
Strategies Targeting Type 2 Inflammation: From Monoclonal Antibodies to JAK-Inhibitors

 and
and
Abstract
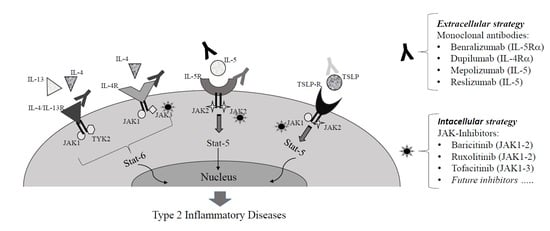
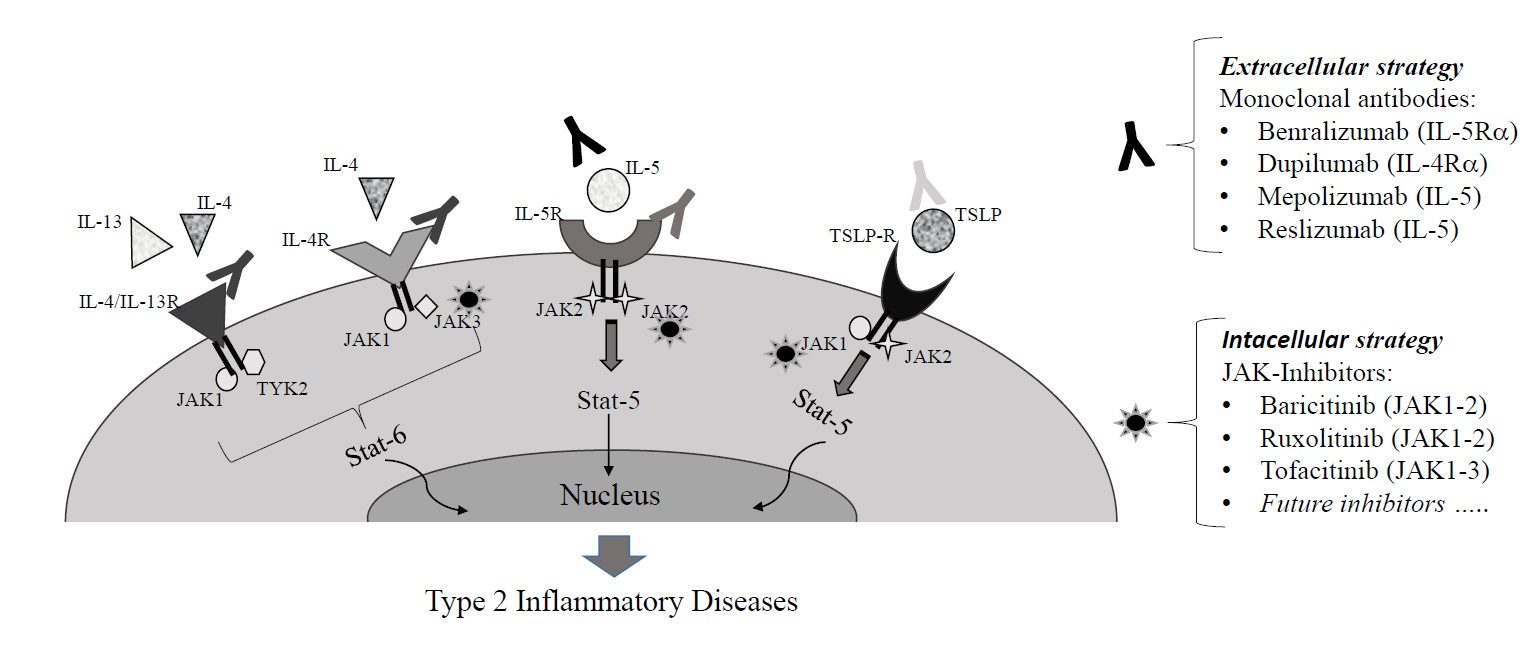
:
{kind=link}
1. Introduction
2. Type 2 Cytokines Signal Pathways
3. Monoclonal Antibodies Targeting Type 2 Inflammation
4. JAK-Inhibitors Targeting Type 2 Cytokine Pathways
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Loza, M.J.; Djukanovic, R.; Chung, K.F.; Horowitz, D.; Ma, K.; Branigan, P.; Barnathan, E.S.; Susulic, V.S.; Silkoff, P.E.; Sterk, P.J.; et al. Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir. Res. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Tomassen, P.; Vandeplas, G.; Van Zele, T.; Cardell, L.-O.; Arebro, J.; Olze, H.; Förster-Ruhrmann, U.; Kowalski, M.L.; Olszewska-Ziąber, A.; Holtappels, G.; et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis ofbiomarkers. J. Allergy Clin. Immunol. 2016, 137, 1449–1456. [Google Scholar] [CrossRef] [Green Version]
- Bel, E.H.; Brinke, A.T. New Anti-Eosinophil Drugs for Asthma and COPD. Chest 2017, 152, 1276–1282. [Google Scholar] [CrossRef]
- Perez-De-Llano, L.; Tran, T.; Al-Ahmad, M.; Alacqua, M.; Bulathsinhala, L.; Busby, J.; Canonica, G.; Carter, V.; Chaudhry, I.; Christoff, G.; et al. Characterization of Eosinophilic and Non-Eosinophilic Severe Asthma Phenotypes and Proportion of Patients with These Phenotypes in the International Severe Asthma Registry (ISAR). Am. J. Respir. Crit. Care Med. 2020, 201, A4525. [Google Scholar] [CrossRef]
- Delemarre, T.; Holtappels, G.; De Ruyck, N.; Zhang, N.; Nauwynck, H.; Bachert, C.; Gevaert, E. Type 2 inflammation in chronic rhinosinusitis without nasal polyps: Another relevant endotype. J. Allergy Clin. Immunol. 2020, 146, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulos, C.; Gogali, A.; Bartziokas, K.; Kostikas, K. Identification and treatment of T2-low asthma in the era of biologics. ERJ Open Res. 2021, 7, 00309–02020. [Google Scholar] [CrossRef]
- Zhu, Z.; Hasegawa, K.; Camargo, C.A.; Liang, L. Investigating asthma heterogeneity through shared and distinct genetics: Insights from genome-wide cross-trait analysis. J. Allergy Clin. Immunol. 2021, 147, 796–807. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Zhu, Z.; Lee, P.H.; Chaffin, M.; Chung, W.; Loh, P.R.; Lu, Q.; Christiani, D.C.; Liang, L. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat. Genet. 2018, 50, 857–864. [Google Scholar] [CrossRef]
- Benjamin, M.R.; Stevens, W.W.; Li, N.; Bose, S.; Grammer, L.C.; Kern, R.C.; Tan, B.K.; Conley, D.B.; Smith, S.S.; Welch, K.C.; et al. Clinical Characteristics of Patients with Chronic Rhinosinusitis without Nasal Polyps in an Academic Setting. J. Allergy Clin. Immunol. Pract. 2019, 7, 1010–1016. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Ordovas-Montanes, J.; Allon, S.J.; Buchheit, K.M.; Vukovic, M.; Derakhshan, T.; Feng, C.; Lai, J.; Hughes, T.K.; Nyquist, S.K.; et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci. Immunol. 2021, 6, eabb7221. [Google Scholar] [CrossRef] [PubMed]
