Gene Regulatory Network Analysis of Perivascular Adipose Tissue of Abdominal Aortic Aneurysm Identifies Master Regulators of Key Pathogenetic Pathways



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Gene Expression Data
2.2. Selection of Differentially Expressed (DE) Genes in Dilated PVAT of AAA
2.3. Identification of Cis-Regulatory Elements and TFs
2.4. Inferring the TFs’ Upstream Regulatory Factors
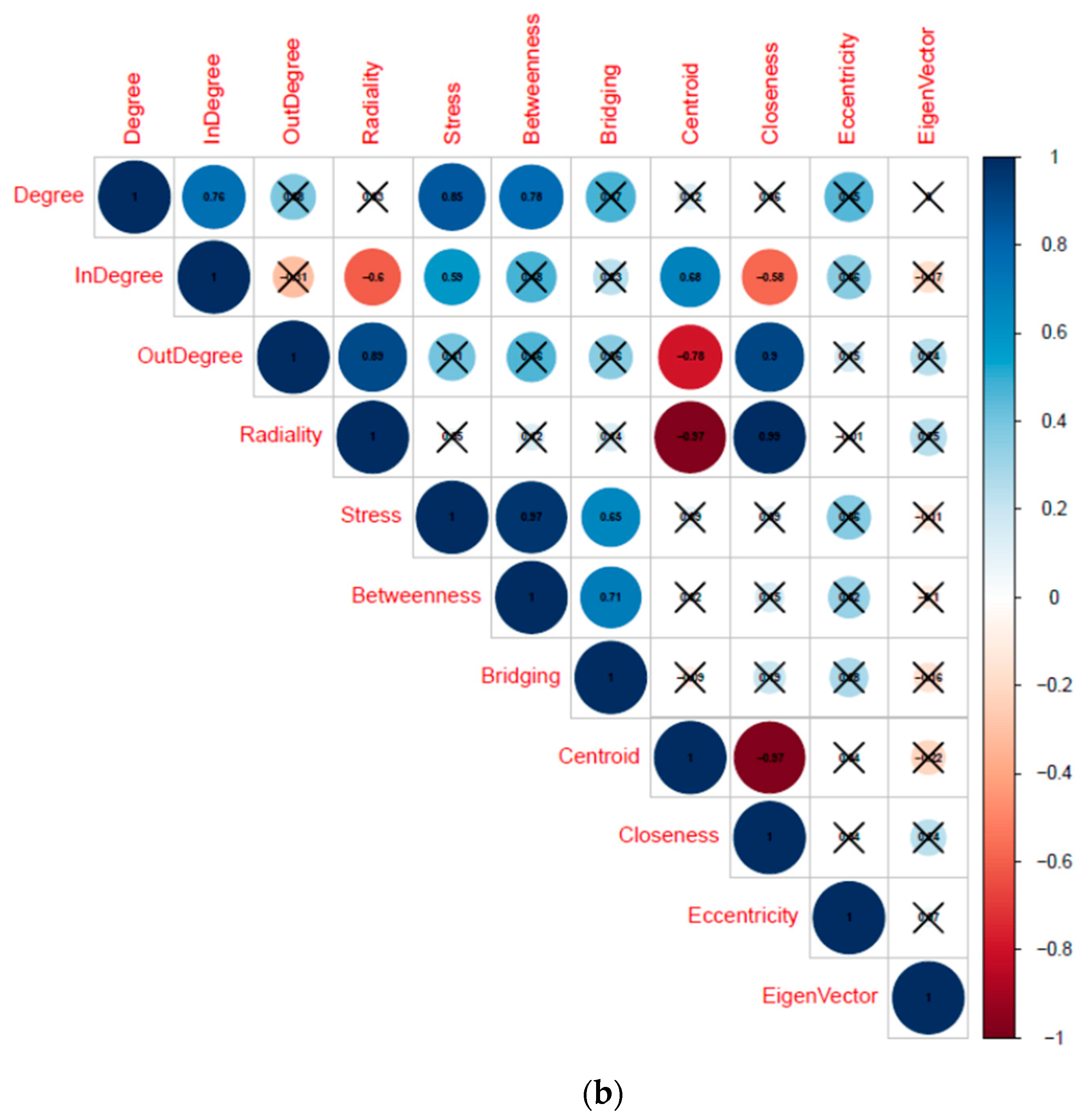
2.5. Topological Analysis
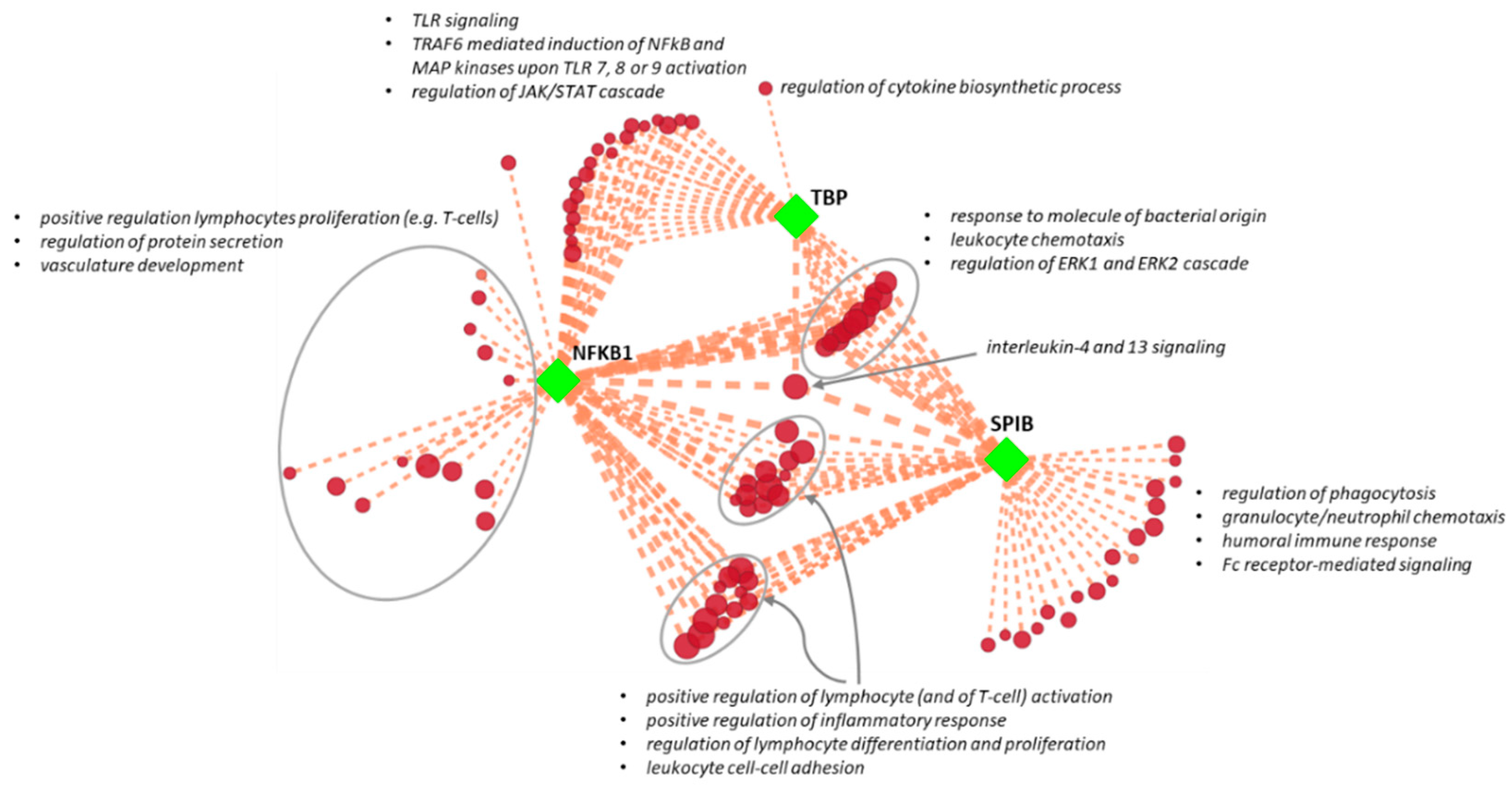
2.6. Linking Transcriptional Complex Clusters with AAA Pathogenetic Biological Functions
3. Results
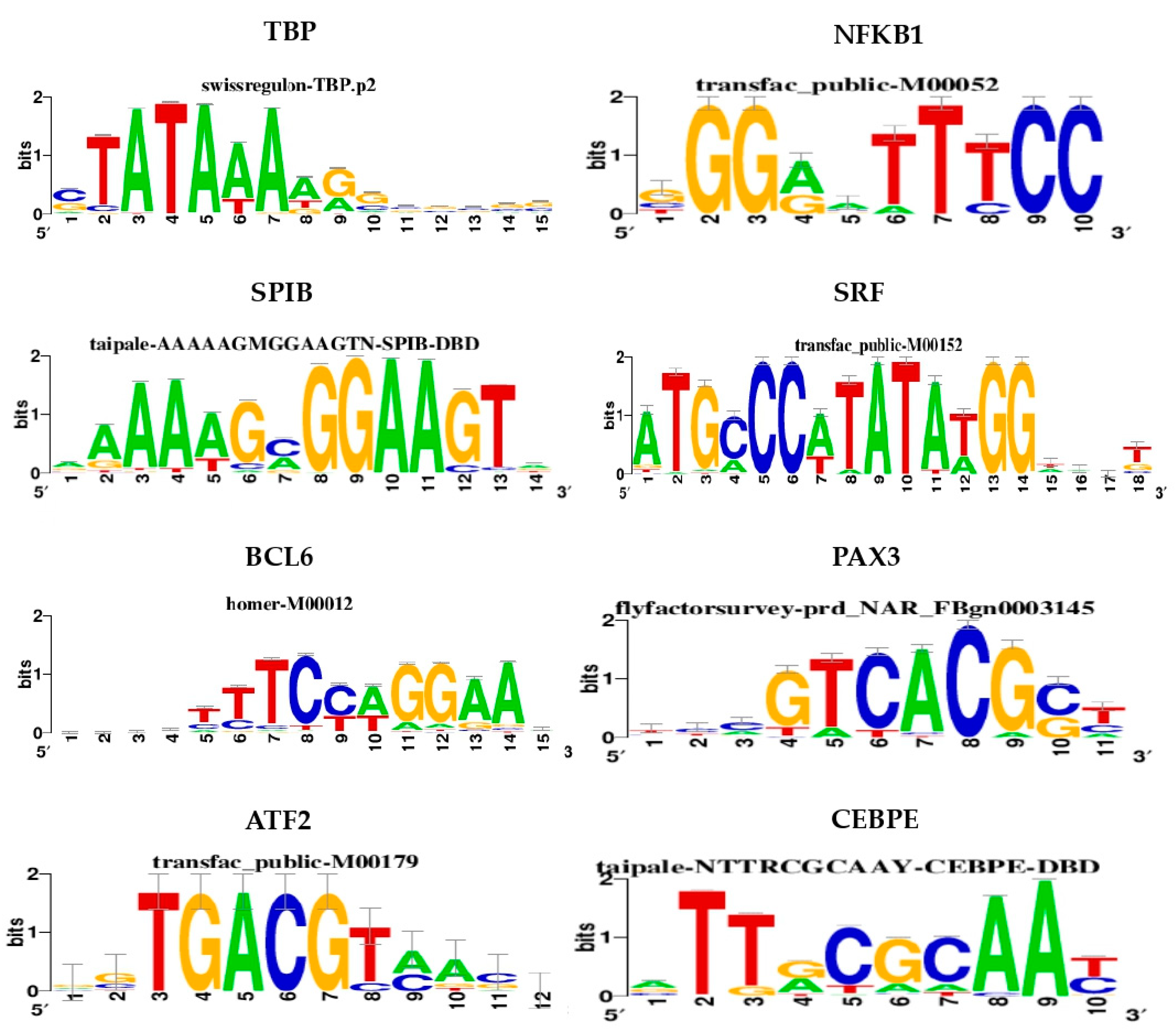
3.1. Identification of Motifs and Tracks of Genes Over-Expressed in Dilated PVAT of AAA Patients
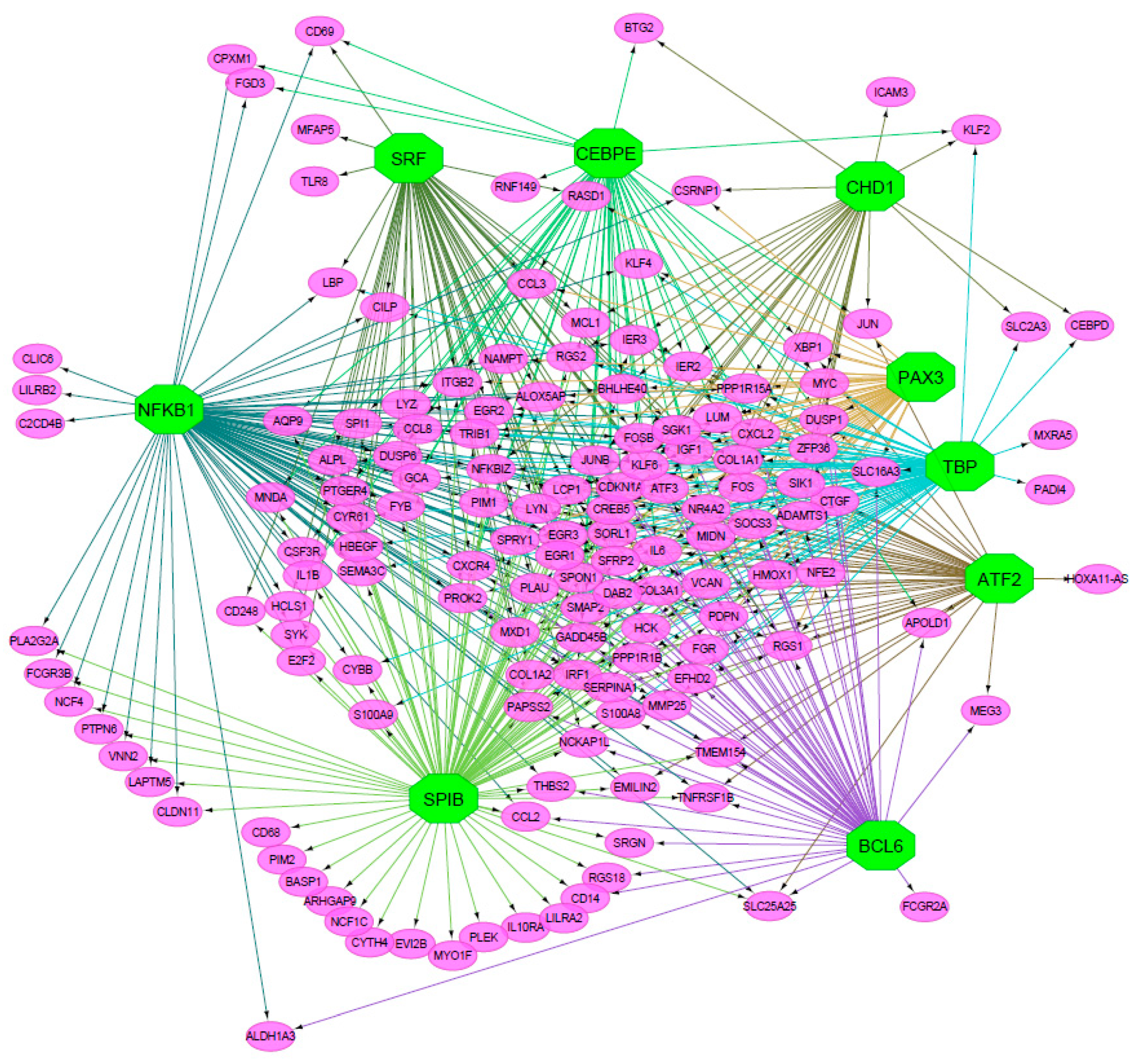
3.1.1. Regulatory Network of TFs and Over-Expressed Genes in Dilated PVAT
3.1.2. Selection of “hub” TFs through Topological Analysis
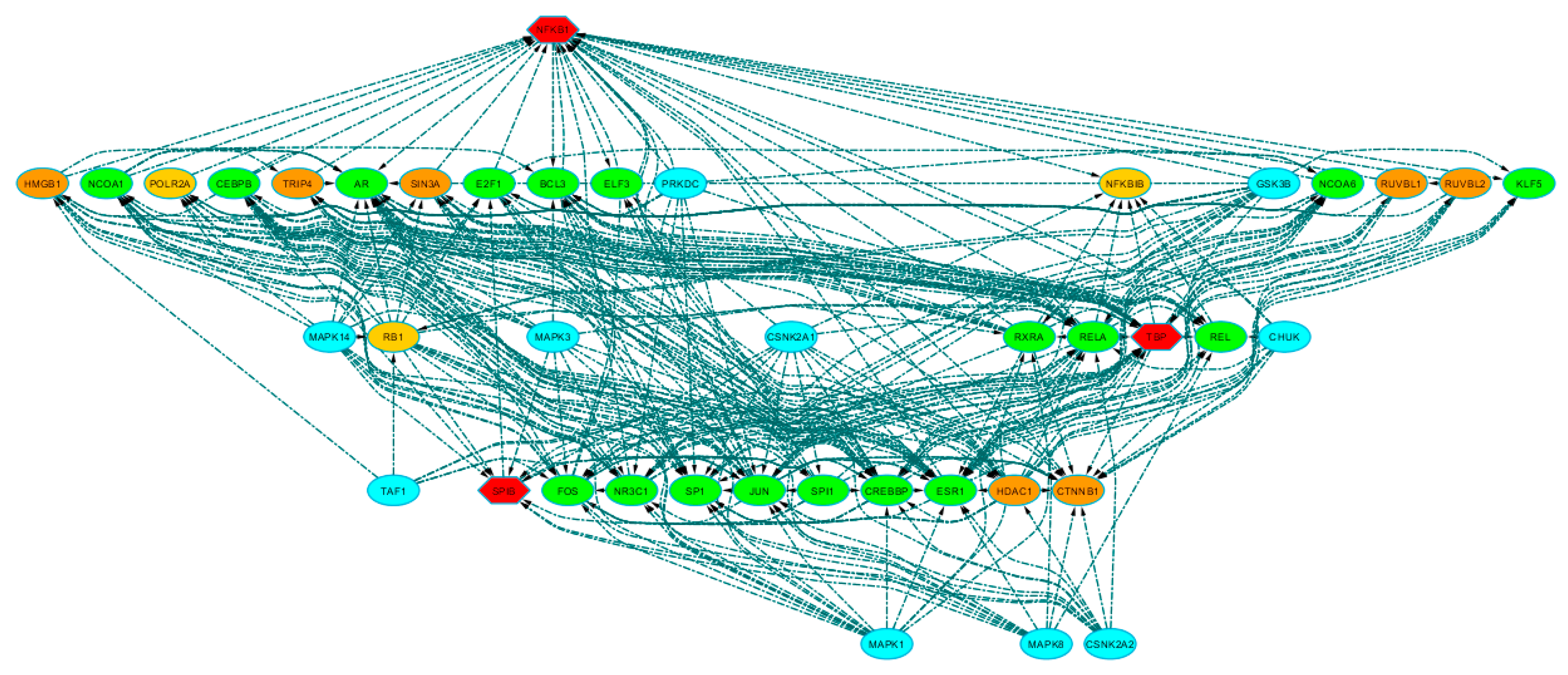
3.2. Identification of Transcriptional Complexes and Upstream Regulators
3.2.1. Connecting Additional Proteins to TFs through Protein–Protein Interactions (PPI)
3.2.2. Identification of Protein Kinases Upstream Transcriptional Complexes.
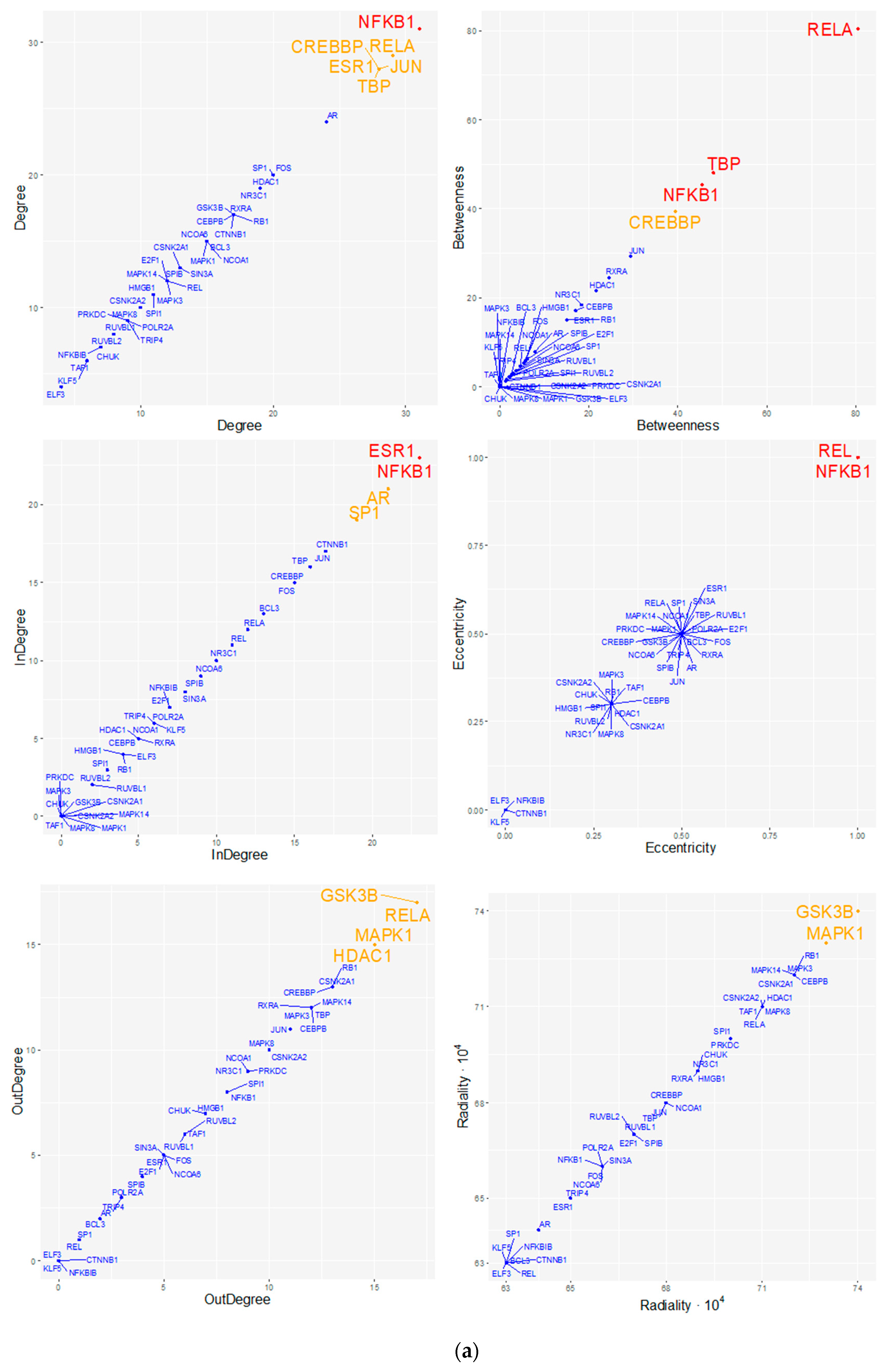
3.2.3. Topological Analysis of Regulatory Transcriptional-Complex
3.3. Association of Regulatory Subnetworks with AAA Pathogenetic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | Abdominal aortic aneurysm |
| AUC | Area under the cumulative recovery curve |
| ChIP | Chromatin immunoprecipitation |
| DE | Differentially expressed |
| GO-BP | Gene Ontology–biological process |
| GSEA | Gene set enrichment analysis |
| NES | Normalized enrichment score |
| PWM | Positional weight matrix |
| PPI | Protein–protein interaction |
| PVAT | Perivascular adipose tissue |
