
MicroRNA and Alternative mRNA Splicing Events in Cancer Drug Response/Resistance: Potent Therapeutic Targets

 ,
,
Abstract
:1. Introduction
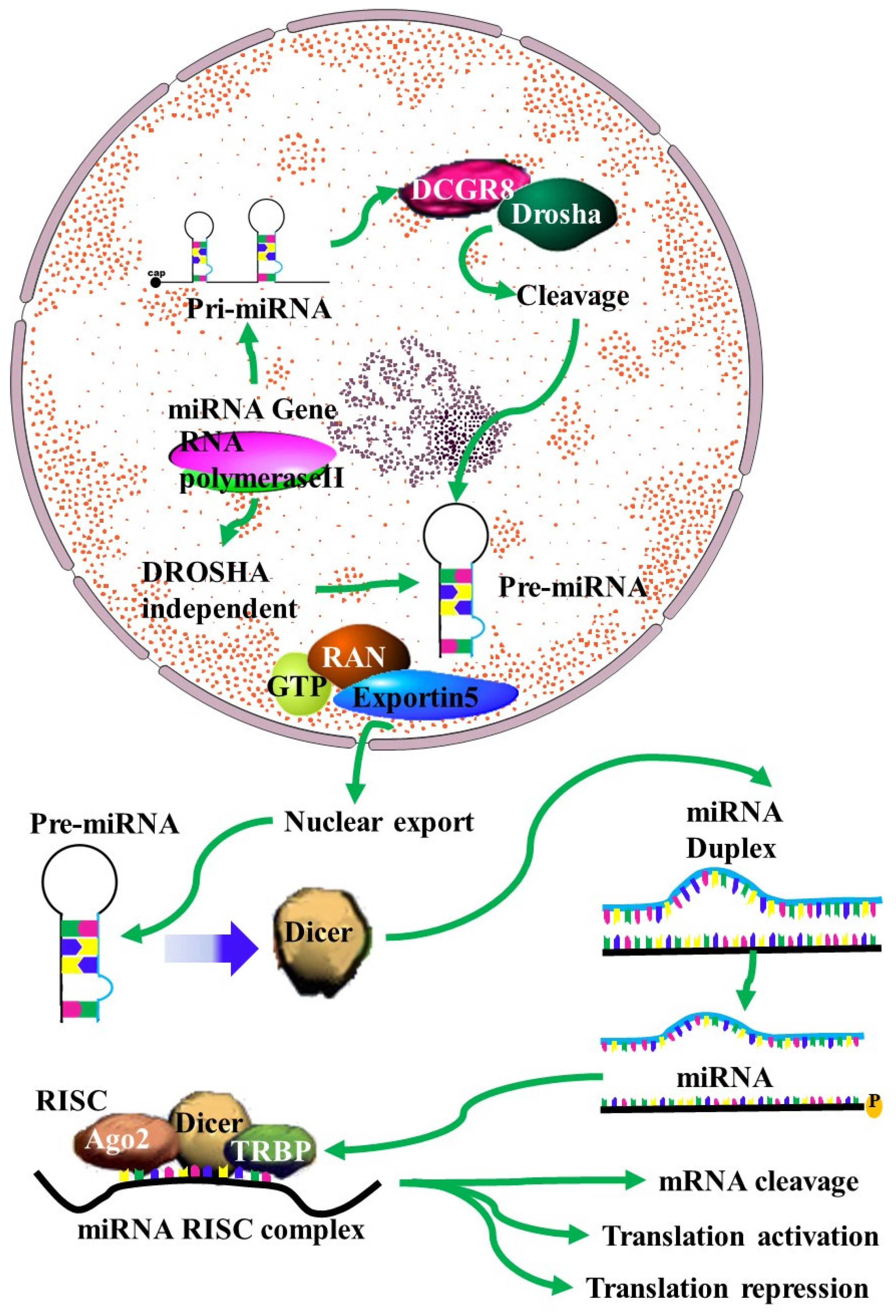
2. Normal and Aberrant Biosynthesis of MiRNAs
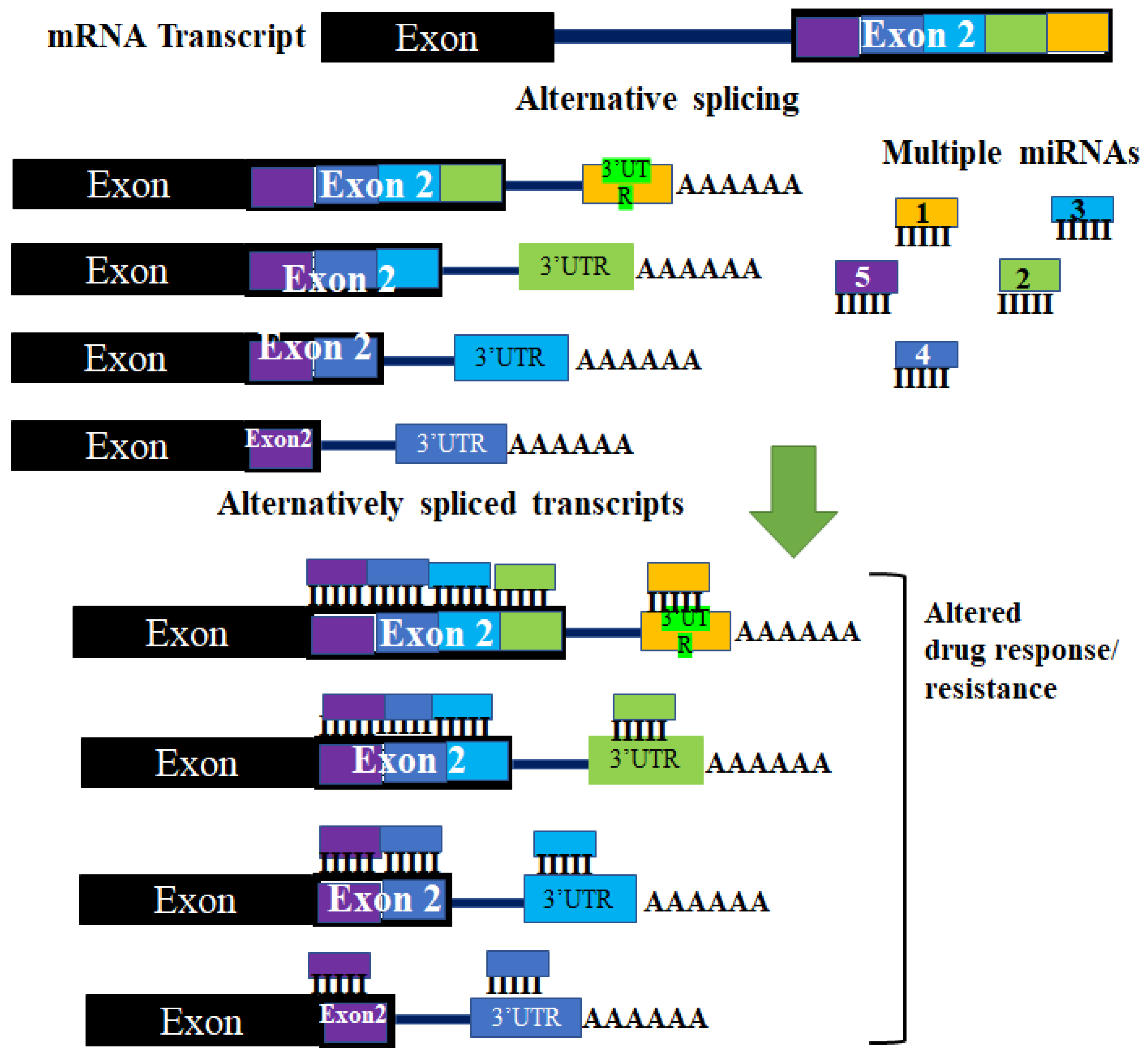
3. Normal and Aberrant mRNA Splicing Events: Targeting the 3′UTR
3.1. Normal and Aberrant Splicing Events in Cancer
3.2. Targeting the 3′UTR of Different Transcripts Generated by Alternative Splicing
4. MiRNA and Alternative Splicing-Induced Drug Resistance
4.1. Breast Cancer
4.1.1. Alternative Splicing and Drug Resistance in Breast Cancer
4.1.2. MiRNA-Induced Drug Resistance in Breast Cancer
4.2. Cervical Cancer
4.2.1. Alternative Splicing in Drug Resistance in Cervical Cancer
4.2.2. MiRNA-Induced Chemo and Radioresistance in Cervical Cancer
4.3. Prostate Cancer
4.3.1. Alternative Splicing in Prostate Cancer
4.3.2. MiRNA-Induced Drug Resistance in Prostate Cancer
4.4. Ovarian Cancer
4.4.1. Alternative Splicing in Drug Resistance in Ovarian Cancer
4.4.2. MiRNA-Induced Drug Resistance in Ovarian Cancer
4.5. Leukemia
4.5.1. Alternative Splicing in Drug Resistance in Leukemia
4.5.2. MiRNA-Induced Drug Resistance in Leukemia
4.6. Colorectal Cancer
4.6.1. Alternative Splicing Induced Drug Resistance in Colorectal Cancer
4.6.2. MiRNA-Induced Drug Resistance in Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
| Colorectal Cancer | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| Splicing of VEGFA | Bevacizumab | VEGFA165b binds to the antibody preventing it from binding to and blocking VEGF | [158] |
| TIA-1 spliced to truncated sTIA-1 | Higher levels of full-length TIA-1 and lower levels of sTIA-1 increase the expression of VEGF-A165b (anti-angiogenic). | [159] | |
| SYK is spliced to long (SYK(L) and short SYK(S) | 5-FU | The increased expression of either of the isoforms leads to increased sensitivity to the drug | [160] |
| miRNA | |||
| miRNA | Drug | Effects | Ref |
| miR-153 | Multidrug | Alteration of the expression of multiple isoforms of specific protein targets. Many of these targets are involved in roles such as cell cycle regulation and cell death | [5] |
| miR-297 | |||
| miR-451 | |||
| miR-222 | |||
| miR-1915 | |||
| miR-10b | 5-FU | Increased resistance to the drug by acting on BIM | [165] |
| miR-21 | Induces resistance to the drug by downregulating hMSH2 expression | [166] | |
| miR-23a | miR-23a enhances sensitivity to the drug by acting through the APAF-1/caspase 9 apoptosis pathway | [167] | |
| miR-34a | miR-34a acts on c-kit by downregulating it (c-Kit) and increasing sensitivity to the drug | [168] | |
| miR-96 | Expression of miR-96 decreased XIAP and p53 stability regulator UBE2N, increased apoptosis and increased sensitivity to the drug | [169] | |
| miR-203 | miR-203 targets and decreases expression of TYMS, increasing sensitivity to the drug. | [170] | |
| miR-497 | miR-497 decreases the expression of Smurf1 leading to increased drug sensitivity. | [171] | |
| miR-587 | Regulates PPP2R1B expression increasing resistance to the drug | [172] | |
| miR-20a | miR-20a targets and downregulates BNIP2, increasing resistance to the drug | [173] | |
| miR-20a | Oxaliplatin | miR-20a targets and downregulates BNIP2, increasing drug resistance | [173] |
| miR-203 | miR-293 negatively regulates ATM increasing resistance to the drug | [174] | |
| miR-503-5p | miR-503-5p targets PUMA leading to decreased expression of PUMA, leading to increased resistance to the drug | [175] | |
| miR-1915 | Inhibits BCL-2 leading to increased sensitivity to the drug | [176] | |
| miR-20a | miR-20a targets BNIP2 downregulating BNIP2 and increasing resistance to the drug | [173] | |
| miR-451 | Irinotecan | miR-451 downregulates MIF which downregulates its target COX-2 increasing sensitivity to the drug | [177] |
| miR-1915 | Doxorubicin | Inhibits BCL-2 expression, increasing sensitivity to the drug | [176] |
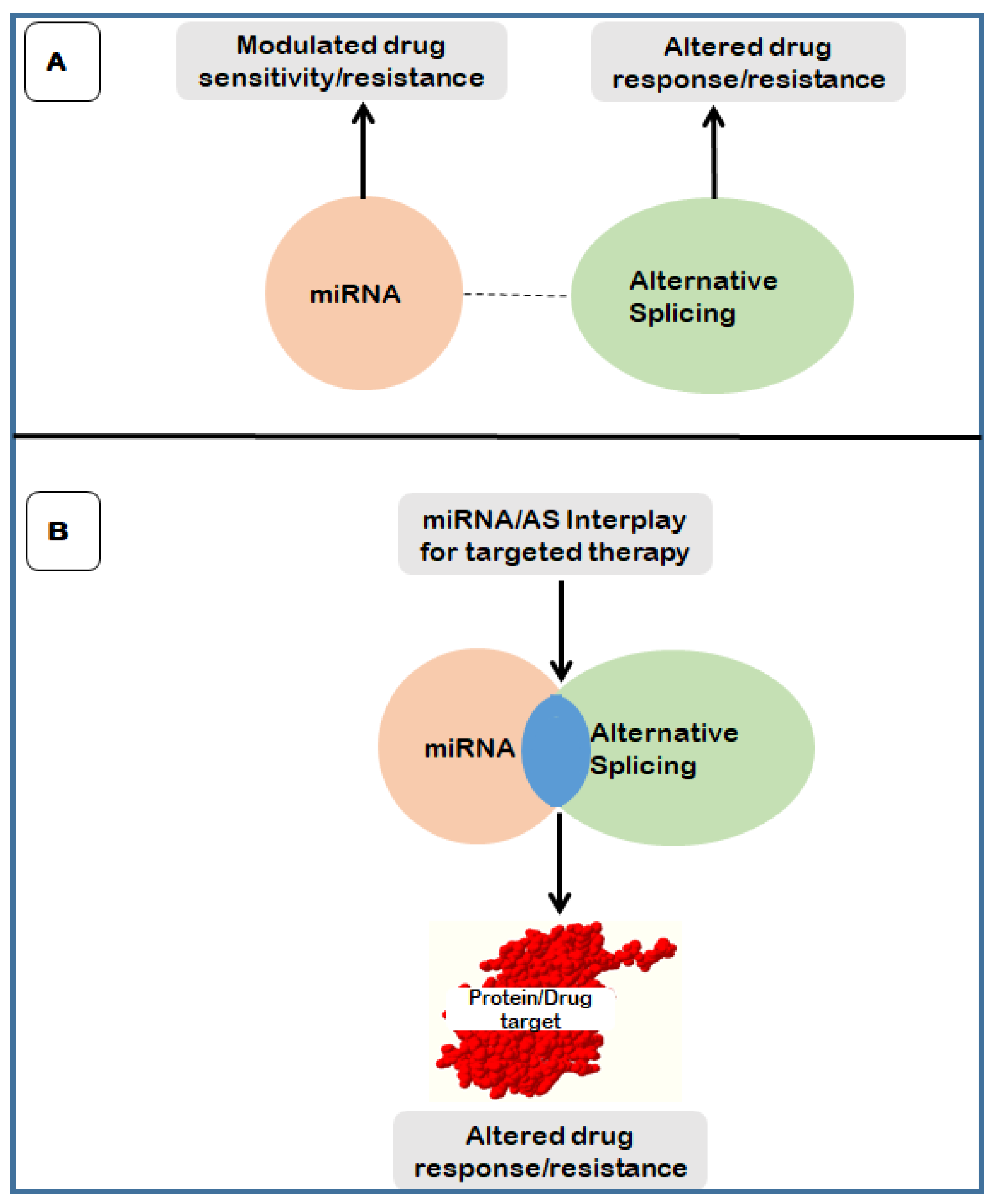
5. The Interplay between Alternative Splicing and MiRNA in Drug Resistance
5.1. mRNA AS/miRNA Interplay
5.2. mRNA AS/miRNA Interplay in Chemoresistance: Targeting the ceRNA Networks
5.3. The Interaction between MiRNAs and Splicing Factors
| miRNA | Splicing Factor | Type of Alteration | Cancer | Effect Produced | Ref |
|---|---|---|---|---|---|
| miR-18a | HnRNP1A1 | HnRNP1A1 suppresses miR-18a | Prostate | Docetaxel resistance. KRAS upregulation | [190] |
| miR-15a-5p | HnRNPA1 downregulated | Pancreatic | Tumor suppression | [190] | |
| miR-25-3p | [190] | ||||
| miR-7 family | SRSF1 | Correct synthesis of miR-7 family | Pancreatic | SRSF1 is involved in the expression of many cancer promoting genes as well as in the expression of pro-apoptotic isoforms of Bcl-x, RON, and MCL-1 | [191] |
| miR-7 family | miR-7 suppresses the expression of SRSF1 | Prostate | [191] | ||
| miR-222 | Downregulation of SRSF1 | Prostate | [191] | ||
| miR-221 | Pancreatic | [191] | |||
| miR-17-92 | Pancreatic | [191] | |||
| miR-10a | Lung | Increased retinoic acid sensitivity | [197] | ||