- Divekar, R.; Rank, M.; Squillace, D.; Kita, H.; Lal, D. Unsupervised network mapping of commercially available immunoassay yields three distinct chronic rhinosinusitis endotypes. Int. Forum Allergy Rhinol. 2017, 7, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Fujieda, S.; Imoto, Y.; Kato, Y.; Ninomiya, T.; Tokunaga, T.; Tsutsumiuchi, T.; Yoshida, K.; Kidoguchi, M.; Takabayashi, T. Eosinophilic chronic rhinosinusitis. Allergol. Int. 2019, 68, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, D.W.; Gevaert, P. Chronic Rhinosinusitis without Nasal Polyps. J. Allergy Clin. Immunol. Pract. 2016, 4, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Schleimer, R.P. Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis. Annu. Rev. Pathol. 2017, 12, 331–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Ouyang, H.; Luo, R. Distinct characteristics of nasal polyps with and without eosinophilia. Braz. J. Otorhinolaryngol. 2017, 83, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Fokkens, W.; Lund, V.; Hopkins, C.; Hellings, P.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J. Executive Summary of EPOS 2020 Including Integrated Care Pathways. Rhinol. J. 2020, 58, 82–111. [Google Scholar] [CrossRef]
- Peters, M.C.; Wenzel, S.E. Intersection of biology and therapeutics: Type 2 targeted therapeutics for adult asthma. Lancet 2020, 395, 371–383. [Google Scholar] [CrossRef]
- Lee, Y.; Quoc, Q.L.; Park, H.S. Biomarkers for Severe Asthma: Lessons From Longitudinal Cohort Studies. Allergy Asthma Immunol. Res. 2021, 13, 375–389. [Google Scholar] [CrossRef]
- Bachert, C.; Han, J.K.; Desrosiers, M.; Hellings, P.W.; Amin, N.; Lee, S.E.; Mullol, J.; Greos, L.S.; Bosso, J.V.; Laidlaw, T.M.; et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): Results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet 2019, 394, 1638–1650. [Google Scholar] [CrossRef] [Green Version]
- Gevaert, P.; Omachi, T.A.; Corren, J.; Mullol, J.; Han, J.; Lee, S.E.; Kaufman, D.; Ligueros-Saylan, M.; Howard, M.; Zhu, R.; et al. Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J. Allergy Clin. Immunol. 2020, 146, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.; Bachert, C.; Fokkens, W.; Desrosiers, M.; Wagenmann, M.; Lee, S.; Sousa, A.; Smith, S.; Martin, N.; Mayer, B.; et al. Late Breaking Abstract-Add-on mepolizumab for chronic rhinosinusitis with nasal polyps: SYNAPSE study. Airw. Pharmacol. Treat. 2020, 56, 4616. [Google Scholar] [CrossRef]
- Lin, Y.J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Casteele, N.V. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, I.E.; Cai, F.; Tom, J.A.; Galanter, J.M.; Owen, R.P.; Zhu, R.; Williams, M.; McGregor, A.G.; Eliahu, A.; Durk, M.R.; et al. Inhaled JAK inhibitor GDC-0214 reduces exhaled nitric oxide in patients with mild asthma: A randomized, controlled, proof-of-activity trial. J. Allergy Clin. Immunol. 2021, 148, 783–789. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. 1JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Padjen, I.; Fanouriakis, A.; Boumpas, D.T. Pathogenic and Therapeutic Relevance of JAK/STAT Signaling in Systemic Lupus Erythematosus: Integration of Distinct Inflammatory Pathways and the Prospect of Their Inhibition with an Oral Agent. Cells 2019, 8, 898. [Google Scholar] [CrossRef] [Green Version]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 2019, 58 (Suppl. S1), i43–i54. [Google Scholar] [CrossRef] [Green Version]
- Zak, M.; Denglerb, H.S.; Rajapaksaa, N.S. Inhaled Janus Kinase (JAK) inhibitors for the treatment of asthma. Bioorganic Med. Chem. Lett. 2019, 29, 126658. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.H.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Winkler, T.; Frey, U. Airway remodeling: Shifting the trigger point for exacerbations in asthma. J. Allergy Clin. Immunol. 2021, 148, 710–712. [Google Scholar] [CrossRef]
