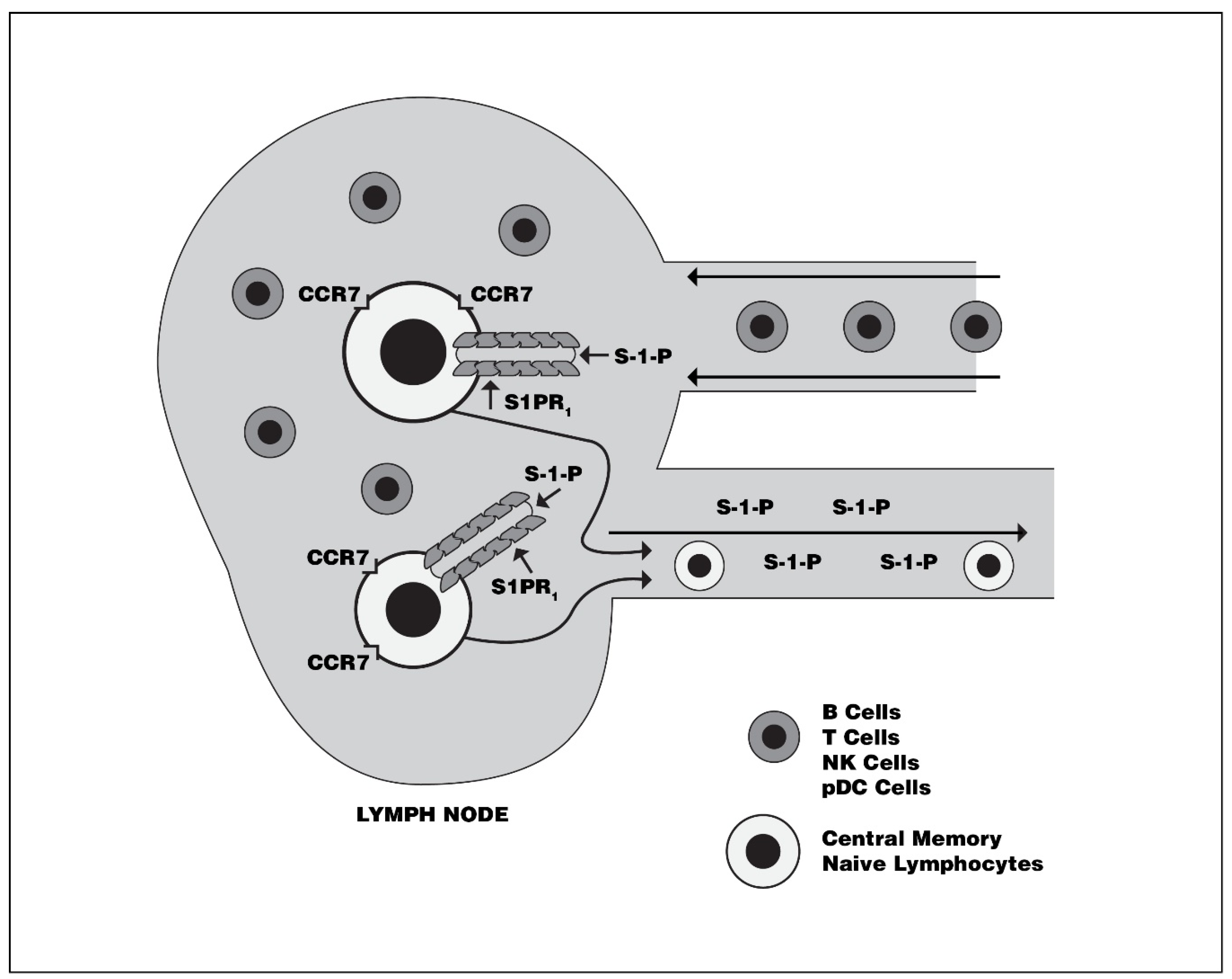
2.1.1. S1PR-1
2.1.2. S1PR-2
2.1.3. S1P Neurotropism and S1PR-1/S1PR-2 Synergy
2.1.4. S1PR-5
2.2. Direct Pharmacological Actions of the S1PR Modulators in the CNS
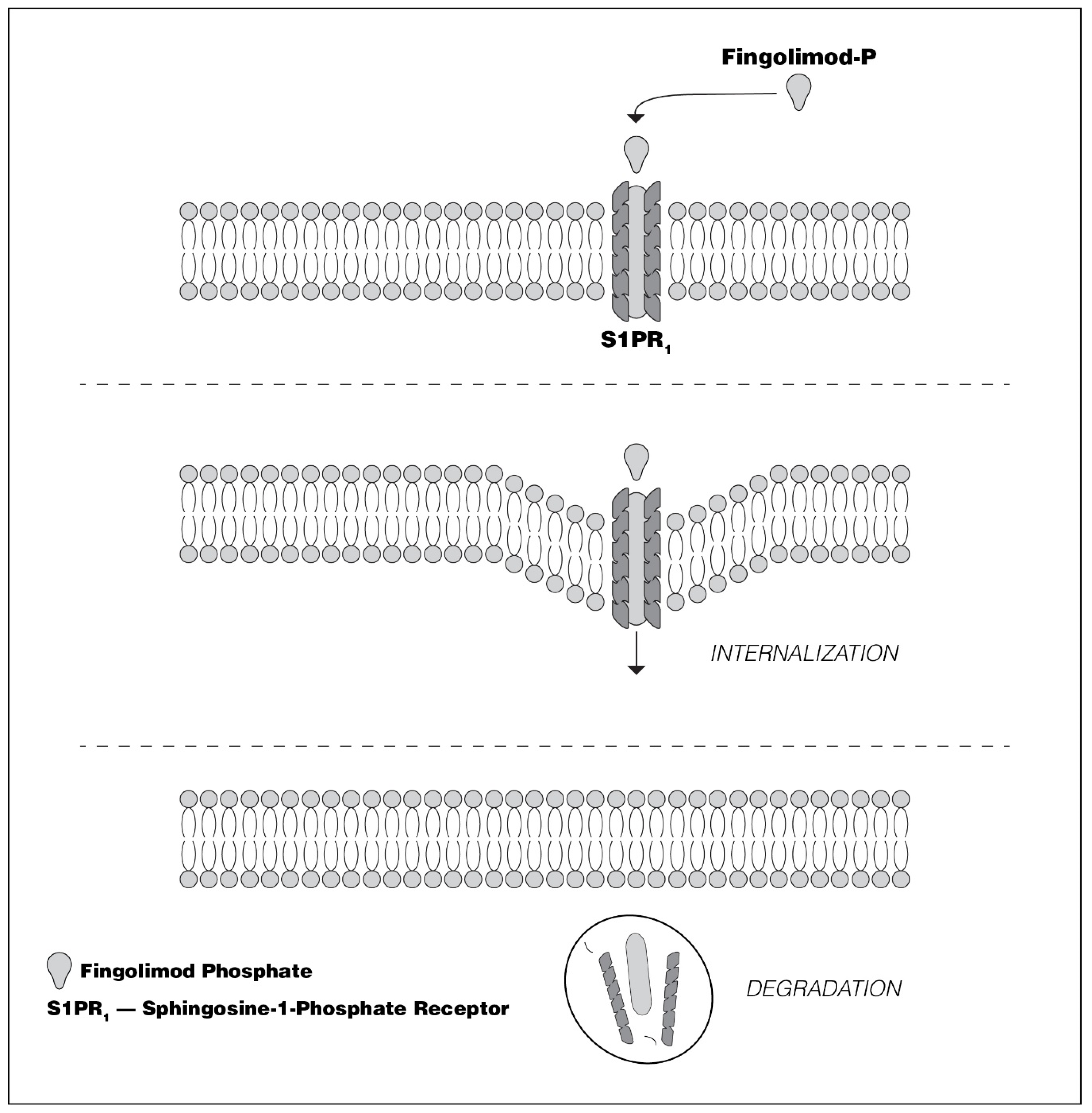
2.2.1. Fingolimod
2.2.2. Siponimod
2.2.3. Ozanimod
2.2.4. Ponesimod
3. Clinical Trial Results
3.1. Fingolimod
3.1.1. FREEDOMS
3.1.2. TRANSFORMS
3.1.3. FREEDOMS II
3.1.4. Extended TRANSFORMS
3.1.5. PARADIGM
3.2. Siponimod
EXPAND
3.3. Ozanimod
3.3.1. RADIANCE Part B
3.3.2. SUNBEAM
3.4. Ponesimod
OPTIMUM
4. Safety
4.1. Fingolimod
4.2. Siponimod
4.3. Ozanimod
4.4. Contraindications and Cautions
4.5. Ponesimod
5. Potential Therapeutic Opportunities
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Boil. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Brunkhorst, R.; Vutukuri, R.; Pfeilschifter, W. Fingolimod for the treatment of neurological diseases-state of play and future perspectives. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The Immune Modulator FTY720 Targets Sphingosine 1-Phosphate Receptors. J. Boil. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [Green Version]
- Webb, M.; Tham, C.-S.; Lin, F.-F.; Lariosa-Willingham, K.; Yu, N.; Hale, J.; Mandala, S.; Chun, J.; Rao, T.S. Sphingosine 1-phosphate receptor agonists attenuate relapsing–remitting experimental autoimmune encephalitis in SJL mice. J. Neuroimmunol. 2004, 153, 108–121. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.-W.; Huang, Y.; Cyster, J.G.; et al. Promotion of Lymphocyte Egress into Blood and Lymph by Distinct Sources of Sphingosine-1-Phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.M.; Reavill, C.; Brown, G.; Brown, J.T.; Cluderay, J.E.; Crook, B.; Davies, C.H.; Dawson, L.A.; Grau, E.; Heidbreder, C.; et al. LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Mol. Cell Neurosci. 2003, 24, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Brinkmann, V. Faculty Opinions recommendation of A mechanistically novel, first oral therapy for multiple sclerosis: The development of fingolimod (FTY720, Gilenya). Fac. Opin. Post-Pub. Peer Rev. Biomed. Lit. 2014, 12, 213–228. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta (BBA)-Mol. Cell Boil. Lipids 2013, 1831, 20–32. [Google Scholar] [CrossRef]
- Giussani, P.C.; Tringali, C.A.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, R.; Anelli, V.V.; Giussani, P.C.; Tettamanti, G.; Viani, P.; Riboni, L. Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia 2006, 53, 621–630. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential Role for Sphingosine Kinases in Neural and Vascular Development. Mol. Cell. Boil. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [Green Version]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.C.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.N.; Kampfl, A.W.; Zhao, X.; Hayes, R.L.; Dash, P.K. Sphingosine-1-phosphate induces apoptosis of cultured hippocampal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 1999, 94, 405–415. [Google Scholar] [CrossRef]