| miR-10b | Neuroblastoma | [197] | |||
| miR-193a | SRSF6 | Downregulation of SRSF1 | Pancreatic | increased metastasis | [189] |
| miR-124 | PTBP1 | Downregulates PTBP1 | Pancreatic | increased expression of an isoform of PKM worse prognosis | [192] |
| Tumor suppressor miRNAs | Rbfox2 | Rbfox2 is downregulated in cancer | Multiple | Rbfox2 promotes cell invasion | [194] |
| miR-103a | SF3B1 | SF3B1 regulates maturation of miR-103a | Pancreatic | Promotes tumor growth | [193] |
| miR-423 | SF3B1 regulates maturation of miR-423 | Ovarian | Inhibits proliferation and metastasis | [193] | |
| miR-193a-5p | SRSF6 | Alternative splicing of OGDHL and ECM1 | Pancreatic | promotes cell metastasis | [189] |
| miR-184 | AF1 | LncRNA UCA1 promotes proliferation by suppressing MiR-184 | Oral squamous cell carcinoma | cisplatin resistance | [198] |
| miR-221 | Quaking (QKI) | miR-221 targets the most abundant isoform of QKI and reduced QKI-5 levels | Colorectal | Increased resistance to cytotoxic agents | [199] |
6. Clinical Implications of Chemoresistance
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J. MicroRNAs are important regulators of drug resistance in colorectal cancer. Biol. Chem. 2017, 398, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 53, 100728. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Du, C.; Chen, D.; Gu, X.; Li, C.; Yao, M.; Pan, J.; Cheng, J.; Jiang, D.; et al. A miR-20a/MAPK1/c-Myc regulatory feedback loop regulates breast carcinogenesis and chemoresistance. Cell Death Differ. 2018, 25, 406–420. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget 2017, 8, 22003–22013. [Google Scholar] [CrossRef] [Green Version]
- Magee, P.; Shi, L.; Garofalo, M. Role of microRNAs in chemoresistance. Ann. Transl. Med. 2015, 3, 332. [Google Scholar] [CrossRef]
- Pouyanrad, S.; Rahgozar, S.; Ghodousi, E.S. Dysregulation of miR-335-3p, targeted by NEAT1 and MALAT1 long non-coding RNAs, is associated with poor prognosis in childhood acute lymphoblastic leukemia. Gene 2019, 692, 35–43. [Google Scholar] [CrossRef]
- Turrini, E.; Haenisch, S.; Laechelt, S.; Diewock, T.; Bruhn, O.; Cascorbi, I. MicroRNA profiling in K-562 cells under imatinib treatment: Influence of miR-212 and miR-328 on ABCG2 expression. Pharm. Genom. 2012, 22, 198–205. [Google Scholar] [CrossRef]
- Ghodousi, E.S.; Rahgozar, S. MicroRNA-326 and microRNA-200c: Two novel biomarkers for diagnosis and prognosis of pediatric acute lymphoblastic leukemia. J. Cell. Biochem. 2018, 119, 6024–6032. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Aquino-Jarquin, G. Reversal of multidrug resistance of leukemia cells is not necessarily induced by direct miR-138/MDR1 promoter interaction. Leuk. Res. 2017, 57, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hua, Y.; Liu, C.; Cheng, S. MiRNA-494 inhibits apoptosis and promotes autophagy of acute myeloid leukemia cells by downregulating FGFR2. Minerva Endocrinol. 2019, 44, 410–413. [Google Scholar] [CrossRef]
- Wang, L.W.; Wang, H.R.; Ji, W.G.; Guo, S.L.; Li, H.X.; Xu, X.Y. MiRNA-485-5p suppresses the proliferation of acute myeloid leukemia via targeting SALL4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4842–4849. [Google Scholar] [CrossRef]
- Fan, S.J.; Li, H.B.; Cui, G.; Kong, X.L.; Sun, L.L.; Zhao, Y.Q.; Li, Y.H.; Zhou, J. MiRNA-149 * promotes cell proliferation and suppresses apoptosis by mediating JunB in T-cell acute lymphoblastic leukemia. Leuk. Res. 2016, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Tang, Y.J.; Zhang, W.G.; Wan, C.C.; Chen, Y.; Zhang, L.J. MiR-143 regulates proliferation and apoptosis of myelocytic leukemia cell HL-60 via modulating ERK1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9237. [Google Scholar] [CrossRef]
- Liu, L.; Ren, W.; Chen, K. MiR-34a Promotes Apoptosis and Inhibits Autophagy by Targeting HMGB1 in Acute Myeloid Leukemia Cells. Cell. Physiol. Biochem. 2017, 41, 1981–1992. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol. 2017, 1509, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karube, Y.; Tanaka, H.; Osada, H.; Tomida, S.; Tatematsu, Y.; Yanagisawa, K.; Yatabe, Y.; Takamizawa, J.; Miyoshi, S.; Mitsudomi, T.; et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005, 96, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Allegra, D.; Bilan, V.; Garding, A.; Döhner, H.; Stilgenbauer, S.; Kuchenbauer, F.; Mertens, D.; Zucknick, M. Defective DROSHA processing contributes to downregulation of MiR-15/-16 in chronic lymphocytic leukemia. Leukemia 2014, 28, 98–107. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Gao, P.; Wu, M. c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 2013, 3, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.; Gi, Y.J.; Lin, Y.L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Trinidad, A.G.; Caswell, P.T.; Norman, J.C.; Vousden, K.H. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J. Biol. Chem. 2014, 289, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wan, G.; Berger, F.G.; He, X.; Lu, X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell 2011, 41, 371–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010, 18, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kushwaha, P.P.; Gupta, S.J.C.D.R. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019, 2, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Holmila, R.; Fouquet, C.; Cadranel, J.; Zalcman, G.; Soussi, T. Splice mutations in the p53 gene: Case report and review of the literature. Hum. Mutat. 2003, 21, 101–102. [Google Scholar] [CrossRef]