| TcoF | Transcription co-factor |
| TF | Transcription factor |
| TLR | Toll-like receptors |
| TSS | Transcription start site |
References
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, J.H.; Matsumura, J.S. Pharmacologic Management of Aneurysms. Circ. Res. 2019, 124, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.L.; Stubbe, J.; Lindholt, J.S. Animal Models Used to Explore Abdominal Aortic Aneurysms: A Systematic Review. Eur. J. Vasc. Endovasc. Surg. 2016, 52, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromp, G.; Kuivaniemi, H. Developments in genomics to improve understanding, diagnosis and management of aneurysms and peripheral artery disease. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacentini, L.; Werba, J.P.; Bono, E.; Saccu, C.; Tremoli, E.; Spirito, R.; Colombo, G.I. Genome-Wide Expression Profiling Unveils Autoimmune Response Signatures in the Perivascular Adipose Tissue of Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2019, 39, 237–249. [Google Scholar] [CrossRef]
- Piacentini, L.; Saccu, C.; Bono, E.; Tremoli, E.; Spirito, R.; Colombo, G.I.; Werba, J.P. Gene-expression profiles of abdominal perivascular adipose tissue distinguish aortic occlusive from stenotic atherosclerotic lesions and denote different pathogenetic pathways. Sci. Rep. 2020, 10, 6245. [Google Scholar] [CrossRef]
- Rajsheker, S.; Manka, D.; Blomkalns, A.L.; Chatterjee, T.K.; Stoll, L.L.; Weintraub, N.L. Crosstalk between perivascular adipose tissue and blood vessels. Curr. Opin. Pharmacol. 2010, 10, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arter. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375. [Google Scholar] [CrossRef]
