
A DNA Methylation-Based Gene Signature Can Predict Triple-Negative Breast Cancer Diagnosis


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Methylome Data Sets
2.2. Bioinformatics Analysis
2.2.1. Generation of a Diagnostic Signature for TNBC
2.2.2. Robustness of the Model
2.3. Validation of the Signature
2.3.1. Patient Samples
2.3.2. DNA and cfDNA Extraction and Bisulphite Conversion
2.3.3. Pyrosequencing
2.4. Statistical Analysis
3. Results
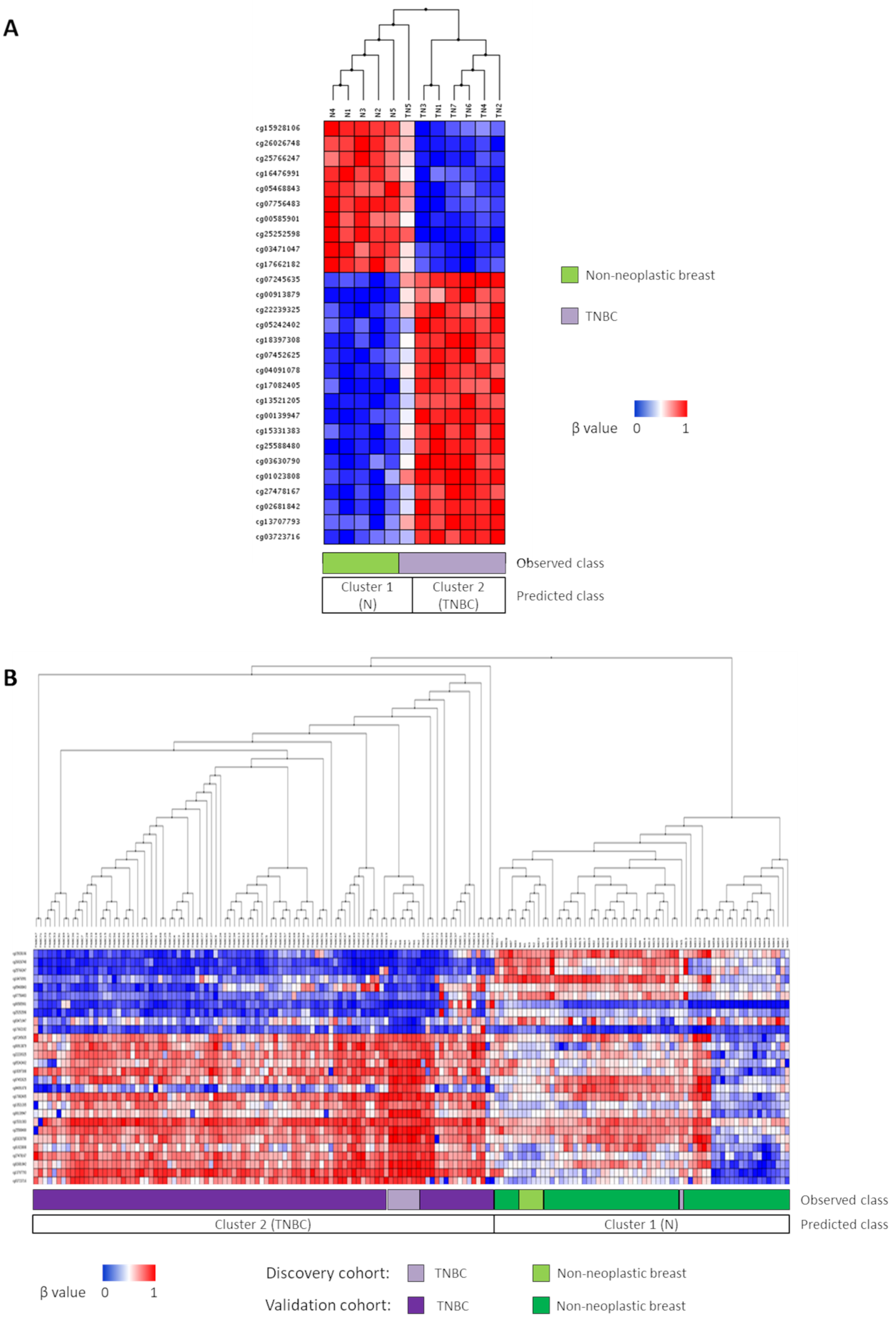
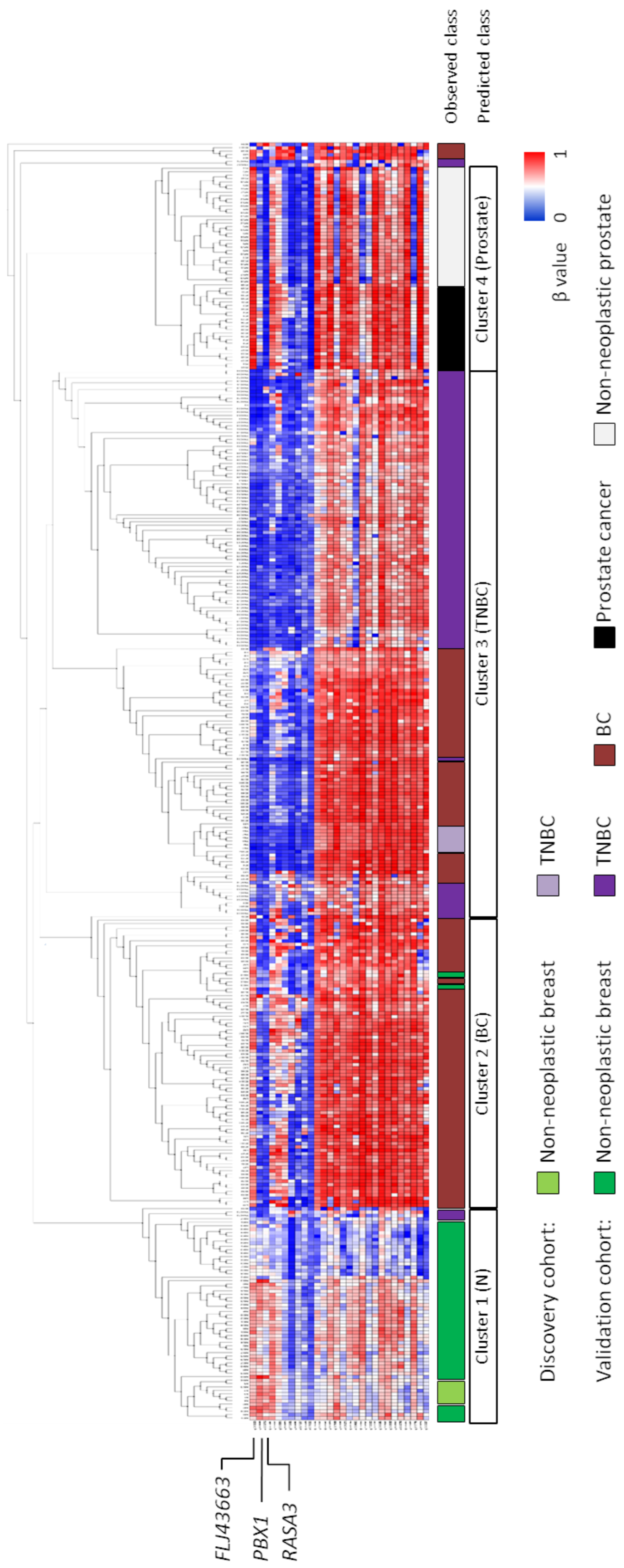
3.1. Novel Diagnostic DNA Methylation Signature for TNBC
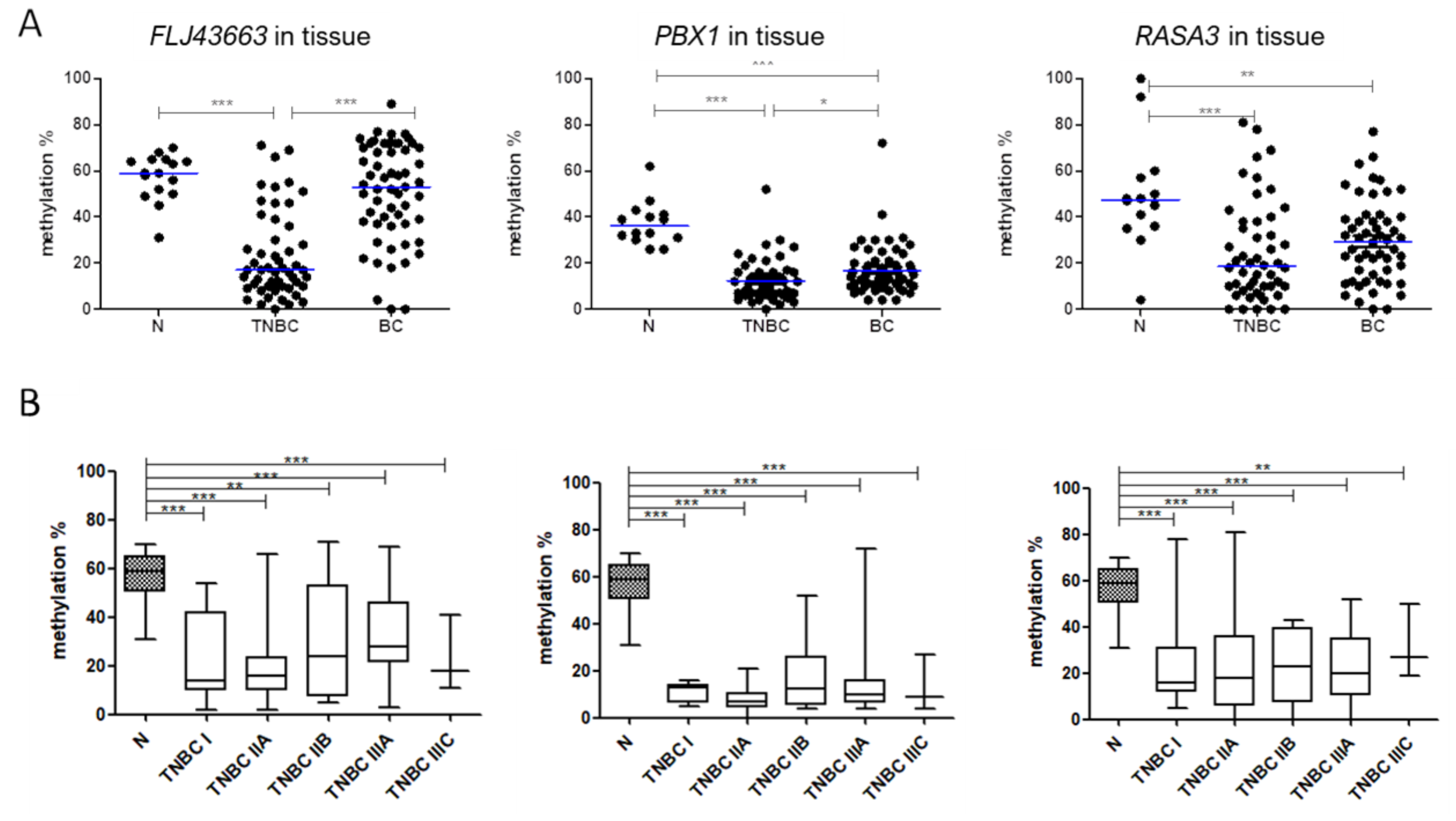
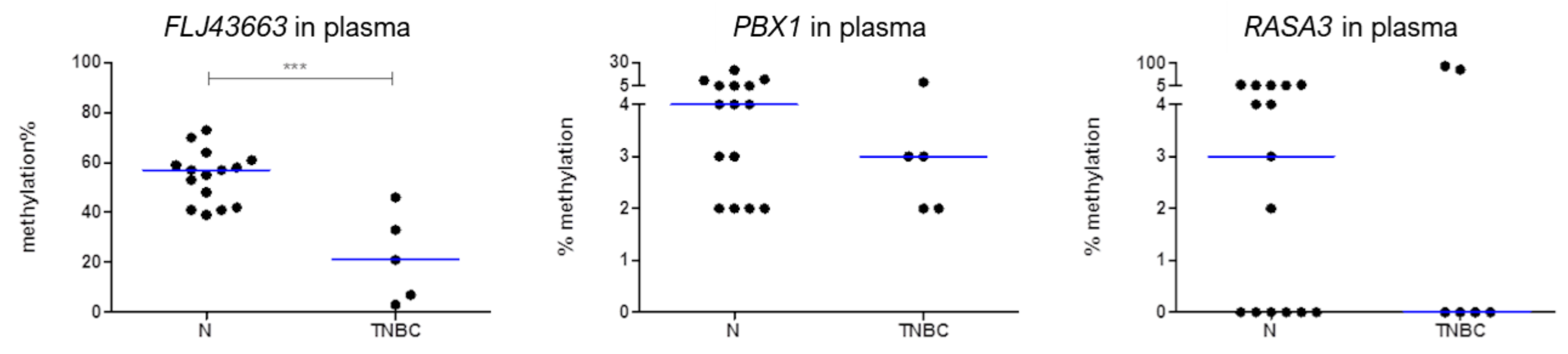
3.2. Methylation Status of FLJ43663, PBX1, and RASA3 in Breast Tissue and Plasma
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untch, M.; Gerber, B.; Harbeck, N.; Jackisch, C.; Marschner, N.; Mobus, V.; von Minckwitz, G.; Loibl, S.; Beckmann, M.W.; Blohmer, J.U.; et al. 13th st. Gallen international breast cancer conference 2013: Primary therapy of early breast cancer evidence, controversies, consensus—Opinion of a german team of experts (zurich 2013). Breast Care 2013, 8, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Rhodes, A.; Jasani, B.; Balaton, A.J.; Miller, K.D. Immunohistochemical demonstration of oestrogen and progesterone receptors: Correlation of standards achieved on in house tumours with that achieved on external quality assessment material in over 150 laboratories from 26 countries. J. Clin. Pathol. 2000, 53, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino-Mathews, A. Novel uses of immunohistochemistry in breast pathology: Interpretation and pitfalls. Mod. Pathol. 2021, 34, 62–77. [Google Scholar] [CrossRef]
- Viale, G.; Slaets, L.; Bogaerts, J.; Rutgers, E.; Van’t Veer, L.; Piccart-Gebhart, M.J.; de Snoo, F.A.; Stork-Sloots, L.; Russo, L.; Dell’Orto, P.; et al. High concordance of protein (by IHC), gene (by FISH.; HER2 only), and microarray readout (by TargetPrint) of ER, PgR, and HER2: Results from the EORTC 10041/BIG 03-04 MINDACT trial. Ann. Oncol. 2014, 25, 816–823. [Google Scholar] [CrossRef]
