Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections

 and
and 
Abstract
:1. Introduction
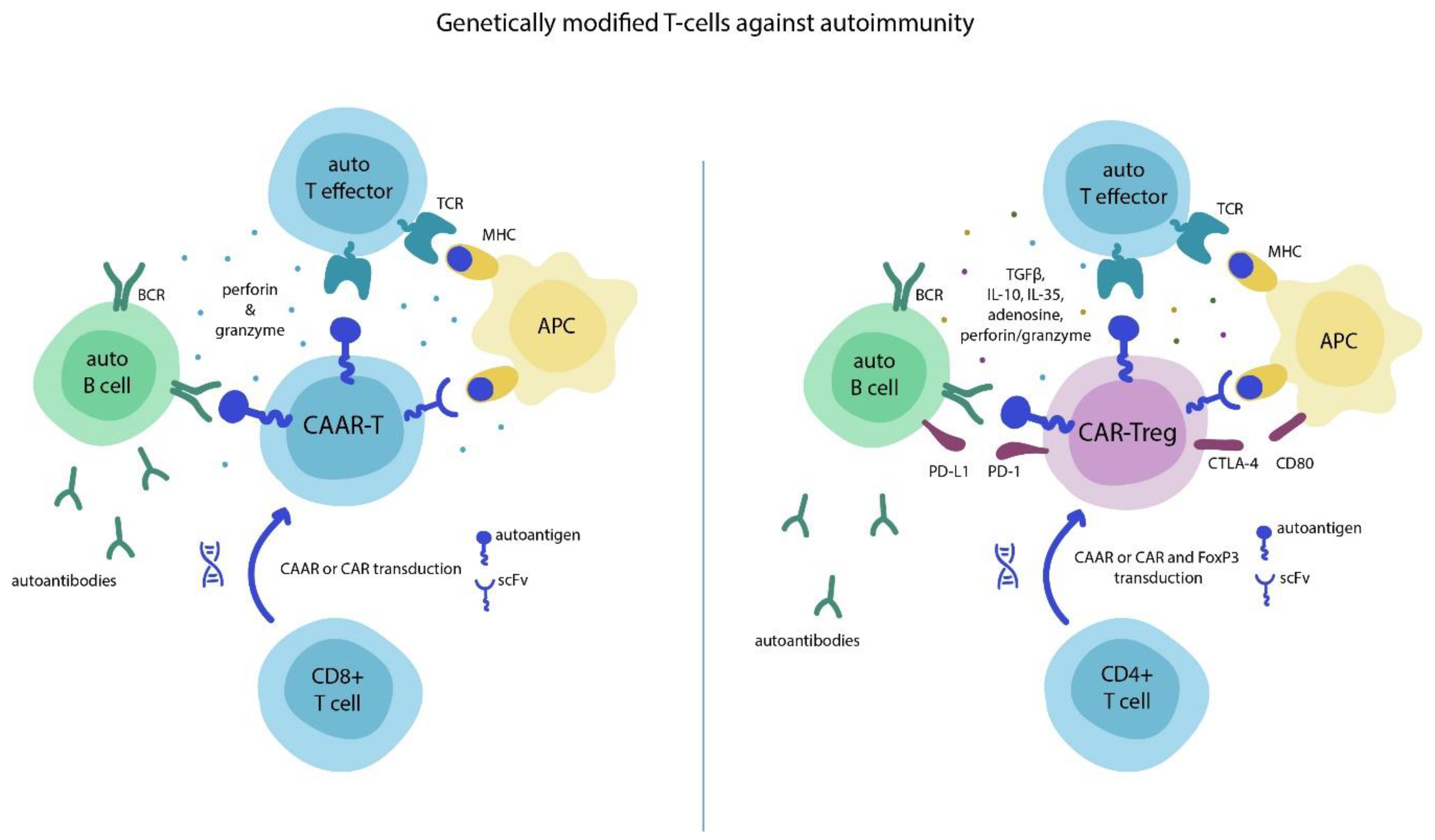
2. Chimeric Antigen Receptor (CAR)-T Cells and Autoimmunity
3. Allergy and Asthma
4. Infectious Diseases
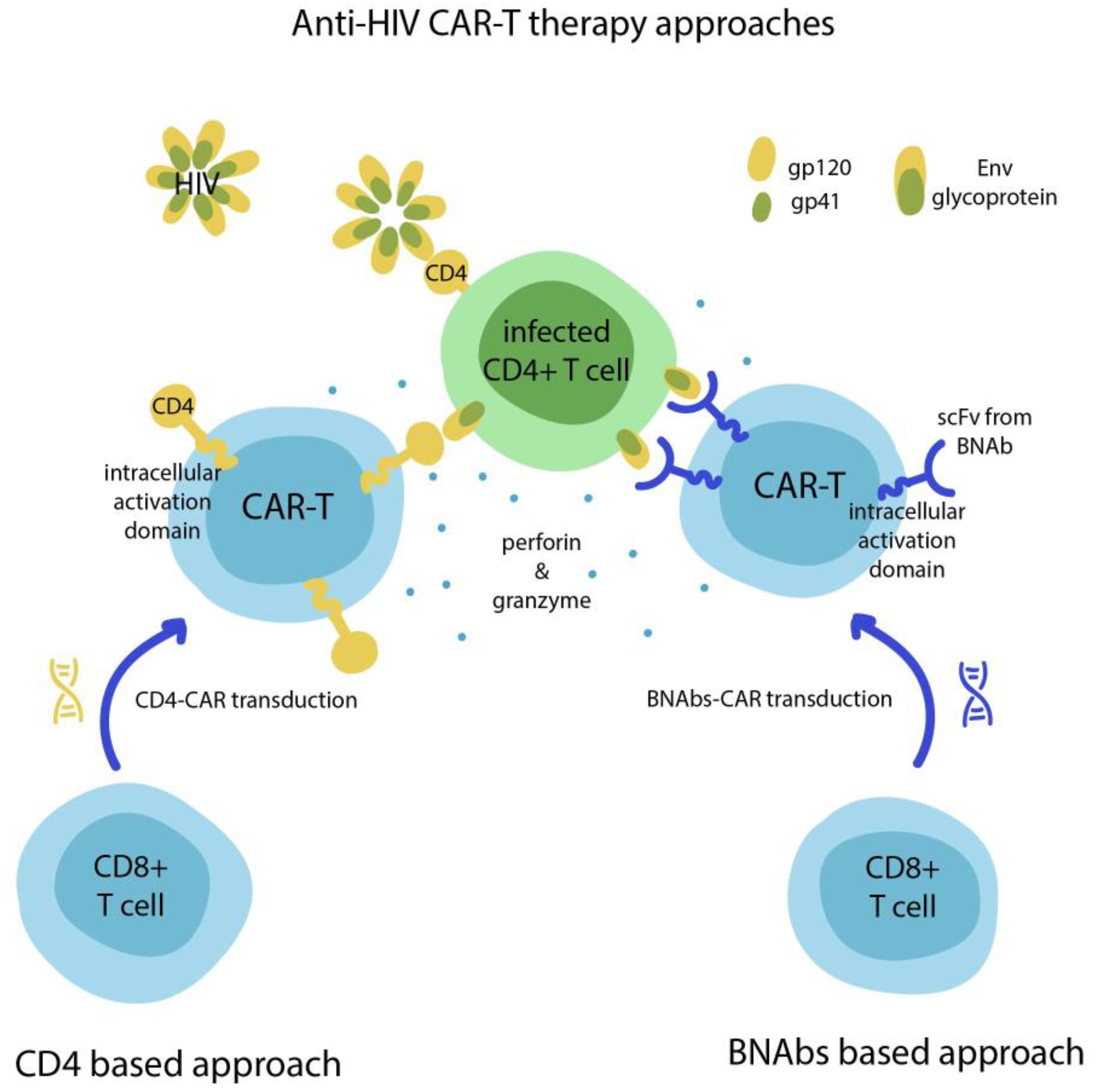
5. HIV
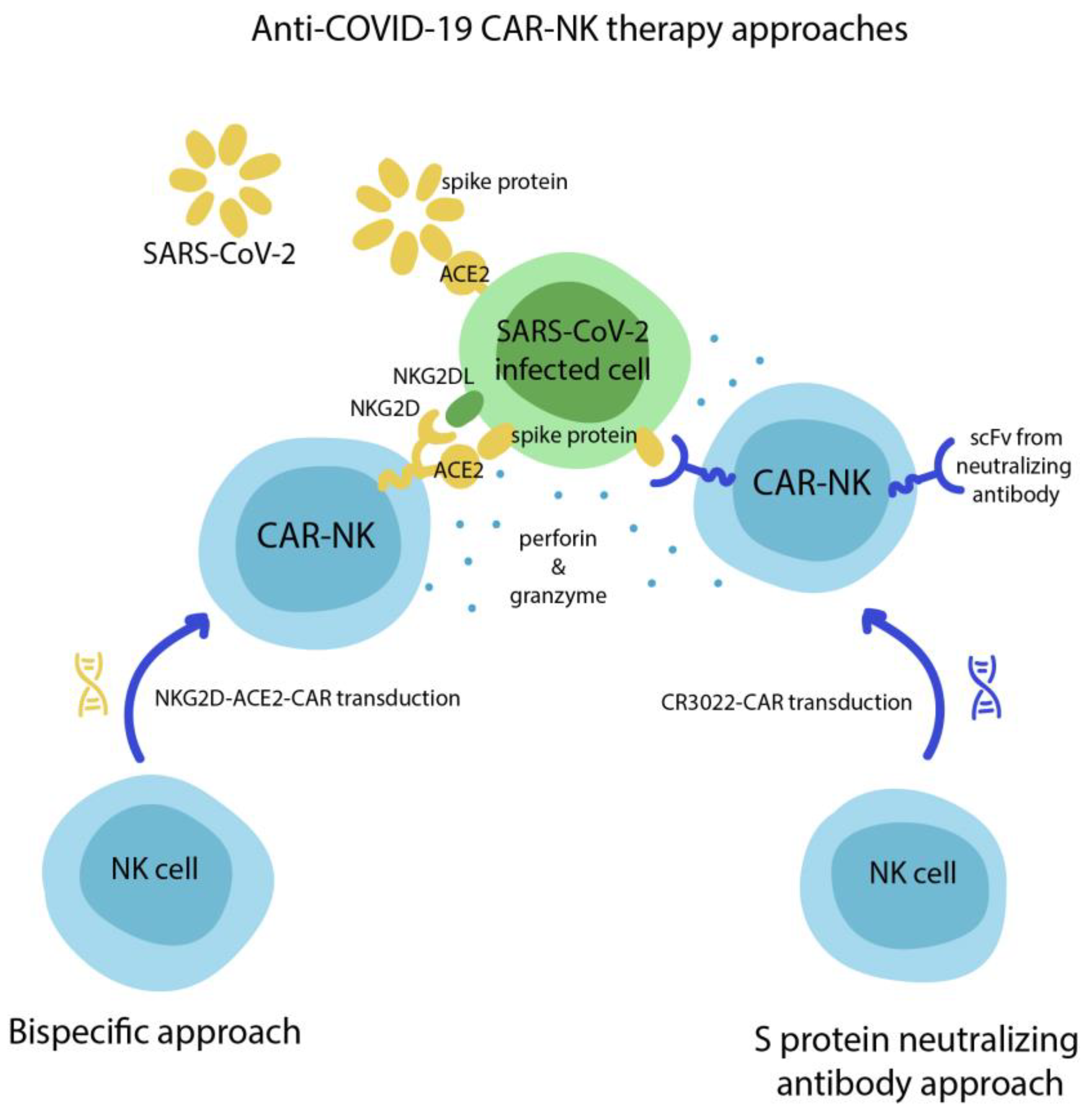
6. COVID-19
7. Cardiac Fibrosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cell Transfer |
| ADs | Allergic Diseases |
| AIDs | Autoimmune Diseases |
| APC | Antigen-Presenting Cell |
| BAR | B cell Antibody-targeting Receptor |
| BCMA | B Cell Maturation Antigen |
| BCR | B Cell Receptor |
| BNAbs | Broadly Neutralizing Antibodies |
| CAAR | Chimeric Autoantibody Receptor |
| CAR | Chimeric Antigen Receptor |
| cART | combination Antiretroviral Therapy |
| CEA | Carcinoembryonic Antigen |
| CNS | Central Nervous System |
| CRD | Carbohydrate Recognition Domain |
| CRS | Cytokine Release Syndrome |
| CSS | Cytokine Storm Syndrome |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| FAP | Fibroblast Activation Protein |
| gB | Glycoprotein B |
| GvHD | graft-versus-host disease |
| HA | Hemophilia A |
| HBV | Hepatitis B Virus |
| HCMV | Human Cytomegalovirus |
| HCV | Hepatitis C Virus |
| HCV/E2 | HCV E2 glycoprotein |
| HIV-1 | Human Immunodeficiency Virus type 1 |
| IFI | Invasive Fungal Infection |
| IFNγ | Interferon gamma |
| MHC | major histocompatibility complex |
| MOG | Myelin Oligodendrocyte Glycoprotein |
| MS | Multiple Sclerosis |
| NK | Natural Killer |
| NMOSD | Neuromyelitis Optica Spectrum Disorder |
| NOD | Non-obese diabetic |
| PID | Primary Immune Deficiency |
| PV | Pemphigus Vulgaris |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| scFv | single-chain Variable fragment |