- Yan, Q.; Forno, E.; Herrera-Luis, E.; Pino-Yanes, M.; Yang, G.; Oh, S.; Acosta-Pérez, E.; Hu, D.; Eng, C.; Huntsman, S.; et al. A genome-wide association study of asthma hospitalizations in adults. J. Allergy Clin. Immunol. 2021, 147, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Kim, H.J.; Park, J.H.; Shin, J.M.; Lee, H.M. Apigenin alleviates TGF-β1-induced nasal mucosa remodeling by inhibiting MAPK / NF-kB signaling pathways in chronic rhinosinusitis. PLoS ONE 2018, 13, e0201595. [Google Scholar]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, Z.; Wang, G.; Zou, J.; Yu, X.; Zhang, D.; Zeng, G.; Tang, C. Interleukin-5 promotes ATP-binding cassette transporter A1 expression through miR-211/JAK2/STAT3 pathways in THP-1-dervied macrophages. Acta Biochim. Biophys. Sin. 2020, 52, 832–841. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef]
- Jin, S.; Yang, X.; Li, J.; Yang, W.; Ma, H.; Zhang, Z. p53-targeted lincRNA-p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol. Cancer 2019, 18, 38–56. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Hettinga, A.; Kale, S.L.; Hu, S.; Xie, M.M.; Dent, A.L.; Ray, A.; Poholek, A.C. Blimp-1 is essential for allergen-induced asthma and Th2 cell development in the lung. J. Exp. Med. 2020, 217, 20190742. [Google Scholar] [CrossRef]
- Karpathiou, G.; Papoudou-Bai, A.; Ferrand, E.; Dumollard, J.M.; Peoc’H, M. STAT6: A review of a signaling pathway implicated in various diseases with a special emphasis in its usefulness in pathology. Pathol. Res. Pract. 2021, 223, 153477. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, H.; Chan, L.S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAK-STAT 2013, 2, e24137. [Google Scholar] [CrossRef] [Green Version]
- Al Tuwaijri, A.; Gagné-Ouellet, V.; Madore, A.-M.; Laprise, C.; Naumova, A.K. Local genotype influences DNA methylation at two asthma-associated regions, 5q31 and 17q21, in a founder effect population. J. Med. Genet. 2016, 53, 232–241. [Google Scholar] [CrossRef]
- Bajbouj, K.; Hachim, M.Y.; Ramakrishnan, R.K.; Fazel, H.; Mustafa, J.; Alzaghari, S.; Eladl, M.; Shafarin, J.; Olivenstein, R.; Hamid, Q. IL-13 Augments Histone Demethylase JMJD2B/KDM4B Expression Levels, Activity, and Nuclear Translocation in Airway Fibroblasts in Asthma. J. Immunol. Res. 2021, 2021, 6629844. [Google Scholar] [CrossRef]
- Penke, L.R.K.; Ouchi, H.; Speth, J.; Lugogo, N.; Huang, Y.J.; Huang, S.K.; Peters-Golden, M. Transcriptional regulation of the IL-13Rα2 gene in human lung fibroblasts. Sci. Rep. 2020, 10, 1083. [Google Scholar] [CrossRef] [PubMed]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef]
- McBrien, C.N.; Menzies-Gow, A. The Biology of Eosinophils and Their Role in Asthma. Front. Med. 2017, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Ford, L.; Pearlman, D.; Spector, S.; Sher, L.; Skobieranda, F.; Wang, L.; Kirkesseli, S.; Rocklin, R.; Bock, B.; et al. Dupilumab in Persistent Asthma with Elevated Eosinophil Levels. N. Engl. J. Med. 2013, 368, 2455–2466. [Google Scholar] [CrossRef]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Bleecker, E.R.; FitzGerald, J.M.; Chanez, P.; Papi, A.; Weinstein, S.F.; Barker, P.; Sproule, S.; Gilmartin, G.; Aurivillius, M.; Werkström, V.; et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): A randomised, multicentre, placebo-controlled phase 3 trial. Lancet 2016, 388, 2115–2127. [Google Scholar] [CrossRef]