- Queiroz, M.; Sena, C.M. Perivascular adipose tissue in age-related vascular disease. Ageing Res. Rev. 2020, 59, 101040. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Dehmer, M.; Haibe-Kains, B. Gene regulatory networks and their applications: Understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, C.; Rieckhof, G.; Califano, A. Reverse-engineering human regulatory networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Moll, F.L.; Powell, J.T.; Fraedrich, G.; Verzini, F.; Haulon, S.; Waltham, M.; van Herwaarden, J.A.; Holt, P.J.E.; van Keulen, J.W.; Rantner, B.; et al. Management of abdominal aortic aneurysms clinical practice guidelines of the European society for vascular surgery. Eur. J. Vasc. Endovasc. Surg. 2011, 41, S1–S58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italian Journal of Vascular and Endovascular Surgery 23 March 2016 (1 Suppl 1)—Minerva Medica Journals. Available online: https://www.minervamedica.it/en/journals/vascular-endovascular-surgery/issue.php?cod=R46Y2016S01 (accessed on 30 July 2020).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Janky, R.; Verfaillie, A.; Imrichová, H.; Van de Sande, B.V.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Sanchez, M.N.; Potier, D.; et al. iRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, A.; Imrichova, H.; Janky, R.; Aerts, S. iRegulon and i-cisTarget: Reconstructing Regulatory Networks Using Motif and Track Enrichment. Curr. Protoc. Bioinform. 2015, 52, 2.16.1–2.16.39. [Google Scholar] [CrossRef]
- Chen, E.Y.; Xu, H.; Gordonov, S.; Lim, M.P.; Perkins, M.H.; Ma’ayan, A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012, 28, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012, 7, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The Importance of Bottlenecks in Protein Networks: Correlation with Gene Essentiality and Expression Dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Dale, M.A.; Ruhlman, M.K.; Baxter, B.T. Inflammatory cell phenotypes in AAAs: Their role and potential as targets for therapy. Arter. Thromb. Vasc. Biol. 2015, 35, 1746–1755. [Google Scholar] [CrossRef] [Green Version]