- Hagen, N.; Van Veldhoven, P.P.; Proia, R.L.; Park, H.; Merrill, A.H.; Van Echten-Deckert, G. Subcellular Origin of Sphingosine 1-Phosphate Is Essential for Its Toxic Effect in Lyase-deficient Neurons. J. Boil. Chem. 2009, 284, 11346–11353. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Hans, M.; Hartmann, D.; Swandulla, D.; Van Echten-Deckert, G. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 2011, 18, 1356–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroi, D.N.; Karunakaran, I.; Gräler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; Van Echten-Deckert, G. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Saba, J.D. Sphingosine phosphate lyase insufficiency syndrome (SPLIS): A novel inborn error of sphingolipid metabolism. Adv. Boil. Regul. 2019, 71, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.C.; Ferraretto, A.; Gravaghi, C.; Bassi, R.; Tettamanti, G.; Riboni, L.; Viani, P. Sphingosine-1-Phosphate and Calcium Signaling in Cerebellar Astrocytes and Differentiated Granule Cells. Neurochem. Res. 2006, 32, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Deutschmann, A.U.; Raucamp, M.; Karunakaran, I.; Glebov, K.; Hans, M.; Walter, J.; Saba, J.; Gräler, M.; Ehninger, D.; et al. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 2016, 6, 37064. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Cyster, J.G.; Hla, T. FTY720: Sphingosine 1-Phosphate Receptor-1 in the Control of Lymphocyte Egress and Endothelial Barrier Function. Arab. Archaeol. Epigr. 2004, 4, 1019–1025. [Google Scholar] [CrossRef]
- Chiba, K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Ther. 2005, 108, 308–319. [Google Scholar] [CrossRef]
- Thangada, S.; Khanna, K.M.; Blaho, V.A.; Oo, M.L.; Im, D.-S.; Guo, C.; Lefrancois, L.; Hla, T. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J. Exp. Med. 2010, 207, 1475–1483. [Google Scholar] [CrossRef]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.A.; Timony, G.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [Green Version]
- Mandala, S. Alteration of Lymphocyte Trafficking by Sphingosine-1-Phosphate Receptor Agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Thangada, S.; Wu, M.T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [Green Version]
- Gräler, M.H.; Goetzl, E.J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G protein-coupled receptors. FASEB J. 2004, 18, 551–553. [Google Scholar] [CrossRef]
- Mullershausen, F.; Zecri, F.; Çetin, C.; Billich, A.; Guerini, D.; Seuwen, K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat. Methods 2009, 5, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subei, A.M.; Cohen, J.A. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 2015, 29, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao-Draayer, Y.; Sarazin, J.; Fox, D.; Schiopu, E. The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin. Immunol. 2016, 175, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2010, 108, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Miron, V.E.; Dukala, D.; Proia, R.L.; Ludwin, S.K.; Traka, M.; Antel, J.P.; Soliven, B. Neurobiological effects of sphingosine 1-phosphate receptor modulation in the cuprizone model. FASEB J. 2011, 25, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Nystad, A.E.; Lereim, R.R.; Wergeland, S.; Oveland, E.; Myhr, K.-M.; Bø, L.; Torkildsen, Ø. Fingolimod downregulates brain sphingosine-1-phosphate receptor 1 levels but does not promote remyelination or neuroprotection in the cuprizone model. J. Neuroimmunol. 2020, 339, 577091. [Google Scholar] [CrossRef]