- FitzGerald, J.M.; Bleecker, E.R.; Nair, P.; Korn, S.; Ohta, K.; Lommatzsch, M.; Ferguson, G.T.; Busse, W.W.; Barker, P.; Sproule, S.; et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 388, 2128–2141. [Google Scholar] [CrossRef]
- Humbert, M.; Beasley, R.; Ayres, J.; Slavin, R.; Hébert, J.; Bousquet, J.; Beeh, K.-M.; Ramos, S.; Canonica, G.W.; Hedgecock, S.; et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005, 60, 309–316. [Google Scholar] [CrossRef]
- Castro, M.; Zangrilli, J.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O’Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef]
- Corren, J.; Weinstein, S.; Janka, L.; Zangrilli, J.; Garin, M. Phase 3 Study of Reslizumab in Patients With Poorly Controlled Asthma. Chest 2016, 150, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjermer, L.; Lemiere, C.; Maspero, J.; Weiss, S.; Zangrilli, J.; Germinaro, M. Reslizumab for Inadequately Controlled Asthma with Elevated Blood Eosinophil Levels: A Randomized Phase 3 Study. Chest 2016, 150, 789–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.F.; Nair, P. Airway Inflammation and Inflammatory Biomarkers. Semin. Respir. Crit. Care Med. 2018, 39, 056–063. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.P.; Arnau, A.M.G.; Saini, S.S. Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy 2017, 72, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, J.T.; Bieneman, A.P.; Chichester, K.L.; Hamilton, R.G.; Xiao, H.; Saini, S.S.; Liu, M.C. Decreases in human dendritic cell–dependent TH2-like responses after acute in vivo IgE neutralization. J. Allergy Clin. Immunol. 2010, 125, 896–901.e6. [Google Scholar] [CrossRef]
- Fahy, J.V.; Fleming, H.E.; Wong, H.H.; Liu, J.T.; Su, J.Q.; Reimann, J.; Fick, R.B.; Boushey, H.A. The effect of an anti-IgE monoclonal antibody on the early- and late-phase responses to allergen inhalation in asthmatic subjects. Am. J. Respir. Crit. Care Med. 1997, 155, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Rossettini, B.; Montaini, G.; Matucci, A.; Vultaggio, A.; Mazzoni, A.; Palterer, B.; Parronchi, P.; Maggi, E.; Liotta, F.; et al. Omalizumab dampens type 2 inflammation in a group of long-term treated asthma patients and detaches IgE from FcεRI. Eur. J. Immunol. 2018, 48, 2005–2014. [Google Scholar] [CrossRef] [Green Version]
- Casale, T.B.; Luskin, A.T.; Busse, W.; Zeiger, R.; Trzaskoma, B.; Yang, M.; Griffin, N.M.; Chipps, B.E. Omalizumab Effectiveness by Biomarker Status in Patients with Asthma: Evidence from PROSPERO, a Prospective Real-World Study. J. Allergy Clin. Immunol. Pract. 2019, 7, 156–164.e1. [Google Scholar] [CrossRef] [PubMed]
- Hanania, N.A.; Wenzel, S.; Rosén, K.; Hsieh, H.-J.; Mosesova, S.; Choy, D.F.; Lal, P.; Arron, J.R.; Harris, J.M.; Busse, W. Exploring the Effects of Omalizumab in Allergic Asthma. Am. J. Respir. Crit. Care Med. 2013, 187, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Özgen, H.; Tepetam, F.M.; Bulut, I.; Örçen, C. The significance of eosinophil and eosinophil lymphocyte ratio (ELR) in predicting response to omalizumab treatment in patients with severe allergic asthma. Tuberk. Toraks 2021, 69, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chipps, B.E.; Lanier, B.; Milgrom, H.; Deschildre, A.; Hedlin, G.; Szefler, S.J.; Kattan, M.; Kianifard, F.; Ortiz, B.; Haselkorn, T.; et al. Omalizumab in children with uncontrolled allergic asthma: Review of clinical trial and real-world experience. J. Allergy Clin. Immunol. 2017, 139, 1431–1444. [Google Scholar] [CrossRef] [Green Version]