- Folkesson, M.; Vorkapic, E.; Gulbins, E.; Japtok, L.; Kleuser, B.; Welander, M.; Länne, T.; Wågsäter, D. Inflammatory cells, ceramides, and expression of proteases in perivascular adipose tissue adjacent to human abdominal aortic aneurysms. J. Vasc. Surg. 2017, 65, 1171–1179.e1. [Google Scholar] [CrossRef] [Green Version]
- Schmeier, S.; Alam, T.; Essack, M.; Bajic, V.B. TcoF-DB v2: Update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017, 45, D145–D150. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.F.H.; Stoffel, E.; Cohen, P. Role of Perivascular Adipose Tissue in Vascular Physiology and Pathology. Hypertension 2017, 69, 770–777. [Google Scholar] [CrossRef]
- Horimatsu, T.; Kim, H.W.; Weintraub, N.L. The Role of Perivascular Adipose Tissue in Non-atherosclerotic Vascular Disease. Front. Physiol. 2017, 8, 969. [Google Scholar] [CrossRef] [PubMed]
- Estrelinha, M.; Hinterseher, I.; Kuivaniemi, H. Gene expression studies in human abdominal aortic aneurysm. Rev. Vasc. Med. 2014, 2, 77–82. [Google Scholar] [CrossRef]
- Hao, T.; Wang, Q.; Zhao, L.; Wu, D.; Wang, E.; Sun, J. Analyzing of Molecular Networks for Human Diseases and Drug Discovery. Curr. Top. Med. Chem. 2018, 18, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Rezaei-Tavirani, M.; Goliaei, B.; Peyvandi, A.A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 2014, 7, 17–31. [Google Scholar]