- Constâncio, V.; Nunes, S.P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9, 624. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Miao, Z.; Wang, K.; Lv, Y.; Qiu, L.; Guo, L. Expression levels and clinical values of miR-92b-3p in breast cancer. World J. Surg. Oncol. 2021, 19, 239. [Google Scholar] [CrossRef]
- Pasha, H.A.; Rezk, N.A.; Riad, M.A. Circulating Cell Free Nuclear DNA, Mitochondrial DNA and Global DNA Methylation: Potential Noninvasive Biomarkers for Breast Cancer Diagnosis. Cancer Investig. 2019, 37, 432–439. [Google Scholar] [CrossRef]
- Andre, F.; Ismaila, N.; Henry, N.L.; Somerfield, M.R.; Bast, R.C.; Barlow, W.; Collyar, D.E.; Hammond, M.E.; Kuderer, N.M.; Liu, M.C.; et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: ASCO clinical practice guideline update-integration of results from TAILORx. J. Clin. Oncol. 2019, 37, 1956–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, V.; Martinez-Cardus, A.; Esteller, M. The Epigenomic Revolution in Breast Cancer: From Single-Gene to Genome-Wide Next-Generation Approaches. Am. J. Pathol. 2017, 187, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Garcia, J.; Osca-Verdegal, R.; Mena-Molla, S.; Garcia-Gimenez, J.L. Epigenetic IVD Tests for Personalized Precision Medicine in Cancer. Front. Genet. 2019, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Costa-Pinheiro, P.; Montezuma, D.; Henrique, R.; Jeronimo, C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics 2015, 7, 1003–1015. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Bhat, S.A.; Majid, S.; Wani, H.A.; Rashid, S. Diagnostic utility of epigenetics in breast cancer—A review. Cancer Treat. Res. Commun. 2019, 19, 100125. [Google Scholar] [CrossRef]
- Kloten, V.; Becker, B.; Winner, K.; Schrauder, M.G.; Fasching, P.A.; Anzeneder, T.; Veeck, J.; Hartmann, A.; Knuchel, R.; Dahl, E. Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res. 2013, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, M.; Yin, H.; Li, J.; Li, X.; Wang, D.; Su, Y.; Niu, M.; Zhong, Z.; Wang, J.; Zhang, X.; et al. Detection of aberrant methylation of a six-gene panel in serum DNA for diagnosis of breast cancer. Oncotarget 2016, 7, 18485–18494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahvar, F.; Salimi, M.; Mozdarani, H. Plasma GBP2 promoter methylation is associated with advanced stages in breast cancer. Genet. Mol. Biol. 2020, 43, e20190230. [Google Scholar] [CrossRef] [PubMed]