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. 2020. Available online: www.ginasthma.org (accessed on 24 November 2020).
- Holguin, F.; Cardet, J.C.; Chung, K.F.; Diver, S.; Ferreira, D.; Fitzpatrick, A.; Gaga, M.; Kellermeyer, L.; Khurana, S.; Knight, S.L.; et al. Management of severe asthma: A European Respiratory Society/American Thoracic Society guideline. Eur. Respir. J. 2019, 55, 1900588. [Google Scholar] [CrossRef] [Green Version]
- Berek, C. Eosinophils can more than kill. J. Exp. Med. 2018, 215, 1967–1969. [Google Scholar] [CrossRef] [Green Version]
- Farne, H.A.; Wilson, A.; Powell, C.; Bax, L.; Milan, S.J. Anti-IL5 therapies for asthma. Cochrane Database Syst. Rev. 2017, 9, CD010834. [Google Scholar] [CrossRef]
- Salter, B.; Ju, X.; Sehmi, R. Eosinophil Lineage-Committed Progenitors as a Therapeutic Target for Asthma. Cells 2021, 10, 412. [Google Scholar] [CrossRef]
- Busse, W.W.; Katial, R.; Gossage, D.; Sari, S.; Wang, B.; Kolbeck, R.; Coyle, A.J.; Koike, M.; Spitalny, G.L.; Kiener, P.A.; et al. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti–IL-5 receptor α antibody, in a phase I study of subjects with mild asthma. J. Allergy Clin. Immunol. 2010, 125, 1237–1244.e2. [Google Scholar] [CrossRef]
- Matucci, A.; Maggi, E.; Vultaggio, A. Eosinophils, the IL-5/IL-5Rα axis, and the biologic effects of benralizumab in severe asthma. Respir. Med. 2019, 160, 105819. [Google Scholar] [CrossRef] [PubMed]
- Bel, E.H.; Wenzel, S.; Thompson, P.J.; Prazma, C.M.; Keene, O.; Yancey, S.W.; Ortega, H.G.; Pavord, I. Oral Glucocorticoid-Sparing Effect of Mepolizumab in Eosinophilic Asthma. N. Engl. J. Med. 2014, 371, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; Fitzgerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab Treatment in Patients with Severe Eosinophilic Asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Ortega, H.G.; Yancey, S.W.; Mayer, B.; Gunsoy, N.B.; Keene, O.; Bleecker, E.R.; Brightling, C.; Pavord, I. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: A secondary analysis of the DREAM and MENSA studies. Lancet Respir. Med. 2016, 4, 549–556. [Google Scholar] [CrossRef]
- Haldar, P.; Brightling, C.; Hargadon, B.; Gupta, S.; Monteiro, W.; Sousa, A.; Marshall, R.P.; Bradding, P.; Green, R.H.; Wardlaw, A.; et al. Mepolizumab and Exacerbations of Refractory Eosinophilic Asthma. N. Engl. J. Med. 2009, 360, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.; Brusselle, G.; Bel, E.H.; FitzGerald, J.M.; Masoli, M.; Korn, S.; Kato, M.; Albers, F.C.; Bradford, E.S.; Gilson, M.J.; et al. Long-term Safety and Clinical Benefit of Mepolizumab in Patients with the Most Severe Eosinophilic Asthma: The COSMEX Study. Clin. Ther. 2019, 41, 2041–2056.e5. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.; Corren, J.; Pavord, I.D.; Maspero, J.; Wenzel, S.; Rabe, K.F.; Busse, W.W.; Ford, L.; Sher, L.; Fitzgerald, J.M.; et al. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N. Engl. J. Med. 2018, 378, 2486–2496. [Google Scholar] [CrossRef]