- Rivas, J.D.L.; Prieto, C. Protein Interactions: Mapping Interactome Networks to Support Drug Target Discovery and Selection. In Bioinformatics and Drug Discovery; Larson, R.S., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 279–296. ISBN 978-1-61779-965-5. [Google Scholar]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-κB Pathway in Lymphocyte Development and Function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [Green Version]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Wong, H.; Banerjee, A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [Green Version]
- Shiraya, S.; Miwa, K.; Aoki, M.; Miyake, T.; Oishi, M.; Kataoka, K.; Ohgi, S.; Ogihara, T.; Kaneda, Y.; Morishita, R. Hypertension Accelerated Experimental Abdominal Aortic Aneurysm Through Upregulation of Nuclear Factor κB and Ets. Hypertension 2006, 48, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Tanaka, T.; Kent, K.C.; Yamanouchi, D. Osteoclastogenic Differentiation of Macrophages in the Development of Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Tieu, B.; Recinos, A.; Tilton, R.G.; Brasier, A.R. RhoA mediates angiotensin II-induced phospho-Ser536 nuclear factor kappaB/RelA subunit exchange on the interleukin-6 promoter in VSMCs. Circ. Res. 2006, 99, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijaz, T.; Sun, H.; Pinchuk, I.V.; Milewicz, D.M.; Tilton, R.G.; Brasier, A.R. Deletion of NF-κB/RelA in Angiotensin II-Sensitive Mesenchymal Cells Blocks Aortic Vascular Inflammation and Abdominal Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2017, 37, 1881–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Miyake, T.; Kurashiki, T.; Morishita, R. Molecular Pharmacological Approaches for Treating Abdominal Aortic Aneurysm. Ann. Vasc. Dis. 2019, 12, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeman, J.H.N.; Rabelink, T.J.; van Bockel, J.H. Immunosuppression and the abdominal aortic aneurysm: Doctor Jekyll or Mister Hyde? Circulation 2011, 124, e463–e465. [Google Scholar] [CrossRef] [Green Version]