| SLE | Systemic Lupus Erythematosus |
| T1D | Type 1 Diabetes |
| TCR | T cell receptor |
| TILs | Tumor-Infiltrating Lymphocytes |
| TNFα | Tumor Necrosis Factor alpha |
| Treg | T regulatory lymphocyte |
References
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Parvathaneni, K.; Scott, D.W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018, 2, 2332–2340. [Google Scholar] [CrossRef] [Green Version]
- Fishman, S.; Lewis, M.D.; Siew, L.K.; De Leenheer, E.; Kakabadse, D.; Davies, J.; Ziv, D.; Margalit, A.; Karin, N.; Gross, G.; et al. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol. Ther. 2017, 25, 456–464. [Google Scholar] [CrossRef]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2019, 96, 50–58. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.F.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S.I. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 2012, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmaeilzadeh, A.; Tahmasebi, S.; Athari, S.S. Chimeric antigen receptor -T cell therapy: Applications and challenges in treatment of allergy and asthma. Biomed. Pharmacother. 2020, 123, 109685. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.E.; Fay, B.L.; Adejuwon, A.; Han, H.; Ma, Z. Chimeric antigen receptors based on low affinity mutants of FcϵRI Re-direct T cell specificity to cells expressing membrane IgE. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric antigen receptor-redirected regulatory T cells suppress experimental allergic airway inflammation, a model of asthma. Front. Immunol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef] [Green Version]