- DiNome, M.L.; Orozco, J.I.J.; Matsuba, C.; Manughian-Peter, A.O.; Ensenyat-Mendez, M.; Chang, S.C.; Jalas, J.R.; Salomon, M.P.; Marzese, D.M. Clinicopathological Features of Triple-Negative Breast Cancer Epigenetic Subtypes. Ann. Surg. Oncol. 2019, 26, 3344–3353. [Google Scholar] [CrossRef]
- Pineda, B.; Diaz-Lagares, A.; Perez-Fidalgo, J.A.; Burgues, O.; Gonzalez-Barrallo, I.; Crujeiras, A.B.; Sandoval, J.; Esteller, M.; Lluch, A.; Eroles, P. A two-gene epigenetic signature for the prediction of response to neoadjuvant chemotherapy in triple-negative breast cancer patients. Clin. Epigenetics 2019, 11, 33. [Google Scholar] [CrossRef]
- Mathe, A.; Wong-Brown, M.; Locke, W.J.; Stirzaker, C.; Braye, S.G.; Forbes, J.F.; Clark, S.J.; Avery-Kiejda, K.A.; Scott, R.J. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci. Rep. 2016, 6, 33435. [Google Scholar] [CrossRef]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [Green Version]
- Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Córdoba, A.; de Azúa, Y.R.; Aguiar, B.; Beloqui, R.; Armendáriz, P.; Arriola, M.; Martín-Sánchez, E.; et al. ADAM12 is a potential therapeutic target regulated by hypomethylation in triple-negative breast cancer. Int. J. Mol. Sci. 2020, 21, 903. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; Christensen, B.C. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017, 19, 81. [Google Scholar] [CrossRef] [Green Version]
- Avery-Kiejda, K.A.; Mathe, A.; Scott, R.J. Genome-wide miRNA, gene and methylation analysis of triple negative breast cancer to identify changes associated with lymph node metastases. Genom. Data 2017, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; Bizet, M.; Desmedt, C.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Koch, A.; Larsimont, D.; Salgado, R.; Van Den Eynden, G.; et al. DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J. Clin. Investig. 