- Willis, S.N.; Tellier, J.; Liao, Y.; Trezise, S.; Light, A.; O’Donnell, K.; Garrett-Sinha, L.A.; Shi, W.; Tarlinton, D.M.; Nutt, S.L. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat. Commun. 2017, 8, 1426. [Google Scholar] [CrossRef]
- Rosales, C. Fcγ Receptor Heterogeneity in Leukocyte Functional Responses. Front. Immunol. 2017, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jönsson, F. Expression, Role, and Regulation of Neutrophil Fcγ Receptors. Front. Immunol. 2019, 10, 1958. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, C.-Q.; Wang, S.-W.; Li, W.; Li, J.; Wang, S.-M. Characterization of Fc gamma receptor IIb expression within abdominal aortic aneurysm. Biochem. Biophys. Res. Commun. 2017, 485, 295–300. [Google Scholar] [CrossRef]
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Ghosh, A.; DiMusto, P.D.; Ehrlichman, L.K.; Sadiq, O.; McEvoy, B.; Futchko, J.S.; Henke, P.K.; Eliason, J.L.; Upchurch, G.R. The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J. Am. Coll. Surg. 2012, 215, 668–680.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neels, J.G.; Hassen-Khodja, R.; Chinetti, G. Nuclear receptors in abdominal aortic aneurysms. Atherosclerosis 2020, 297, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ichiki, T.; Funakoshi, Y.; Ito, K.; Takeshita, A. Downregulation of angiotensin II type 1 receptor by all-trans retinoic acid in vascular smooth muscle cells. Hypertension 2000, 35, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez, V.; Alameda, D.; Rico, D.; Mota, R.; Gonzalo, P.; Cedenilla, M.; Fischer, T.; Boscá, L.; Glass, C.K.; Arroyo, A.G.; et al. Retinoid X receptor α controls innate inflammatory responses through the up-regulation of chemokine expression. Proc. Natl. Acad. Sci. USA 2010, 107, 10626–10631. [Google Scholar] [CrossRef] [Green Version]