- Tanko, Q.; Franklin, B.; Lynch, H.; Knezetic, J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat. Res. 2002, 503, 37–42. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Hallam, S.E.; Venne, V.L.; Lyon, E.; Ward, K. Implications of a novel cryptic splice site in the BRCA1 gene. Am. J. Med. Genet. 1998, 80, 140–144. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Colapietro, P.; Gervasini, C.; Natacci, F.; Rossi, L.; Riva, P.; Larizza, L. NF1 exon 7 skipping and sequence alterations in exonic splice enhancers (ESEs) in a neurofibromatosis 1 patient. Hum. Genet. 2003, 113, 551–554. [Google Scholar] [CrossRef]
- Damm, F.; Thol, F.; Kosmider, O.; Kade, S.; Löffeld, P.; Dreyfus, F.; Stamatoullas-Bastard, A.; Tanguy-Schmidt, A.; Beyne-Rauzy, O.; de Botton, S.; et al. SF3B1 mutations in myelodysplastic syndromes: Clinical associations and prognostic implications. Leukemia 2012, 26, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Ogawa, S. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 2014, 5, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Guo, J.; Huang, Q.; Chen, X.; Li-Ling, J.; Li, Q.; Ma, F. Retained introns increase putative microRNA targets within 3’ UTRs of human mRNA. FEBS Lett. 2007, 581, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Tian, B. Reprogramming of 3’ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 2009, 4, e8419. [Google Scholar] [CrossRef] [Green Version]
- Meyers-Needham, M.; Ponnusamy, S.; Gencer, S.; Jiang, W.; Thomas, R.J.; Senkal, C.E.; Ogretmen, B. Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 2012, 4, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francies, F.Z.; Hull, R.; Khanyile, R.; Dlamini, Z. Breast cancer in low-middle income countries: Abnormality in splicing and lack of targeted treatment options. Am. J. Cancer Res. 2020, 10, 1568–1591. [Google Scholar]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Oliveira, M.; Reis-Mendes, A.; Carvalho, F.; Remião, F.; Bastos, M.L.; Costa, V.M. Doxorubicin Is Key for the Cardiotoxicity of FAC (5-Fluorouracil + Adriamycin + Cyclophosphamide) Combination in Differentiated H9c2 Cells. Biomolecules 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhou, Z.; Subhramanyam, C.S.; Cao, Q.; Heng, Z.S.L.; Liu, W.; Fu, X.; Hu, Q. SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells. Commun. Biol. 2020, 3, 268. [Google Scholar] [CrossRef]
- Tanaka, I.; Chakraborty, A.; Saulnier, O.; Benoit-Pilven, C.; Vacher, S.; Labiod, D.; Lam, E.W.F.; Bièche, I.; Delattre, O.; Pouzoulet, F.; et al. ZRANB2 and SYF2-mediated splicing programs converging on ECT2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020, 48, 2676–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagnoli, L.; Ladomery, M.; Tagliabue, E.; Pupa, S.M. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers 2019, 11, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, V.; Gautrey, H.; Kirby, J.; Tyson-Capper, A. HER2 splice variants in breast cancer: Investigating their impact on diagnosis and treatment outcomes. Oncotarget 2020, 11, 4338–4357. [Google Scholar] [CrossRef]
- Ren, Y.Q.; Wang, H.J.; Zhang, Y.Q.; Liu, Y.B. WBP2 modulates G1/S transition in ER+ breast cancer cells and is a direct target of miR-206. Cancer Chemother. Pharmacol. 2017, 79, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, H.; Huang, Y.F.; Li, M.L.; Cheng, J.H.; Hu, P.; Lu, C.H.; Zhang, Y.; Liu, N.; Tzeng, C.M.; et al. WW domain-binding protein 2: An adaptor protein closely linked to the development of breast cancer. Mol. Cancer 2017, 16, 128. [Google Scholar] [CrossRef] [Green Version]
- Jansen, M.P.; Reijm, E.A.; Sieuwerts, A.M.; Ruigrok-Ritstier, K.; Look, M.P.; Rodríguez-González, F.G.; Heine, A.A.; Martens, J.W.; Sleijfer, S.; Foekens, J.A.; et al. High miR-26a and low CDC2 levels associate with decreased EZH2 expression and with favorable outcome on tamoxifen in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 133, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Z.; Wang, Q.; Xing, X.J.; Zhao, Y. Overexpression of microRNA-365 inhibits breast cancer cell growth and chemo-resistance through GALNT4. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4710–4718. [Google Scholar] [PubMed]
- Sun, R.; Liu, Z.; Ma, G.; Lv, W.; Zhao, X.; Lei, G.; Xu, C. Associations of deregulation of mir-365 and its target mRNA TTF-1 and survival in patients with NSCLC. Int. J. Clin. Exp. Pathol. 2015, 8, 2392–2399. [Google Scholar] [PubMed]