- Seyedsadr, M.S.; Weinmann, O.; Amorim, A.; Ineichen, B.V.; Egger, M.; Mirnajafi-Zadeh, J.; Becher, B.; Javan, M.; Schwab, M.E. Inactivation of sphingosine-1-phosphate receptor 2 (S1PR2) decreases demyelination and enhances remyelination in animal models of multiple sclerosis. Neurobiol. Dis. 2019, 124, 189–201. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.-G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Brana, C.; Frossard, M.J.; Gobert, R.P.; Martinier, N.; Boschert, U.; Seabrook, T.J. Immunohistochemical detection of sphingosine-1-phosphate receptor 1 and 5 in human multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2014, 40, 564–578. [Google Scholar] [CrossRef]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Hof, B.V.H.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bielawski, J.; Yang, H.; Kong, Y.; Zhou, B.; Li, J. Functional antagonism of sphingosine-1-phosphate receptor 1 prevents cuprizone-induced demyelination. Glia 2017, 66, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Assi, E.; Cazzato, D.; De Palma, C.; Perrotta, C.; Clementi, E.; Cervia, D. Sphingolipids and Brain Resident Macrophages in Neuroinflammation: An Emerging Aspect of Nervous System Pathology. Clin. Dev. Immunol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Koyrakh, L.; Roman, M.I.; Brinkmann, V.; Wickman, K. The Heart Rate Decrease Caused by Acute FTY720 Administration Is Mediated by the G Protein-Gated Potassium Channel I KACh. Arab. Archaeol. Epigr. 2005, 5, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Bünemann, M.; Brandts, B.; Zu Heringdorf, D.M.; Van Koppen, C.J.; Jakobs, K.H.; Pott, L. Activation of muscarinic K+ current in guinea-pig atrial myocytes by sphingosine-1-phosphate. J. Physiol. 1995, 489, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanna, M.G.; Liao, J.; Jo, E.; Alfonso, C.; Ahn, M.-Y.; Peterson, M.S.; Webb, B.; Lefebvre, S.; Chun, J.; Gray, N.; et al. Sphingosine 1-Phosphate (S1P) Receptor Subtypes S1P1and S1P3, Respectively, Regulate Lymphocyte Recirculation and Heart Rate. J. Boil. Chem. 2004, 279, 13839–13848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhang, F. Implication of sphingosin-1-phosphate in cardiovascular regulation. Front. Biosci. 2016, 21, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, A.S.; El-Alwani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 2007, 21, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.; Canals, D.; Hannun, Y.A.; Obeid, L.M. Sphingosine-1-phosphate receptor 2. FEBS J. 2013, 280, 6354–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Orengo, L.; Daniels, B.P.; Dorsey, D.; Basak, S.A.; Grajales-Reyes, J.G.; McCandless, E.E.; Piccio, L.; Schmidt, R.E.; Cross, A.H.; Crosby, S.D.; et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J. Clin. Investig. 2014, 124, 2571–2584. [Google Scholar] [CrossRef] [Green Version]