- Krebs, K.; Böttinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T Cells Expressing a Chimeric Antigen Receptor That Binds Hepatitis B Virus Envelope Proteins Control Virus Replication in Mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Festag, M.M.; Festag, J.; Fräßle, S.P.; Asen, T.; Sacherl, J.; Schreiber, S.; Mück-Häusl, M.A.; Busch, D.H.; Wisskirchen, K.; Protzer, U. Evaluation of a Fully Human, Hepatitis B Virus-Specific Chimeric Antigen Receptor in an Immunocompetent Mouse Model. Mol. Ther. 2019, 27, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Kruse, R.L.; Shum, T.; Tashiro, H.; Barzi, M.; Yi, Z.; Whitten-Bauer, C.; Legras, X.; Bissig-Choisat, B.; Garaigorta, U.; Gottschalk, S.; et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 2018, 20, 697–705. [Google Scholar] [CrossRef]
- Sautto, G.A.; Wisskirchen, K.; Clementi, N.; Castelli, M.; Diotti, R.A.; Graf, J.; Clementi, M.; Burioni, R.; Protzer, U.; Mancini, N. Chimeric antigen receptor (CAR)-engineered t cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016, 65, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Proff, J.; Brey, C.U.; Ensser, A.; Holter, W.; Lehner, M. Turning the tables on cytomegalovirus: Targeting viral Fc receptors by CARs containing mutated CH2-CH3 IgG spacer domains. J. Transl. Med. 2018, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, S.J. An Influenza Virus M2 Protein Specific Chimeric Antigen Receptor Modulates Influenza A/WSN/33 H1N1 Infection In Vivo. Open Virol. J. 2013, 7, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, S.; Del Vecchio, C.; Gavioli, R.; Mattiuzzo, G.; Cusi, M.G.; Micheli, L.; Gennari, F.; Siccardi, A.; Marasco, W.A.; Palù, G.; et al. T-cell engineering by a chimeric T-cell receptor with antibody-type specificity for the HIV-1 gp120. Gene Ther. 2005, 12, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zhang, W.; Zhang, H. Development of CAR-T cells for long-term eradication and surveillance of HIV-1 reservoir. Curr. Opin. Virol. 