- Song, Y.K.; Wang, Y.; Wen, Y.Y.; Zhao, P.; Bian, Z.J. MicroRNA-22 Suppresses Breast Cancer Cell Growth and Increases Paclitaxel Sensitivity by Targeting NRAS. Technol. Cancer Res. Treat. 2018, 17, 1533033818809997. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Picard, D. MiR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell. Biol. 2009, 29, 3783–3790. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, X.; Wang, M.; Xiao, G.; Yang, G.; Wang, H.; Li, Y.; Sun, X.; Qin, S.; Du, N.; et al. A miR-26a/E2F7 feedback loop contributes to tamoxifen resistance in ER-positive breast cancer. Int. J. Oncol. 2018, 53, 1601–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Zhang, X.; Li, M.; Chen, Q.; Ye, M.; Liang, W.; Ding, L.; Cai, H.; Fu, D.; Lv, Z. MiR-26a and its target CKS2 modulate cell growth and tumorigenesis of papillary thyroid carcinoma. PLoS ONE 2013, 8, e67591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Zhu, Y.; Liu, Y.; Sun, L.; Lv, X.; Wu, Y.; Hu, P.; Su, F.; Gong, C.; Song, E.; et al. E2F7 overexpression leads to tamoxifen resistance in breast cancer cells by competing with E2F1 at miR-15a/16 promoter. Oncotarget 2015, 6, 31944–31957. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Ge, S.; Lin, D.; Han, J.; Ying, G.; Ba, Y. Identification of HGF as a novel target of miR-15a/16/195 in gastric cancer. Investig. New Drugs 2020, 38, 922–933. [Google Scholar] [CrossRef]
- Hu, W.; Tan, C.; He, Y.; Zhang, G.; Xu, Y.; Tang, J. Functional miRNAs in breast cancer drug resistance. Oncotargets Ther. 2018, 11, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Wang, X.; Zhang, L.; Li, D.; Liu, X.; Song, G.; Cao, H.; Zhu, J.; Li, H. Plasma microRNAs Predict Chemoresistance in Patients with Metastatic Breast Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819828709. [Google Scholar] [CrossRef]
- Renthal, N.E.; Chen, C.C.; Williams, K.C.; Gerard, R.D.; Prange-Kiel, J.; Mendelson, C.R. MiR-200 family and targets, ZEB1 and ZEB2, modulate uterine quiescence and contractility during pregnancy and labor. Proc. Natl. Acad. Sci. USA 2010, 107, 20828–20833. [Google Scholar] [CrossRef] [Green Version]
- Gasparello, J.; Fabbri, E.; Bianchi, N.; Breveglieri, G.; Zuccato, C.; Borgatti, M.; Gambari, R.; Finotti, A. BCL11A mRNA Targeting by miR-210: A Possible Network Regulating γ-Globin Gene Expression. Int. J. Mol. Sci. 2017, 18, 2530. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.W.; Calses, P.; Kemp, C.J.; Taniguchi, T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012, 72, 4037–4046. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Deng, C.; Lu, W.; Xiao, J.; Ma, D.; Guo, M.; Recker, R.R.; Gatalica, Z.; Wang, Z.; Xiao, G.G. let-7 microRNAs induce tamoxifen sensitivity by downregulation of estrogen receptor α signaling in breast cancer. Mol. Med. 2011, 17, 1233–1241. [Google Scholar] [CrossRef]
- Bai, W.D.; Ye, X.M.; Zhang, M.Y.; Zhu, H.Y.; Xi, W.J.; Huang, X.; Zhao, J.; Gu, B.; Zheng, G.X.; Yang, A.G.; et al. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int. J. Cancer 2014, 135, 1356–1368. [Google Scholar] [CrossRef]
- Ye, X.; Bai, W.; Zhu, H.; Zhang, X.; Chen, Y.; Wang, L.; Yang, A.; Zhao, J.; Jia, L. MiR-221 promotes trastuzumab-resistance and metastasis in HER2-positive breast cancers by targeting PTEN. BMB Rep. 2014, 47, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Shen, H.; Cao, Y.; Li, H.; Qin, R.; Chen, Q.; Long, L.; Zhu, X.L.; Xie, C.J.; Xu, W.L. Involvement of miR-30c in resistance to doxorubicin by regulating YWHAZ in breast cancer cells. Braz. J. Med. Biol. Res. 2014, 47, 60–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.Y.; Chang, E.; Lee, E.J.; Lee, H.W.; Kang, H.G.; Chun, K.H.; Woo, Y.M.; Kong, H.K.; Ko, J.Y.; Suzuki, H.; et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. 2014, 74, 7573–7582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Zheng, W.; Li, N.; Su, Z.; Zhao, L.; Zhou, H.; Jia, L. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 41942. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Y.; Shen, H.; Li, H.; Long, L.; Hui, L.; Xu, W. MiR-137 restoration sensitizes multidrug-resistant MCF-7/ADM cells to anticancer agents by targeting YB-1. Acta Biochim. Biophys. Sin. 2013, 45, 80–86. [Google Scholar] [CrossRef] [Green Version]
- He, D.X.; Gu, X.T.; Li, Y.R.; Jiang, L.; Jin, J.; Ma, X. Methylation-regulated miR-149 modulates chemoresistance by targeting GlcNAc N-deacetylase/N-sulfotransferase-1 in human breast cancer. FEBS J. 2014, 281, 4718–4730. [Google Scholar] [CrossRef]
- Yang, G.; Wu, D.; Zhu, J.; Jiang, O.; Shi, Q.; Tian, J.; Weng, Y. Upregulation of miR-195 increases the sensitivity of breast cancer cells to Adriamycin treatment through inhibition of Raf-1. Oncol. Rep. 2013, 30, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Oak, P.S.; Wagner, E.; Roidl, A. MiR-200c sensitizes breast cancer cells to doxorubicin treatment by decreasing TrkB and Bmi1 expression. PLoS ONE 2012, 7, e50469. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503. [Google Scholar] [CrossRef] [Green Version]