- Kempf, A.; Tews, B.; Arzt, M.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The Sphingolipid Receptor S1PR2 Is a Receptor for Nogo-A Repressing Synaptic Plasticity. PLoS Boil. 2014, 12, e1001763. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.C.; Rosenberg, S.S.; Fancy, S.P.J.; Zhao, C.; Shen, Y.-A.A.; Hahn, A.T.; McGee, A.W.; Xu, X.; Zheng, B.; Zhang, L.I.; et al. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. USA 2011, 109, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Kikuta, J.; Shimazu, Y.; Meier-Schellersheim, M.; Germain, R.N. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J. Exp. Med. 2010, 207, 2793–2798. [Google Scholar] [CrossRef] [Green Version]
- Thurnherr, T.; Benninger, Y.; Wu, X.; Chrostek, A.; Krause, S.M.; Nave, K.-A.; Franklin, R.; Brakebusch, C.; Suter, U.; Relvas, J.B. Cdc42 and Rac1 Signaling Are Both Required for and Act Synergistically in the Correct Formation of Myelin Sheaths in the CNS. J. Neurosci. 2006, 26, 10110–10119. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.S.; Hofereiter, J.; Rübsamen, H.; Melms, J.; Schwarz, S.; Faber, H.; Weber, P.; Pütz, B.; Loleit, V.; Weber, F.; et al. Fingolimod induces neuroprotective factors in human astrocytes. J. Neuroinflamm. 2015, 12, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Heng, B.; Teo, J.D.; Humphrey, S.J.; Qi, Y.; Couttas, T.A.; Stefen, H.; Brettle, M.; Fath, T.; Guillemin, G.J.; et al. Sphingosine 1-phosphate but not Fingolimod protects neurons against excitotoxic cell death by inducing neurotrophic gene expression in astrocytes. J. Neurochem. 2019, 153, 173–188. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: An oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.; Barron, S.; Diaz, J.; Lubetzki, C.; Zalc, B.; Schwartz, J.C.; Sokoloff, P. A rat G protein-coupled receptor selectively expressed in myelin-forming cells. J. Neurosci. 1998, 10, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Im, N.-S.; Clemens, J.; Macdonald, T.L.; Lynch, K.R. Characterization of the Human and Mouse Sphingosine 1-Phosphate Receptor, S1P5(Edg-8): Structure−Activity Relationship of Sphingosine1-Phosphate Receptors. Biochemistry 2001, 40, 14053–14060. [Google Scholar] [CrossRef]
- Terai, K.; Soga, T.; Takahashi, M.; Kamohara, M.; Ohno, K.; Yatsugi, S.; Okada, M.; Yamaguchi, T. Edg-8 receptors are preferentially expressed in oligodendrocyte lineage cells of the rat CNS. Neuroscice 2003, 116, 1053–1062. [Google Scholar] [CrossRef]
- Yu, N.; Lariosa-Willingham, K.D.; Lin, F.-F.; Webb, M.; Rao, T.S. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 2003, 45, 17–27. [Google Scholar] [CrossRef]