- Wenzel, S.; Castro, M.; Corren, J.; Maspero, J.; Wang, L.; Zhang, B.; Pirozzi, G.; Sutherland, E.R.; Evans, R.R.; Joish, V.N.; et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: A randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 2016, 388, 31–44. [Google Scholar] [CrossRef]
- Busse, W.; Maspero, J.F.; Katelaris, C.H.; Saralaya, D.; Guillonneau, S.; Zhang, B.; Taniou, C.; Staudinger, H.; Chao, J.; Amin, N.; et al. Dupilumab improves SNOT-22 scores in asthma patients with chronic rhinosinusitis or nasal polypsosis (CRS/NP) in LIBERTY ASTHMA QUEST. Allergy Immunol. 2018, 52, PA1125. [Google Scholar] [CrossRef]
- Blauvelt, A.; de Bruin-Weller, M.; Gooderham, M.; Cather, J.C.; Weisman, J.; Pariser, D.; Simpson, E.L.; Papp, K.A.; Hong, H.C.-H.; Rubel, D.; et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): A 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017, 389, 2287–2303. [Google Scholar] [CrossRef]
- Kavanagh, J.E.; D’Ancona, G.; Elstad, M.; Green, L.; Fernandes, M.; Thomson, L.; Roxas, C.; Dhariwal, J.; Nanzer, A.M.; Kent, B.D.; et al. Real-World Effectiveness and the Characteristics of a “Super-Responder” to Mepolizumab in Severe Eosinophilic Asthma. Chest 2020, 158, 491–500. [Google Scholar] [CrossRef]
- Pelaia, C.; Pelaia, G.; Longhini, F.; Crimi, C.; Calabrese, C.; Gallelli, L.; Sciacqua, A.; Vatrella, A. Monoclonal Antibodies Targeting Alarmins: A New Perspective for Biological Therapies of Severe Asthma. Biomedicines 2021, 9, 1108. [Google Scholar] [CrossRef]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Corren, J.; Gil, E.G.; Griffiths, J.M.; Parnes, J.R.; van der Merwe, R.; Sałapa, K.; O’Quinn, S. Tezepelumab improves patient-reported outcomes in patients with severe, uncontrolled asthma in PATHWAY. Ann. Allergy Asthma Immunol. 2021, 126, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Allinne, J.; Scott, G.; Lim, W.K.; Birchard, D.; Erjefält, J.S.; Sandén, C.; Ben, L.-H.; Agrawal, A.; Kaur, N.; Kim, J.H.; et al. IL-33 blockade affects mediators of persistence and exacerbation in a model of chronic airway inflammation. J. Allergy Clin. Immunol. 2019, 144, 1624–1637.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, W.J.; Tyldesley, S.; Rodda, S.; Halperin, R.; Pai, H.; McKenzie, M.; Duncan, G.; Morton, G.; Hamm, J.; Murray, N. Androgen Suppression Combined with Elective Nodal and Dose Escalated Radiation Therapy (the ASCENDE-RT Trial): An Analysis of Survival Endpoints for a Randomized Trial Comparing a Low-Dose-Rate Brachytherapy Boost to a DoseEscalated External Beam Boost for High- and Intermediate-risk Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 275–285. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, C.-S.; Lim, D.H.; Ahn, S.-H.; Son, B.K.; Kim, J.H.; Jang, T.Y. Beneficial Effect of Anti-Interleukin-33 on the Murine Model of Allergic Inflammation of the Lower Airway. J. Asthma 2012, 49, 738–743. [Google Scholar] [CrossRef]
- Ballantyne, S.J.; Barlow, J.L.; Jolin, H.E.; Nath, P.; Williams, A.S.; Chung, K.F.; Sturton, G.; Wong, S.H.; McKenzie, A.N. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 2007, 120, 1324–1331. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.C.; Campbell, G.A.; McKenzie, A.N.J.; Lloyd, C.M. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Marone, G.; Granata, F.; Pucino, V.; Pecoraro, A.; Heffler, E.; Loffredo, S.; Scadding, G.W.; Varricchi, G. The Intriguing Role of Interleukin 13 in the Pathophysiology of Asthma. Front. Pharmacol. 2019, 10, 1387. [Google Scholar] [CrossRef] [Green Version]