- Galán, M.; Varona, S.; Orriols, M.; Rodríguez, J.A.; Aguiló, S.; Dilmé, J.; Camacho, M.; Martínez-González, J.; Rodriguez, C. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. Dis. Model. Mech. 2016, 9, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Akimova, T.; Beier, U.H.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases and T-cell immune responses. Blood 2012, 119, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Kim, H.W.; de Chantemèle, E.J.B.; Weintraub, N.L. Perivascular Adipocytes in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2220–2227. [Google Scholar] [CrossRef]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between Adipocytes and Immune Cells in Adipose Tissue Inflammation and Metabolic Dysregulation in Obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar] [CrossRef]
- Wang, S.K.; Murphy, M.P. Immune Modulation as a Treatment for Abdominal Aortic Aneurysms. Circ. Res. 2018, 122, 925–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.W.; Gracon, A.S.A.; Murphy, M.P.; Wilkes, D.S. Exploring autoimmunity in the pathogenesis of abdominal aortic aneurysms. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H719–H727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluestone, J.A.; Bour-Jordan, H. Current and future immunomodulation strategies to restore tolerance in autoimmune diseases. Cold Spring Harb. Perspect. Biol. 2012, 4, a007542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagan, A.; Mikolajczyk, T.P.; Mrowiecki, W.; MacRitchie, N.; Daly, K.; Meldrum, A.; Migliarino, S.; Delles, C.; Urbanski, K.; Filip, G.; et al. T Cells Are Dominant Population in Human Abdominal Aortic Aneurysms and Their Infiltration in the Perivascular Tissue Correlates With Disease Severity. Front. Immunol. 2019, 10, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | TF | NES | AUC | # Targets | # Motifs/Tracks |