- Van Doorn, R.; Pinheiro, M.A.L.; Kooij, G.; Lakeman, K.; Hof, B.V.H.; Van Der Pol, S.M.A.; Geerts, D.; Van Horssen, J.; Van Der Valk, P.; Van Der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflamm. 2012, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Pinschewer, D.; Chiba, K.; Feng, L. FTY720: A novel transplantation drug that modulates lymphocyte traffic rather than activation. Trends Pharmacol. Sci. 2000, 21, 49–52. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.-P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Farez, M.F.; Correale, J.; Information, P.E.K.F.C. Sphingosine 1-phosphate signaling in astrocytes: Implications for progressive multiple sclerosis. J. Neurol. Sci. 2016, 361, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; De Lima, K.A.; Borucki, D.M.; Chao, C.-C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) Enhances Remyelination Following Demyelination of Organotypic Cerebellar Slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.; Baharvand, H.; Javan, M. Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015, 311, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Li, Y.; Ding, X.; Shang, X.; Lü, M.; Elias, S.B.; Chopp, M. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2015, 76, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Muthukumarana, A.; Harrison, P.C.; Nodop, M.S.; Chen, R.R.; Harrington, K.E.; Dinallo, R.M.; Horan, J.C.; Patnaude, L.; Modis, L.K.; et al. The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P(1)) and hypertension (S1P(3)) in rat. PLoS ONE 2012, 7, e52985. [Google Scholar] [CrossRef] [Green Version]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.; Hinterding, K.; Cooke, N.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef]
- Pan, S.; Gray, N.S.; Gao, W.; Mi, Y.; Fan, Y.; Wang, X.; Tuntland, T.; Che, J.; Lefebvre, S.; Chen, Y.; et al. Discovery of BAF312 (Siponimod), a Potent and Selective S1P Receptor Modulator. ACS Med. Chem. Lett. 2013, 4, 333–337. [Google Scholar] [CrossRef]
- Forrest, M.; Sun, S.-Y.; Hajdu, R.; Bergstrom, J.; Card, D.; Doherty, G.; Hale, J.; Keohane, C.; Meyers, C.; Milligan, J.; et al. Immune Cell Regulation and Cardiovascular Effects of Sphingosine 1-Phosphate Receptor Agonists in Rodents Are Mediated via Distinct Receptor Subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Coelho, R.P.; Payne, S.G.; Bittman, R.; Spiegel, S.; Sato-Bigbee, C. The Immunomodulator FTY720 Has a Direct Cytoprotective Effect in Oligodendrocyte Progenitors. J. Pharmacol. Exp. Ther. 2007, 323, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.G.; Kim, H.J.; Miron, V.E.; Cook, S.; Kennedy, T.E.; Foster, C.A.; Antel, J.P.; Soliven, B. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 2007, 55, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflamm. 2016, 13, 31. [Google Scholar]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.A.; Comi, G.; Arnold, D.L. Efficacy and safety of ozanimod in multiple sclerosis: Dose-blinded extension of a randomized phase II study. Mult. Scler. 2019, 25, 1255–1262. [Google Scholar] [CrossRef]
- ZEPOSIA (Ozanimod) (Package Insert). Celgene Corporation: Summit, NJ, USA. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209899s000lbl.pdf (accessed on 18 July 2020).