- Ao, X.; Nie, P.; Wu, B.; Xu, W.; Zhang, T.; Wang, S.; Chang, H.; Zou, Z. Decreased expression of microRNA-17 and microRNA-20b promotes breast cancer resistance to taxol therapy by upregulation of NCOA3. Cell Death Dis. 2016, 7, e2463. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, J.; Li, S.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Tang, J. The function role of miR-181a in chemosensitivity to adriamycin by targeting Bcl-2 in low-invasive breast cancer cells. Cell. Physiol. Biochem. 2013, 32, 1225–1237. [Google Scholar] [CrossRef]
- Kastl, L.; Brown, I.; Schofield, A.C. MiRNA-34a is associated with docetaxel resistance in human breast cancer cells. Breast Cancer Res. Treat. 2012, 131, 445–454. [Google Scholar] [CrossRef]
- Wang, H.; Tan, G.; Dong, L.; Cheng, L.; Li, K.; Wang, Z.; Luo, H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS ONE 2012, 7, e34210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Xu, K.; Yagüe, E. MiR-218 targets survivin and regulates resistance to chemotherapeutics in breast cancer. Breast Cancer Res. Treat. 2015, 151, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, R.; Katsumata, N.; Shibata, T.; Kamura, T.; Kasamatsu, T.; Nakanishi, T.; Nishimura, S.; Ushijima, K.; Takano, M.; Satoh, T.; et al. Paclitaxel Plus Carboplatin Versus Paclitaxel Plus Cisplatin in Metastatic or Recurrent Cervical Cancer: The Open-Label Randomized Phase III Trial JCOG0505. J. Clin. Oncol. 2015, 33, 2129–2135. [Google Scholar] [CrossRef]
- Moore, K.N.; Herzog, T.J.; Lewin, S.; Giuntoli, R.L.; Armstrong, D.K.; Rocconi, R.P.; Spannuth, W.A.; Gold, M.A. A comparison of cisplatin/paclitaxel and carboplatin/paclitaxel in stage IVB, recurrent or persistent cervical cancer. Gynecol. Oncol. 2007, 105, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Cui, H.; Yu, H.; Ji, Q.; Kang, L.; Han, B.; Wang, J.; Dong, Q.; Li, Y.; Yan, Z.; et al. MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression. Oncogenesis 2016, 5, e197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Cai, H.; Xiao, Z.X.; Wang, H.; Yang, P. Effect of radiotherapy on the survival of cervical cancer patients: An analysis based on SEER database. Medicine 2019, 98, e16421. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat. Genet. 2013, 45, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, J.; Ren, Z.; Chen, Y.; Li, J.; Miao, X.; Song, Y.; Zhao, T.; Li, Y.; Shi, Y.; et al. A specific miRNA signature promotes radioresistance of human cervical cancer cells. Cancer Cell Int. 2013, 13, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, C.; Rani, S.; Breslin, S.; Gogarty, M.; Ghobrial, I.M.; Crown, J.; O’Driscoll, L. MiR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol. Cancer 2014, 13, 71. [Google Scholar] [CrossRef] [Green Version]
- Pedroza-Torres, A.; Campos-Parra, A.D.; Millan-Catalan, O.; Loissell-Baltazar, Y.A.; Zamudio-Meza, H.; de León, D.C.; Montalvo-Esquivel, G.; Isla-Ortiz, D.; Herrera, L.A.; Ángeles-Zaragoza, Ó.; et al. MicroRNA-125 modulates radioresistance through targeting p21 in cervical cancer. Oncol. Rep. 2018, 39, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.X.; Zhang, S.M.; Li, J.; Yang, B.; Ouyang, W.; Mei, Z.J.; Chen, J.; Dai, J.; Ke, S.; Zhou, F.X.; et al. MicroRNA-320 regulates the radiosensitivity of cervical cancer cells C33AR by targeting β-catenin. Oncol. Lett. 2016, 12, 4983–4990. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, L.; Chen, Z. Transcriptome profiling of cervical cancer cells acquired resistance to cisplatin by deep sequencing. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2820–2829. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Wu, L.; Ding, H.; Wang, Y.; Zhang, Y.; Chen, X.; Chen, X.; Zhang, C.Y.; Zhang, Q.; Zen, K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J. Biol. Chem. 2012, 287, 4148–4156. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, M.; Li, X.; Tang, H. MiR-214 reduces cell survival and enhances cisplatin-induced cytotoxicity via down-regulation of Bcl2l2 in cervical cancer cells. FEBS Lett. 2013, 587, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Mehterov, N.; Kazakova, M.; Sbirkov, Y.; Vladimirov, B.; Belev, N.; Yaneva, G.; Todorova, K.; Hayrabedyan, S.; Sarafian, V. Alternative RNA Splicing—The Trojan Horse of Cancer Cells in Chemotherapy. Genes 2021, 12, 1085. [Google Scholar] [CrossRef]
- Phillips, J.W.; Pan, Y.; Tsai, B.L.; Xie, Z.; Demirdjian, L.; Xiao, W.; Yang, H.T.; Zhang, Y.; Lin, C.H.; Cheng, D.; et al. Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc. Natl. Acad. Sci. USA 2020, 117, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Melnyk, J.E.; Steri, V.; Nguyen, H.G.; Hann, B.; Feng, F.Y.; Shokat, K.M. The splicing modulator sulfonamide indisulam reduces AR-V7 in prostate cancer cells. Bioorg. Med. Chem. 2020, 28, 115712. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, N.; Nimura, K.; Saga, K.; Ishibashi, A.; Kitamura, K.; Nagano, H.; Yoshikawa, Y.; Ishida, K.; Nonomura, N.; Arisawa, M.; et al. SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression. Cancer Res. 2019, 79, 5204–5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, K.; Fujita, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate 2010, 70, 1501–1512. [Google Scholar] [CrossRef]