2019, 38, 21–30. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Sahu, G.K.; Sango, K.; Selliah, N.; Ma, Q.; Skowron, G.; Junghans, R.P. Anti-HIV designer T cells progressively eradicate a latently infected cell line by sequentially inducing HIV reactivation then killing the newly gp120-positive cells. Virology 2013, 446, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Leibman, R.S.; Richardson, M.W.; Ellebrecht, C.T.; Maldini, C.R.; Glover, J.A.; Secreto, A.J.; Kulikovskaya, I.; Lacey, S.F.; Akkina, S.R.; Yi, Y.; et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 2017, 13, 1–30. [Google Scholar] [CrossRef]
- Ali, A.; Kitchen, S.G.; Chen, I.S.Y.; Ng, H.L.; Zack, J.A.; Yang, O.O. HIV-1-Specific Chimeric Antigen Receptors Based on Broadly Neutralizing Antibodies. J. Virol. 2016, 90, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zou, F.; Lu, L.; Chen, C.; He, D.; Zhang, X.; Tang, X.; Liu, C.; Li, L.; Zhang, H. Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4 + T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive. J. Virol. 2016, 90, 9712–9724. [Google Scholar] [CrossRef] [Green Version]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, M.H.; Bolivar-Wagers, S.; Dey, B.; Hajduczki, A.; Vargas-Inchaustegui, D.A.; Danielson, D.T.; Bundoc, V.; Liu, L.; Berger, E.A. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti–human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy 2018, 20, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Golchin, A. Cell-Based Therapy for Severe COVID-19 Patients: Clinical Trials and Cost-Utility. Stem Cell Rev. Rep. 2020, 1–7. [Google Scholar] [CrossRef]