- Bolli, M.H.; Abele, S.; Binkert, C.; Bravo, R.; Buchmann, S.; Bur, D.; Gatfield, J.; Hess, P.; Kohl, C.; Mangold, C.; et al. 2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1Receptor Agonists. J. Med. Chem. 2010, 53, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.; Freedman, M.S.; Prinz, J. Ponesimod, a selective S1P1 receptor modulator: A potential treatment for multiple sclerosis and other immune-mediated diseases. Ther. Adv. Chronic Dis. 2016, 7, 18–33. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Piali, L.; Kuhn, C.; Steiner, B.; Sauvaget, V.; Valette, F.; Clozel, M.; Bach, J.-F.; Chatenoud, L. Therapeutic Use of a Selective S1P1 Receptor Modulator Ponesimod in Autoimmune Diabetes. PLoS ONE 2013, 8, e77296. [Google Scholar] [CrossRef]
- Piali, L.; Froidevaux, S.; Hess, P.; Nayler, O.; Bolli, M.H.; Schlosser, E.; Kohl, C.; Steiner, B.; Clozel, M. The Selective Sphingosine 1-Phosphate Receptor 1 Agonist Ponesimod Protects against Lymphocyte-Mediated Tissue Inflammation. J. Pharmacol. Exp. Ther. 2011, 337, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Brossard, P.; Derendorf, H.; Xu, J.; Maatouk, H.; Halabi, A.; Dingemanse, J. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br. J. Clin. Pharmacol. 2013, 76, 888–896. [Google Scholar] [CrossRef] [Green Version]
- GILENYA (Fingolimod) (Package Insert). Novartis Pharmaceuticals Coporation: East Hanover, NJ, USA. 2019. Available online: https://www.novartis.us/sites/www.novartis.us/files/gilenya.pdf (accessed on 18 July 2020).
- Chitnis, T.; Arnold, U.L.; Banwell, B.; Brück, W.; Ghezzi, A.; Giovannoni, G.; Greenberg, B.M.; Krupp, L.; Rostásy, K.; Tardieu, M.; et al. Trial of Fingolimod versus Interferon Beta-1a in Pediatric Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 1017–1027. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral Fingolimod or Intramuscular Interferon for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.K.B.; Stuve, O.; Hartung, H.-P.; Freedman, M.S.; Hemmer, B.; Rieckmann, P.; Montalban, X.; Ziemssen, F.; Hunter, B.; et al. Safety and Efficacy of Siponimod (BAF312) in Patients with Relapsing-Remitting Multiple Sclerosis. JAMA Neurol. 2016, 73, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Khatri, B.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Montalban, X.; Pelletier, J.; Stites, T.; Ritter, S.; Von Rosenstiel, P.; et al. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: Results from the extension of the randomised TRANSFORMS study. J. Neurol. Neurosurg. Psychiatry 2015, 87, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Cohen, J.; Comi, G.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.-P.; Montalban, X.; Havrdová, E.K.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): A multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019, 18, 1021–1033. [Google Scholar] [CrossRef]
- Comi, G.; Kappos, L.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.-P.; Montalban, X.; Havrdová, E.K.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): A multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019, 18, 1009–1020. [Google Scholar] [CrossRef]
- Deluca, J.; Huang, D.; Cohen, J.A.; Cree, B.A.C.; Chen, Y.; Campanolo, D.; Sheffield, J.K.; Comi, G.; Kappos, L. Ozanimod-Treated Patients Exhibited Improvements in Cognitive Processing Speed in the Phase 3 SUNBEAM Trial of Relapsing Multiple Sclerosis (RMS); ECTRIMS: Berlin, Germany, 2018. [Google Scholar]
- Kappos, L.; Burcklen, M.; Freedman, M.S.; Fox, R.; Havrdová, E.K.; Hennessy, B.; Hohlfeld, R.; Lublin, F.; Montalban, X.; Pozzilli, C.; et al. Efficacy and Safety of Ponesimod Compared to Teriflunomide in Patients with Relapsing Multiple Sclerosis: Results of the Randomized, Active-Controlled, Double-Blind, Parallel-Group Phase 3 OPTIMUM Study; ECTRIMS: Stockholm, Sweden, 2019. [Google Scholar]