- Xu, B.; Niu, X.; Zhang, X.; Tao, J.; Wu, D.; Wang, Z.; Li, P.; Zhang, W.; Wu, H.; Feng, N.; et al. MiR-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of KRAS. Mol. Cell. Biochem. 2011, 350, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Kojima, K.; Ohhashi, R.; Hamada, N.; Nozawa, Y.; Kitamoto, A.; Sato, A.; Kondo, S.; Kojima, T.; Deguchi, T.; et al. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J. Biol. Chem. 2010, 285, 19076–19084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eringyte, I.; Losada, J.N.Z.; Powell, S.M.; Bevan, C.L.; Fletcher, C.E. Coordinated AR and microRNA regulation in prostate cancer. Asian J. Urol. 2020, 7, 233–250. [Google Scholar] [CrossRef]
- Sun, T.; Yang, M.; Chen, S.; Balk, S.; Pomerantz, M.; Hsieh, C.L.; Brown, M.; Lee, G.M.; Kantoff, P.W. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate 2012, 72, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Lu, Y.; Cui, D.; Li, E.; Zhu, Y.; Zhao, Y.; Zhao, F.; Xia, S. MiR-200b suppresses cell proliferation, migration and enhances chemosensitivity in prostate cancer by regulating Bmi-1. Oncol. Rep. 2014, 31, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Mihanfar, A.; Fattahi, A.; Nejabati, H.R. MicroRNA-mediated drug resistance in ovarian cancer. J. Cell. Physiol. 2019, 234, 3180–3191. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wang, J.; Li, H.; Yu, Y.; Wang, X.; Lu, L.; Lv, C.; Chang, B.; Jin, W.; Guo, W.; et al. Extracellular matrix protein-1 secretory isoform promotes ovarian cancer through increasing alternative mRNA splicing and stemness. Nat. Commun. 2021, 12, 4230. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Q.; Qin, R.; Zhang, K.; Li, H. MicroRNA-449a reduces cell survival and enhances cisplatin-induced cytotoxicity via downregulation of NOTCH1 in ovarian cancer cells. Tumour Biol. 2014, 35, 12369–12378. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Park, S.M.; Murmann, A.E.; Gwin, K.; Montag, A.G.; Zillhardt, M.; Hua, Y.J.; Lengyel, E.; Peter, M.E. Let-7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int. J. Cancer 2012, 130, 1787–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Li, N.; Yang, Z.; Zhou, B.; He, Y.; Weng, D.; Fang, Y.; Wu, P.; Chen, P.; Yang, X.; et al. MiR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J. Natl. Cancer Inst. 2013, 105, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.P.; Chen, Y.G.; Lan, J.Y.; Shen, Z.J. MicroRNA-370 suppresses proliferation and promotes endometrioid ovarian cancer chemosensitivity to cDDP by negatively regulating ENG. Cancer Lett. 2014, 353, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Siu, M.K.; Liu, S.S.; Yam, J.W.; Ngan, H.Y.; Chan, D.W. Epigenetic silencing of microRNA-199b-5p is associated with acquired chemoresistance via activation of JAG1-Notch1 signaling in ovarian cancer. Oncotarget 2014, 5, 944–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Xiao, Z.; Zhang, H.; Wang, K.; Liu, W.; Hao, Q. MiR-489 modulates cisplatin resistance in human ovarian cancer cells by targeting Akt3. Anti Cancer Drugs 2014, 25, 799–809. [Google Scholar] [CrossRef]
- Chan, J.K.; Blansit, K.; Kiet, T.; Sherman, A.; Wong, G.; Earle, C.; Bourguignon, L.Y. The inhibition of miR-21 promotes apoptosis and chemosensitivity in ovarian cancer. Gynecol. Oncol. 2014, 132, 739–744. [Google Scholar] [CrossRef]
- Fu, X.; Tian, J.; Zhang, L.; Chen, Y.; Hao, Q. Involvement of microRNA-93, a new regulator of PTEN/Akt signaling pathway, in regulation of chemotherapeutic drug cisplatin chemosensitivity in ovarian cancer cells. FEBS Lett. 2012, 586, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, Z.; Pei, L.; Sun, J. MicroRNA miR-106a-5p targets forkhead box transcription factor FOXC1 to suppress the cell proliferation, migration, and invasion of ectopic endometrial stromal cells via the PI3K/Akt/mTOR signaling pathway. Bioengineered 2021, 12, 2203–2213. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, M.; Huang, J.; Li, Y.; Wang, S.; Harrington, C.A.; Qian, D.Z.; Sun, X.X.; Dai, M.S. MicroRNA-130a suppresses breast cancer cell migration and invasion by targeting FOSL1 and upregulating ZO-1. J. Cell. Biochem. 2018, 119, 4945–4956. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Spoelstra, N.S.; Howe, E.N.; Nordeen, S.K.; Richer, J.K. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol. Cancer Ther. 2009, 8, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, D.R.; Howe, E.N.; Spoelstra, N.S.; Richer, J.K. Loss of miR-200c: A Marker of Aggressiveness and Chemoresistance in Female Reproductive Cancers. J. Oncol. 2010, 2010, 821717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cittelly, D.M.; Dimitrova, I.; Howe, E.N.; Cochrane, D.R.; Jean, A.; Spoelstra, N.S.; Post, M.D.; Lu, X.; Broaddus, R.R.; Spillman, M.A.; et al. Restoration of miR-200c to ovarian cancer reduces tumor burden and increases sensitivity to paclitaxel. Mol. Cancer Ther. 2012, 11, 2556–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Nag, A.; Mandal, C.C. A Comprehensive Review on miR-200c, a Promising Cancer Biomarker with Therapeutic Potential. Curr. Drug Targets 2015, 16, 1381–1403. [Google Scholar] [CrossRef]