- Ma, M.; Badeti, S.; Geng, K.; Liu, D. Efficacy of Targeting SARS-CoV-2 by CAR-NK Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- England, J.T.; Abdulla, A.; Biggs, C.M.; Lee, A.Y.Y.; Hay, K.A.; Hoiland, R.L.; Wellington, C.L.; Sekhon, M.; Jamal, S.; Shojania, K.; et al. Weathering the COVID-19 storm: Lessons from hematologic cytokine syndromes. Blood Rev. 2020, 100707. [Google Scholar] [CrossRef]
- Hoiland, R.L.; Stukas, S.; Cooper, J.; Thiara, S.; Chen, L.Y.C.; Biggs, C.M.; Hay, K.A.; Lee, A.Y.Y.; Shojania, K.; Abdulla, A.; et al. Amelioration of COVID-19-related cytokine storm syndrome: Parallels to chimeric antigen receptor-T cell cytokine release syndrome. Br. J. Haematol. 2020, 190, e126–e156. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Johansen, A.K.Z.; Molkentin, J.D. CARdiac Immunotherapy: T Cells Engineered to Treat the Fibrotic Heart. Mol. Ther. 2019, 27, 1869–1871. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Keller, M.D.; Bollard, C.M. Virus-specific T-cell therapies for patients with primary immune deficiency. Blood 2020, 135, 620–628. [Google Scholar] [CrossRef]
- Mylvaganam, G.; Yanez, A.G.; Maus, M.; Walker, B.D. Toward T Cell-Mediated Control or Elimination of HIV Reservoirs: Lessons From Cancer Immunology. Front. Immunol. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Target/Approach | NCT, Current Status |
|---|---|---|
| Mucosal-Dominant Pemphigus Vulgaris | Clone-specific anti-Dsg3 CAAR-T | NCT04422912, Phase I recruiting |
| Generalized Myasthenia Gravis | Non-specific anti-BCMA CAR-T | NCT04146051, Phase I, II recruiting |
| Systemic Lupus Erythematosus | Non-specific anti-CD19 CAR-T | NCT03030976, Phase I, unknown |
| Neuromyelitis Optica Spectrum Disorder | Non-specific tandem anti-CD19 and anti-CD20 CAR-T | NCT03605238, Phase I, withdrawn |
| Non-specific anti-BCMA CAR-T | NCT04561557, Phase I recruiting | |