- Ontaneda, D.; Moore, A.; Bakshi, R.; Zajicheck, A.; Kattan, M.; Fox, R. Risk Estimates of Progressive Multifocal Leukoencephalopathy Related to Fingolimod; ECTRIMS: Berlin, Germany, 2018. [Google Scholar]
- Boffa, G.; Bruschi, N.; Cellerino, M.; Lapucci, C.; Novi, G.; Sbragia, E.; Capello, E.; Uccelli, A.; Inglese, M. Fingolimod and Dimethyl-Fumarate-Derived Lymphopenia is not Associated with Short-Term Treatment Response and Risk of Infections in a Real-Life MS Population. CNS Drugs 2020, 34, 425–432. [Google Scholar] [CrossRef]
- Conzett, K.B.; Kolm, I.; Jelčić, I.; Kamarachev, J.; Dummer, R.; Braun, R.; French, L.; Linnebank, M.; Hofbauer, G.F.L. Melanoma Occurring During Treatment with Fingolimod for Multiple Sclerosis: A Case Report. Arch. Dermatol. 2011, 147, 991–992. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.L.; Guo, M. Fingolimod (Gilenya) and melanoma. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef]
- Hatcher, S.E.; Waubant, E.; Nourbakhsh, B.; Crabtree-Hartman, E.; Graves, J.S. Rebound Syndrome in Patients with Multiple Sclerosis After Cessation of Fingolimod Treatment. JAMA Neurol. 2016, 73, 790. [Google Scholar] [CrossRef] [Green Version]
- Frau, J.; Sormani, M.P.; Signori, A.; Realmuto, S.; Baroncini, D.; Annovazzi, P.; Signoriello, E.; Maniscalco, G.T.; La Gioia, S.; Cordioli, C.; et al. Clinical activity after fingolimod cessation: Disease reactivation or rebound? Eur. J. Neurol. 2018, 25, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Uygunoğlu, U.; Tütüncü, M.; Altintas, A.; Saip, S.; Siva, A. Factors Predictive of Severe Multiple Sclerosis Disease Reactivation After Fingolimod Cessation. Neurology 2018, 23, 12–16. [Google Scholar] [CrossRef]
- Bobes, N.S. Package inserts. N. Engl. J. Med. 1968, 278, 282. [Google Scholar]
- Linda, H.; von Heijne, A. A case of posterior reversible encephalopathy syndrome associated with gilenya((R)) (fingolimod) treatment for multiple sclerosis. Front. Neurol. 2015, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, C.; Robertson, D.; Casady, L.; Maldonado, J. A Rare Complication of Fingolimod: Case Report of Posterior Reversible Encephalopathy Syndrome. Neurology 2019, 92, 2–16. [Google Scholar]
- Fischer, A.; Prüfer, K.; Good, J.M.; Halbwax, M.; Wiebe, V.; Andre, C.; Atencia, R.; Mugisha, L.; Ptak, S.E.; Pääbo, S. Bonobos Fall within the Genomic Variation of Chimpanzees. PLoS ONE 2011, 6, e21605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Fernandes, M.J.; Turgeon, M.; Tancrède, S.; Di Battista, J.; Poubelle, P.E.; Bourgoin, S.G. Specific and overlapping sphingosine-1-phosphate receptor functions in human synoviocytes: Impact of TNF-α. J. Lipid Res. 2008, 49, 2323–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunemi, S.; Iwasaki, T.; Kitano, S.; Imado, T.; Miyazawa, K.; Sano, H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin. Immunol. 2010, 136, 197–204. [Google Scholar] [CrossRef]
- Militsakh, O.; Day, T.; Hornig, J.; Lentsch, E.; Skoner, J.; Gillespie, M.B.; Sharma, A.; Neville, B.; Rumboldt, Z.; James, R.; et al. Sphingosine-1-Phosphate. Encyclopedia Cancer 2011, 38, 3485. [Google Scholar] [CrossRef]
- Clark, D.N.; Markham, J.L.; Sloan, C.S.; Poole, B. Cytokine inhibition as a strategy for treating systemic lupus erythematosus. Clin. Immunol. 2013, 148, 335–343. [Google Scholar] [CrossRef]
- Gottschalk, T.; Tsantikos, E.; Hibbs, M.L. Pathogenic Inflammation and Its Therapeutic Targeting in Systemic Lupus Erythematosus. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danko, K.; Vencovsky, J.; Lundberg, I.; Amato, A.; Oddis, C.; Malnar, M.; Moher, A.; Colin, L. The Selective Sphingosine-1-Phosphate Receptor 1/5 Modulator Siponimod (BAF312) Shows Beneficial Effects in Patients with Active, Treatment Refractory Polymyositis and Dermatomyositis: A Phase IIa Proof-of-Concept, Double-Blind, Randomized Trial; American College of Rheumatology: Boston, MA, USA, 2014. [Google Scholar]