- Mitamura, T.; Watari, H.; Wang, L.; Kanno, H.; Hassan, M.K.; Miyazaki, M.; Katoh, Y.; Kimura, T.; Tanino, M.; Nishihara, H.; et al. Downregulation of miRNA-31 induces taxane resistance in ovarian cancer cells through increase of receptor tyrosine kinase MET. Oncogenesis 2013, 2, e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.H.; Kim, T.H.; Kim, K.; Song, J.A.; Jung, Y.J.; Jeong, J.Y.; Lee, M.J.; Kim, Y.K.; Lee, D.H.; An, H.J. Dysregulation of miR-106a and miR-591 confers paclitaxel resistance to ovarian cancer. Br. J. Cancer 2013, 109, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, N.; Wang, H.; Jia, X.; Wang, X.; Luo, J. Altered microRNA expression in cisplatin-resistant ovarian cancer cells and upregulation of miR-130a associated with MDR1/P-glycoprotein-mediated drug resistance. Oncol. Rep. 2012, 28, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, B.; Sun, L.; Yan, Q.; Zhang, Y.; Zhang, Z.; Su, Y.; Wang, C. MicroRNA-130b targets PTEN to induce resistance to cisplatin in lung cancer cells by activating Wnt/β-catenin pathway. Cell Biochem. Funct. 2018, 36, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, C.; Yang, Q.; Ding, H.; Jia, J.; Guo, J.; Wang, J.; Wang, Z. Deregulation of let-7e in epithelial ovarian cancer promotes the development of resistance to cisplatin. Oncogenesis 2013, 2, e75. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, H.; Shen, H.; Li, H. MicroRNA-106a modulates cisplatin sensitivity by targeting PDCD4 in human ovarian cancer cells. Oncol. Lett. 2014, 7, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Cai, J.; Wang, Q.; Tang, H.; Cao, J.; Wu, L.; Wang, Z. Epigenetic silencing of miR-130b in ovarian cancer promotes the development of multidrug resistance by targeting colony-stimulating factor 1. Gynecol. Oncol. 2012, 124, 325–334. [Google Scholar] [CrossRef]
- Yang, H.; Kong, W.; He, L.; Zhao, J.J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: MiR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.Y.; Kang, H.; Kim, T.H.; Kim, G.; Heo, J.H.; Kwon, A.Y.; Kim, S.; Jung, S.G.; An, H.J. MicroRNA-136 inhibits cancer stem cell activity and enhances the anti-tumor effect of paclitaxel against chemoresistant ovarian cancer cells by targeting Notch3. Cancer Lett. 2017, 386, 168–178. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Y.; Xie, C.; Yin, X.; Liu, Y.; Cao, Y.; Fang, Y.; Lin, X.; Xu, Y.; Xu, W.; et al. MiR-145 sensitizes ovarian cancer cells to paclitaxel by targeting Sp1 and Cdk6. Int. J. Cancer 2014, 135, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Wang, R.; Chen, L.B. MiR-100 resensitizes docetaxel-resistant human lung adenocarcinoma cells (SPC-A1) to docetaxel by targeting Plk1. Cancer Lett. 2012, 317, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Wang, D.; Li, R.; Tang, Y.; Yuan, L.; Long, X.; Zhou, Q. MiR-197 induces Taxol resistance in human ovarian cancer cells by regulating NLK. Tumour Biol. 2015, 36, 6725–6732. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Walter, W.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; et al. Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 2021, 35, 1108–1120. [Google Scholar] [CrossRef]
- Mäkelä, E.; Pavic, K.; Varila, T.; Salmenniemi, U.; Löyttyniemi, E.; Nagelli, S.G.; Ammunét, T.; Kähäri, V.M.; Clark, R.E.; Elo, L.L.; et al. Discovery of a Novel CIP2A Variant (NOCIVA) with Clinical Relevance in Predicting TKI Resistance in Myeloid Leukemias. Clin. Cancer Res. 2021, 27, 2848–2860. [Google Scholar] [CrossRef] [PubMed]
- Veuger, M.J.; Heemskerk, M.H.; Honders, M.W.; Willemze, R.; Barge, R.M. Functional role of alternatively spliced deoxycytidine kinase in sensitivity to cytarabine of acute myeloid leukemic cells. Blood 2002, 99, 1373–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepis, K.; De Braekeleer, E.; Aspris, D.; Barbieri, I.; Vijayabaskar, M.S.; Liu, W.H.; Gozdecka, M.; Metzakopian, E.; Toop, H.D.; Dudek, M.; et al. SRPK1 maintains acute myeloid leukemia through effects on isoform usage of epigenetic regulators including BRD4. Nat. Commun. 2018, 9, 5378. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz-Rokah, O.; Modai, S.; Pasmanik-Chor, M.; Toren, A.; Shomron, N.; Raanani, P.; Shpilberg, O.; Granot, G. MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR-ABL protein. Cancer Lett. 2015, 356, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.; Tao, K.; Wang, X.; Xiao, Q.; Huang, Z.; Zhong, L.; Cao, W.; Wen, J.; Feng, W. Inhibition of BCR/ABL protein expression by miR-203 sensitizes for imatinib mesylate. PLoS ONE 2013, 8, e61858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Qi, H.W.; Dong, H.G.; Bai, P.; Sun, M.; Liu, H.Y. Targeting deubiquitinating enzyme USP26 by microRNA-203 regulates Snail1’s pro-metastatic functions in esophageal cancer. Cancer Cell Int. 2020, 20, 355. [Google Scholar] [CrossRef]