2017, 127, 3090–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, R.; Salavert, F.; Garcia-Garcia, F.; Carbonell-Caballero, J.; Bleda, M.; Garcia-Alonso, L.; Sanchis-Juan, A.; Perez-Gil, D.; Marin-Garcia, P.; Sanchez, R.; et al. Babelomics 5.0: Functional interpretation for new generations of genomic data. Nucleic Acids Res. 2015, 43, W117–W121. [Google Scholar] [CrossRef] [Green Version]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.X.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 Index, HER2 Status, and Prognosis of Patients with Luminal B Breast Cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [Green Version]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 2002, 41, 154–161. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Martin-Sanchez, E.; Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Blanco-Luquin, I.; Cordoba, A.; Vicente-Garcia, F.; Perez-Janices, N.; Escors, D.; Megias, D.; et al. CHL1 hypermethylation as a potential biomarker of poor prognosis in breast cancer. Oncotarget 2017, 8, 15789–15801. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.; Niu, L.; Zhang, Y.; Wang, X.; Ma, K.; Yin, H.; Dai, J.; Zhou, W.; Pan, Y. Defining optimal cutoff value of MGMT promoter methylation by ROC analysis for clinical setting in glioblastoma patients. J. Neurooncol. 2017, 133, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- De Jesus, J.; Rosa, M. Suboptimal concordance in testing and retesting results of triple-negative breast carcinoma cases among laboratories: One institution experience. Cancer Cell Int. 2019, 19, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Grützmann, R.; Molnar, B.; Pilarsky, C.; Habermann, J.K.; Schlag, P.M.; Saeger, H.D.; Miehlke, S.; Stolz, T.; Model, F.; Roblick, U.J.; et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS ONE 2008, 3, e3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Wang, X.; Jin, P. Developing DNA methylation-based diagnostic biomarkers. J. Genet. Genom. 2018, 45, 87–97. [Google Scholar] [CrossRef]