- Hanania, N.A.; Korenblat, P.; Chapman, K.R.; Bateman, E.D.; Kopecky, P.; Paggiaro, P.; Yokoyama, A.; Olsson, J.; Gray, S.; Holweg, C.T.J.; et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): Replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir. Med. 2016, 4, 781–796. [Google Scholar] [CrossRef]
- Panettieri, R.A.; Sjöbring, U.; Péterffy, A.; Wessman, P.; Bowen, K.; Piper, E.; Colice, G.; Brightling, C. Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): Two randomised, double-blind, placebo-controlled, phase 3 clinical trials. Lancet Respir. Med. 2018, 6, 511–525. [Google Scholar] [CrossRef] [Green Version]
- De Boever, E.H.; Ashman, C.; Cahn, A.P.; Locantore, N.W.; Overend, P.; Pouliquen, I.J.; Serone, A.P.; Wright, T.J.; Jenkins, M.M.; Panesar, I.S.; et al. Efficacy and safety of an anti–IL-13 mAb in patients with severe asthma: A randomized trial. J. Allergy Clin. Immunol. 2014, 133, 989–996.e4. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Leigh, R.; McLaurin, K.K.; Kim, K.; Hultquist, M.; Molfino, N.A. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir. Res. 2013, 14, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matucci, A.; Bormioli, S.; Nencini, F.; Chiccoli, F.; Vivarelli, E.; Maggi, E.; Vultaggio, A. Asthma and Chronic Rhinosinusitis: How Similar Are They in Pathogenesis and Treatment Responses? Int. J. Mol. Sci. 2021, 22, 3340. [Google Scholar] [CrossRef]
- Bissonnette, R.; Papp, K.A.; Poulin, Y.; Gooderham, M.; Raman, M.; Mallbris, L.; Wang, C.; Purohit, V.; Mamolo, C.; Papacharalambous, J.; et al. Topical tofacitinib for atopic dermatitis: A phase II a randomized trial. Br. J. Dermatol. 2016, 175, 902–911. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Gadina, M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp. Dermatol. 2014, 23, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S. Placebo-Controlled Trial of Tofacitinib Monotherapy in Rheumatoid Arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [Green Version]
- De Ávila Machado, M.A.; Moura, C.S.; Guerra, S.F.; Curtis, J.R.; Abrahamowicz, M.; Bernatsky, S. Effectiveness and safety of tofacitinib in rheumatoid arthritis: A cohort study. Arthritis Res. 2018, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bertoldi, I.; Caporali, R. Tofacitinib: Real-World Data and Treatment Persistence in Rheumatoid Arthritis. Open Access Rheumatol. Res. Rev. 2021, 13, 221–237. [Google Scholar] [CrossRef]
- Hosking, A.-M.; Juhasz, M.; Mesinkovska, N.A. Topical Janus kinase inhibitors: A review of applications in dermatology. J. Am. Acad. Dermatol. 2018, 79, 535–544. [Google Scholar] [CrossRef]
- Mease, P.; Hall, S.; FitzGerald, O.; Van Der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Su, C.; Panés, J.; Dai, C.; Jiang, M.; Sun, M.-J. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 377, 496–497. [Google Scholar] [CrossRef] [PubMed]