| Human Immunodeficiency Virus | Anti-gp120 BNAbs based CAR-T | NCT03240328, Phase I recruiting |
| NCT03980691, Phase I recruiting | ||
| Anti-gp120 dual CAR-T | NCT04648046, Phase I not yet recruiting | |
| COVID-19 | Bispecific anti-ACE2 and anti-NKG2D CAR-NK | NCT04324996, Phase I, II recruiting |
| Disease | Target for CAR-T Cell |
|---|---|
| Autoimmunity and allergy | |
| Pemphigus vulgaris | Keratinocyte adhesion protein Dsg3 [6] |
| Hemophilia A | Anti-FVIII antibody [7] |
| Type 1 diabetes | Insulin-B chain [8,9,10] Islet-specific glucose-6-phosphatase catalytic subunit-related protein [8] |
| Multiple sclerosis | Myelin oligodendrocyte glycoprotein [11] |
| Ulcerative colitis | Carcinoembryonic antigen [12] |
| Allergy | Transmembrane form of IgE [14] |
| Allergic asthma | Carcinoembryonic antigen [15] |
| Infectious diseases | |
| HBV | S domain of HBsAg [17,18,19] |
| HCV | Glycoprotein E2 [20] |
| HCMV | Glycoprotein B [21] |
| Aspergillus | Carbohydrate of cell surface [22] |
| Influenza A | M2e protein [23] |
| HIV | gp120 [24,26,27,28,29,30,32] gp41 [29,30] Oligomannose patch on Envs [33] |
| SARS-CoV-2 | S protein [35] |
| Other | |
| Cardiac fibrosis | Fibroblast activation protein [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010059
Zmievskaya E, Valiullina A, Ganeeva I, Petukhov A, Rizvanov A, Bulatov E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines. 2021; 9(1):59. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010059
Chicago/Turabian StyleZmievskaya, Ekaterina, Aygul Valiullina, Irina Ganeeva, Alexey Petukhov, Albert Rizvanov, and Emil Bulatov. 2021. "Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections" Biomedicines 9, no. 1: 59. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9010059