- Taryma-Leśniak, O.; Sokolowska, K.E.; Wojdacz, T.K. Correction to: Current status of development of methylation biomarkers for in vitro diagnostic IVD applications. Clin. Epigenetics 2020, 12, 107. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Bernardini, S.; Miano, R.; Iori, R.; Finazzi-Agrò, E.; Palmieri, G.; Ballerini, S.; Angeloni, C.; Orlandi, A.; Bellincampi, L.; Cortese, C.; et al. Hypermethylation of the CpG islands in the promoter region of the GSTP1 gene in prostate cancer: A useful diagnostic and prognostic marker? Clin. Chim. Acta 2004, 350, 181–188. [Google Scholar] [CrossRef]
- Nikas, J.B.; Nikas, E.G. Genome-Wide DNA Methylation Model for the Diagnosis of Prostate Cancer. ACS Omega 2019, 4, 14895–14901. [Google Scholar] [CrossRef] [Green Version]
- Goh, L.K.; Liem, N.; Vijayaraghavan, A.; Chen, G.; Lim, P.L.; Tay, K.J.; Chang, M.; Low, J.S.W.; Joshi, A.; Huang, H.H.; et al. Diagnostic and prognostic utility of a DNA hypermethylated gene signature in prostate cancer. PLoS ONE 2014, 9, e91666. [Google Scholar] [CrossRef]
- Cheng, J.; Wei, D.; Ji, Y.; Chen, L.; Yang, L.; Li, G.; Wu, L.; Hou, T.; Xie, L.; Ding, G.; et al. Integrative analysis of DNA methylation and gene expression reveals hepatocellular carcinoma-specific diagnostic biomarkers. Genome Med. 2018, 10, 42. [Google Scholar] [CrossRef]
- Sabedot, T.; Malta, T.; Snyder, J.; Nelson, K.; Wells, M.; deCarvalho, A.; Mukherjee, A.; Chitale, D.; Mosella, M.; Sokolov, A.; et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro-Oncology 2021, 23, 1494–1508. [Google Scholar] [CrossRef]
- Martin-Sanchez, E.; Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Cordoba, A.; Vicente-Garcia, F.; Blanco-Luquin, I.; De La Cruz, S.; Aramendia, A.; Guerrero-Setas, D. CDH22 hypermethylation is an independent prognostic biomarker in breast cancer. Clin. Epigenetics 2017, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.; Brown, R.E. Field effect in cancer-an update. Ann. Clin. Lab. Sci. 2009, 39, 331–337. [Google Scholar]
- Baba, Y.; Ishimoto, T.; Kurashige, J.; Iwatsuki, M.; Sakamoto, Y.; Yoshida, N.; Watanabe, M.; Baba, H. Epigenetic field cancerization in gastrointestinal cancers. Cancer Lett. 2016, 375, 360–366. [Google Scholar] [CrossRef]