- Sandborn, W.J.; Feagan, B.G.; Mutneja, H.; Arora, S.; Vij, A. Ozanimod Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 375, e17. [Google Scholar] [CrossRef] [PubMed]
- Vaclavkova, A.; Chimenti, S.; Arenberger, P.; Hollo, P.; Sator, P.G.; Burcklen, M.; Stefani, M.; D’Ambrosio, D. Oral ponesimod in patients with chronic plaque psoriasis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 384, 2036–2045. [Google Scholar] [CrossRef]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Nat. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degagne, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Guo, Y.; Zhang, C.; Fan, F.; Yang, W. Sphingosine Kinase 1 and Sphingosine-1-Phosphate Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 2109. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liang, Y.; Chang, W.; Hu, B.; Zhang, Y. Triple Negative Breast Cancer Depends on Sphingosine Kinase 1 (SphK1)/Sphingosine-1-Phosphate (S1P)/Sphingosine 1-Phosphate Receptor 3 (S1PR3)/Notch Signaling for Metastasis. Med. Sci. Monit. 2018, 24, 1912–1923. [Google Scholar] [CrossRef]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef]
- Ohotski, J.; Long, J.S.; Orange, C.; Elsberger, B.; Mallon, E.; Doughty, J.; Pyne, S.; Pyne, N.J.; Edwards, J. Expression of sphingosine 1-phosphate receptor 4 and sphingosine kinase 1 is associated with outcome in oestrogen receptor-negative breast cancer. Br. J. Cancer 2012, 106, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, J.; Lee, J.F.; Zhang, W.; Kandouz, M.; VanHecke, G.C.; Chen, S.; Ahn, Y.H.; Lonardo, F.; Lee, M.J. TGF-beta/SMAD3 Pathway Stimulates Sphingosine-1 Phosphate Receptor 3 Expression: Implication of sphingosine-1 phosphate receptor 3 in lung adenocarcinoma progression. J. Biol. Chem. 2016, 291, 27343–27353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, A.; Zhang, W.; Lee, J.F.; An, J.; Ekambaram, P.; Liu, J.; Honn, K.V.; Klinge, C.M.; Lee, M.J. Sphingosine-1-phosphate receptor-3 signaling up-regulates epidermal growth factor receptor and enhances epidermal growth factor receptor-mediated carcinogenic activities in cultured lung adenocarcinoma cells. Int. J. Oncol. 2012, 40, 1619–1626. [Google Scholar] [PubMed] [Green Version]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atherosc. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arteriosc. Thromb. Vasc. Biol. 2011, 31, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S1PR | G Protein | Downstream Signaling Pathways |
|---|---|---|
| S1PR 1–5 | Gαi/o→ | Akt→Ras→MAPK→Rac |
| S1PR 2–5 | Gαq11→ | DAG→PKC→Ca2+ |
| S1PR 2 and 3 | Gα12/13→ | Rho→ROCK |
| S1PR Antagonist | T ½ Elimination | Time to Max Concentration | Median Decrease in Maximum Lymphocyte Count | Maximum Decrease in Steady State Lymphocyte Count | Median recovery Time to Normal Lymphocyte Count |
|---|---|---|---|---|---|
| fingolimod | 6–9 days * | 12–26 h | 60% of baseline in 4–6 h | 18–30% of baseline | 1–2 months |
| siponimod | 30 h | 4 h | 20–30% of baseline | 20–30% of baseline | 10 days, but up to 3–4 weeks for some patients |
| ozanimod CC112273 ** | 21 h 11 days | 6–8 h | 30% of baseline | 45% | 30 days *** |
| ponesimod | 21–33 h | 2.5–5 h | Not available | 70% | 4 days |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohan, S.; Lucassen, E.; Smoot, K.; Brink, J.; Chen, C. Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article. Biomedicines 2020, 8, 227. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070227
Cohan S, Lucassen E, Smoot K, Brink J, Chen C. Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article. Biomedicines. 2020; 8(7):227. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070227
Chicago/Turabian StyleCohan, Stanley, Elisabeth Lucassen, Kyle Smoot, Justine Brink, and Chiayi Chen. 2020. "Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article" Biomedicines 8, no. 7: 227. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8070227