- Verbsky, J.W.; Randolph, D.A.; Shornick, L.; Chaplin, D. Nonhematopoietic Expression of Janus Kinase 3 Is Required for Efficient Recruitment of Th2 Lymphocytes and Eosinophils in OVA-Induced Airway Inflammation. J. Immunol. 2002, 168, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.; Cho, H.; Jeon, Y.J.; Kim, J.H.; Ms, M.H.J.; Jeon, S.; Suh, Y.S.; Park, J.J.; Kim, S. Therapeutic Effects of Intranasal Tofacitinib on Chronic Rhinosinusitis with Nasal Polyps in Mice. Laryngoscope 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.L.; Urban, J.; King, B.A. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J. Am. Acad. Dermatol. 2015, 73, 395–399. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Silverberg, J.I.; Nemoto, O.; Forman, S.B.; Wilke, A.; Prescilla, R.; de la Peña, A.; Nunes, F.P.; Janes, J.; Gamalo, M.; et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 2019, 80, 913–921.e9. [Google Scholar] [CrossRef]
- Bieber, T.; Simpson, E.L.; Silverberg, J.I.; Thaçi, D.; Paul, C.; Pink, A.E.; Kataoka, Y.; Chu, C.-Y.; DiBonaventura, M.; Rojo, R.; et al. Abrocitinib versus Placebo or Dupilumab for Atopic Dermatitis. N. Engl. J. Med. 2021, 384, 1101–1112. [Google Scholar] [CrossRef]
- Mümmler, C.; Dünzelmann, K.; Kneidinger, N.; Barnikel, M.; Munker, D.; Gröger, M.; Canis, M.; Behr, J.; Koch, A.; Haubner, F.; et al. Real-life effectiveness of biological therapies on symptoms in severe asthma with comorbid CRSwNP. Clin. Transl. Allergy 2021, 11, e12049. [Google Scholar] [CrossRef]
- Pfaller, B.; Yepes-Nuñez, J.J.; Agache, I.; Akdis, C.A.; Alsalamah, M.; Bavbek, S.; Bossios, A.; Boyman, O.; Chaker, A.; Chan, S.; et al. Biologicals in atopic disease in pregnancy: An EAACI position paper. Allergy 2021, 76, 71–89. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.B.; Tanaka, Y.; Mariette, X.; Curtis, J.R.; Lee, E.B.; Nash, P.; Winthrop, K.L.; Charles-Schoeman, C.; Thirunavukkarasu, K.; Demasi, R.; et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: Integrated analysis of data from the global clinical trials. Ann. Rheum. Dis. 2017, 76, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Curtis, J.R.; Xie, F.; Yun, H.; Bernatsky, S.; Winthrop, K.L. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1843–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörner, T. Lessons from tofacitinib in patients with cardiovascular risk factors: Increased pulmonary embolism or isolated (thrombotic) pulmonary occlusion rates? Ann. Rheum. Dis. 2020, 79, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nakayamada, S.; Sakata, K.; Kitanaga, Y.; Ma, X.; Lee, S.; Ishii, A.; Yamagata, K.; Nakano, K.; Tanaka, Y. Janus Kinase Inhibitor Baricitinib Modulates Human Innate and Adaptive Immune System. Front. Immunol. 2018, 9, 1510. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matucci, A.; Vivarelli, E.; Nencini, F.; Maggi, E.; Vultaggio, A. Strategies Targeting Type 2 Inflammation: From Monoclonal Antibodies to JAK-Inhibitors. Biomedicines 2021, 9, 1497. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101497
Matucci A, Vivarelli E, Nencini F, Maggi E, Vultaggio A. Strategies Targeting Type 2 Inflammation: From Monoclonal Antibodies to JAK-Inhibitors. Biomedicines. 2021; 9(10):1497. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101497
Chicago/Turabian StyleMatucci, Andrea, Emanuele Vivarelli, Francesca Nencini, Enrico Maggi, and Alessandra Vultaggio. 2021. "Strategies Targeting Type 2 Inflammation: From Monoclonal Antibodies to JAK-Inhibitors" Biomedicines 9, no. 10: 1497. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101497