|---|---|---|---|---|---|
| M1 | TBP | 6.20 | 0.098 | 74 | 5 |
| M2 | NFKB1 | 5.91 | 0.095 | 91 | 7 |
| T1 | CHD1 | 5.23 | 0.130 | 29 | 1 |
| M3 | SPIB | 5.12 | 0.085 | 104 | 4 |
| M4 | SRF | 4.66 | 0.080 | 53 | 6 |
| M5 | BCL6 | 4.14 | 0.074 | 55 | 3 |
| M6 | PAX3 | 4.07 | 0.073 | 48 | 2 |
| M7 | ATF2 | 3.58 | 0.068 | 46 | 1 |
| M8 | CEBPE | 3.53 | 0.067 | 50 | 2 |
| TF (Cluster) | Degree | Betweenness Centrality | Radiality |
|---|---|---|---|
| SPIB (M3) | 104 | 0.36 | 3.41 |
| NFKB1 (M2) | 91 | 0.22 | 3.24 |
| TBP (M1) | 74 | 0.14 | 3.02 |
| BCL6 (M5) | 55 | 0.08 | 2.77 |
| SRF (M4) | 53 | 0.08 | 2.75 |
| CEBPE (M8) | 50 | 0.08 | 2.71 |
| PAX3 (M6) | 48 | 0.05 | 2.68 |
| ATF2 (M7) | 46 | 0.06 | 2.65 |
| CHD1 (T1) | 29 | 0.04 | 2.43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piacentini, L.; Chiesa, M.; Colombo, G.I. Gene Regulatory Network Analysis of Perivascular Adipose Tissue of Abdominal Aortic Aneurysm Identifies Master Regulators of Key Pathogenetic Pathways. Biomedicines 2020, 8, 288. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8080288
Piacentini L, Chiesa M, Colombo GI. Gene Regulatory Network Analysis of Perivascular Adipose Tissue of Abdominal Aortic Aneurysm Identifies Master Regulators of Key Pathogenetic Pathways. Biomedicines. 2020; 8(8):288. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8080288
Chicago/Turabian StylePiacentini, Luca, Mattia Chiesa, and Gualtiero Ivanoe Colombo. 2020. "Gene Regulatory Network Analysis of Perivascular Adipose Tissue of Abdominal Aortic Aneurysm Identifies Master Regulators of Key Pathogenetic Pathways" Biomedicines 8, no. 8: 288. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8080288