- Patel, A.; Tripathi, G.; Gopalakrishnan, K.; Williams, N.; Arasaradnam, R.P. Field cancerisation in colorectal cancer: A new frontier or pastures past? World J. Gastroenterol. 2015, 21, 3763–3772. [Google Scholar] [CrossRef]
- Dotto, G.P. Multifocal epithelial tumors and field cancerization: Stroma as a primary determinant. J. Clin. Investig. 2014, 124, 1446–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.L.; Magalhaes, L.; Moreira, F.C.; Reis-das-Merces, L.; Vidal, A.F.; Ribeiro-Dos-Santos, A.M.; Demachki, S.; Anaissi, A.K.M.; Burbano, R.M.R.; Albuquerque, P.; et al. Epigenetic Field Cancerization in Gastric Cancer: microRNAs as Promising Biomarkers. J. Cancer 2019, 10, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Spitzwieser, M.; Holzweber, E.; Pfeiler, G.; Hacker, S.; Cichna-Markl, M. Applicability of HIN-1, MGMT and RASSF1A promoter methylation as biomarkers for detecting field cancerization in breast cancer. Breast Cancer Res. 2015, 17, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitzwieser, M.; Entfellner, E.; Werner, B.; Pulverer, W.; Pfeiler, G.; Hacker, S.; Cichna-Markl, M. Hypermethylation of CDKN2A exon 2 in tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. BMC Cancer 2017, 17, 260. [Google Scholar] [CrossRef] [Green Version]
- Marín-Béjar, O.; Mas, A.M.; González, J.; Martinez, D.; Athie, A.; Morales, X.; Galduroz, M.; Raimondi, I.; Grossi, E.; Guo, S.; et al. The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 2017, 18, 202. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhao, D.; Zhao, Q.; Dong, D.; Mu, L.; Zhao, X.; Guo, M.; Xu, A.; Fang, L.; Liu, Q.; et al. Microarray profiling and co-expression network analysis of lncRNAs and mRNAs in ovarian cancer. Cell Death Discov. 2019, 5, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, G.-Q.; Chen, H.; Zhao, Z.-J.; Chen, H.-Z.; Liu, H.; Wang, G.; Jia, Y.-H.; Pan, S.-H.; Kong, R.; et al. Plasma and tumor levels of Linc-pint are diagnostic and prognostic biomarkers for pancreatic cancer. Oncotarget 2016, 7, 71773–71781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Gong, C.; Li, J.; Tang, J. Downregulation of long non-coding RNA LINC-PINT serves as a diagnostic and prognostic biomarker in patients with non-small cell lung cancer. Oncol. Lett. 2021, 21, 210. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, M.; Zou, L.; Xia, J.; Huang, J.; Deng, Q.; Xu, R. Long non-coding RNA LINC-PINT attenuates paclitaxel resistance in triple-negative breast cancer cells via targeting the RNA-binding protein NONO. Acta Biochim. Biophys. Sin. 2020, 52, 801–809. [Google Scholar] [CrossRef]
- Schurmans, S.; Polizzi, S.; Scoumanne, A.; Sayyed, S.; Molina-Ortiz, P. The Ras/Rap GTPase activating protein RASA3: From gene structure to in vivo functions. Adv. Biol. Regul. 2015, 57, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Fan, X.; He, L.; Zhou, D. Methylation patterns of RASA3 associated with clinicopathological factors in hepatocellular carcinoma. J. Cancer 2018, 9, 2116–2122. [Google Scholar] [CrossRef]
- Nourse, J.; Mellentin, J.D.; Galili, N.; Wilkinson, J.; Stanbridge, E.; Smith, S.D.; Cleary, M.L. Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor. Cell 1990, 60, 535–545. [Google Scholar] [CrossRef]
- Magnani, L.; Patten, D.K.; Nguyen, V.T.; Hong, S.P.; Steel, J.H.; Patel, N.; Lombardo, Y.; Faronato, M.; Gomes, A.R.; Woodley, L.; et al. The pioneer factor PBX1 is a novel driver of metastatic progression in ERα-positive breast cancer. Oncotarget 2015, 6, 21878–21891. [Google Scholar] [CrossRef]
- Ao, X.; Ding, W.; Ge, H.; Zhang, Y.; Ding, D.; Liu, Y. PBX1 is a valuable prognostic biomarker for patients with breast cancer. Exp. Ther. Med. 2020, 20, 385–394. [Google Scholar] [CrossRef]
- Qiu, Y.; Lu, G.; Wu, Y. Coexpression of PBX1 and EMP2 as Prognostic Biomarkers in Estrogen Receptor-Negative Breast Cancer via Data Mining. J. Comput. Biol. 2020, 27, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Eslami, S.Z.; Cortes-Hernandez, L.E.; Cayrefourcq, L.; Alix-Panabieres, C. The Different Facets of Liquid Biopsy: A Kaleidoscopic View. Cold Spring Harb. Perspect. Med. 2019, 10, a037333. [Google Scholar] [CrossRef]
- Chimonidou, M.; Tzitzira, A.; Strati, A.; Sotiropoulou, G.; Sfikas, C.; Malamos, N.; Georgoulias, V.; Lianidou, E. CST6 promoter methylation in circulating cell-free DNA of breast cancer patients. Clin. Biochem. 2013, 46, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Konecny, M.; Markus, J.; Waczulikova, I.; Dolesova, L.; Kozlova, R.; Repiska, V.; Novosadova, H.; Majer, I. The value of SHOX2 methylation test in peripheral blood samples used for the differential diagnosis of lung cancer and other lung disorders. Neoplasma 2016, 63, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Chimonidou, M.; Milaki, G.; Tsaroucha, E.; Kakolyris, S.; Georgoulias, V.; Lianidou, E. SOX17 promoter methylation in plasma circulating tumor DNA of patients with non-small cell lung cancer. Clin. Chem. Lab. Med. 2016, 54, 1385–1393. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Sequencing Primer (5′-3′) | Size (bp) | Tm (°C) |
|---|---|---|---|---|---|
| FLJ43663 | TTGTTTTGAAGGTGGTAAATTAGATT | Btn-ATCCCCTTAATAAATAAAACTACACATC | AAGGTGGTAAATTAGATTTT | 108 | 58 |
| PBX1 | AGAAGGAAGTGGTTTTGTTTAGA | Btn-CTATCAACCAAAAAAAACAAACAATAACA | TTGTTTAGAGGTTATATTTAGTG | 83 | 60 |
| RASA3 | ATAGATGGGGAGATTGAGGTT | Btn-ATCTTCAAACCAAACCCAAAAACTCAATAA | AGTTGTGAGTTTTAGTTTAG | 118 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendaza, S.; Guerrero-Setas, D.; Monreal-Santesteban, I.; Ulazia-Garmendia, A.; Cordoba Iturriagagoitia, A.; De la Cruz, S.; Martín-Sánchez, E. A DNA Methylation-Based Gene Signature Can Predict Triple-Negative Breast Cancer Diagnosis. Biomedicines 2021, 9, 1394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101394
Mendaza S, Guerrero-Setas D, Monreal-Santesteban I, Ulazia-Garmendia A, Cordoba Iturriagagoitia A, De la Cruz S, Martín-Sánchez E. A DNA Methylation-Based Gene Signature Can Predict Triple-Negative Breast Cancer Diagnosis. Biomedicines. 2021; 9(10):1394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101394
Chicago/Turabian StyleMendaza, Saioa, David Guerrero-Setas, Iñaki Monreal-Santesteban, Ane Ulazia-Garmendia, Alicia Cordoba Iturriagagoitia, Susana De la Cruz, and Esperanza Martín-Sánchez. 2021. "A DNA Methylation-Based Gene Signature Can Predict Triple-Negative Breast Cancer Diagnosis" Biomedicines 9, no. 10: 1394. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101394