- Wang, L.S.; Li, L.; Li, L.; Chu, S.; Shiang, K.D.; Li, M.; Sun, H.Y.; Xu, J.; Xiao, F.J.; Sun, G.; et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 2015, 125, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Sims, E.K.; Lakhter, A.J.; Anderson-Baucum, E.; Kono, T.; Tong, X.; Evans-Molina, C. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia 2017, 60, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löwenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, X.Q.; Feng, D.D.; Zhang, X.J.; Wu, J.; Zheng, Y.S.; Chen, X.; Xu, L.; Chen, Y.Q. Upregulation of microRNA-125b contributes to leukemogenesis and increases drug resistance in pediatric acute promyelocytic leukemia. Mol. Cancer 2011, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, R.; Francies, F.Z.; Oyomno, M.; Dlamini, Z. Colorectal Cancer Genetics, Incidence and Risk Factors: In Search for Targeted Therapies. Cancer Manag. Res. 2020, 12, 9869–9882. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Yaffee, P.; Osipov, A.; Tan, C.; Tuli, R.; Hendifar, A. Review of systemic therapies for locally advanced and metastatic rectal cancer. J. Gastrointest. Oncol. 2015, 6, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Tsunekuni, K.; Konno, M.; Asai, A.; Koseki, J.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; Ishii, H. MicroRNA profiles involved in trifluridine resistance. Oncotarget 2017, 8, 53017–53027. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, M.A.H.; Amin, E.M.; Hoareau-Aveilla, C.; Domingo, E.; Symonds, K.E.; Ye, X.; Heesom, K.J.; Salmon, A.; D’Silva, O.; Betteridge, K.B.; et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol. Oncol. 2015, 9, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Pentheroudakis, G.; Mavroeidis, L.; Papadopoulou, K.; Koliou, G.A.; Bamia, C.; Chatzopoulos, K.; Samantas, E.; Mauri, D.; Efstratiou, I.; Pectasides, D.; et al. Angiogenic and Antiangiogenic VEGFA Splice Variants in Colorectal Cancer: Prospective Retrospective Cohort Study in Patients Treated With Irinotecan-Based Chemotherapy and Bevacizumab. Clin. Colorectal Cancer 2019, 18, e370–e384. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Valcárcel, J. Two isoforms of the T-cell intracellular antigen 1 (TIA-1) splicing factor display distinct splicing regulation activities. Control of TIA-1 isoform ratio by TIA-1-related protein. J. Biol. Chem. 2007, 282, 19410–19417. [Google Scholar] [CrossRef] [Green Version]
- Ni, B.; Hu, J.; Chen, D.; Li, L.; Chen, D.; Wang, J.; Wang, L. Alternative splicing of spleen tyrosine kinase differentially regulates colorectal cancer progression. Oncol. Lett. 2016, 12, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Geng, L.; Yi, H.; Huo, W.; Talmon, G.; Kim, Y.C.; Wang, S.M.; Wang, J. Transforming Growth Factor β Mediates Drug Resistance by Regulating the Expression of Pyruvate Dehydrogenase Kinase 4 in Colorectal Cancer. J. Biol. Chem. 2016, 291, 17405–17416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Yang, W.; Feng, W.; Cao, L.; Wang, X.; Niu, L.; Li, Y.; Zhou, W.; Zhang, Y.; Liu, J.; et al. Molecular mechanisms and clinical implications of miRNAs in drug resistance of colorectal cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920947342. [Google Scholar] [CrossRef]
- Nishida, N.; Yamashita, S.; Mimori, K.; Sudo, T.; Tanaka, F.; Shibata, K.; Yamamoto, H.; Ishii, H.; Doki, Y.; Mori, M. MicroRNA-10b is a prognostic indicator in colorectal cancer and confers resistance to the chemotherapeutic agent 5-fluorouracil in colorectal cancer cells. Ann. Surg. Oncol. 2012, 19, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Yang, F.; Wang, Y.; Wang, Y.; Xue, G.; Mei, Q.; Wang, F.; Sun, S. MicroRNA-23a antisense enhances 5-fluorouracil chemosensitivity through APAF-1/caspase-9 apoptotic pathway in colorectal cancer cells. J. Cell. Biochem. 2014, 115, 772–784. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Kaller, M.; Hermeking, H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 2013, 4, 1399–1415. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Kim, I.; Yoon, S.K.; Lee, E.K.; Kuh, H.J. Indirect modulation of sensitivity to 5-fluorouracil by microRNA-96 in human colorectal cancer cells. Arch. Pharmacal Res. 2015, 38, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gao, F.; Zhang, X.P. MiR-203 enhances chemosensitivity to 5-fluorouracil by targeting thymidylate synthase in colorectal cancer. Oncol. Rep. 2015, 33, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zheng, W.; Song, Y.; Du, X.; Tang, Y.; Nie, J.; Han, W. MiRNA-497 Enhances the Sensitivity of Colorectal Cancer Cells to Neoadjuvant Chemotherapeutic Drug. Curr. Protein Pept. Sci. 2015, 16, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talmon, G.; Wang, J. MicroRNA-587 antagonizes 5-FU-induced apoptosis and confers drug resistance by regulating PPP2R1B expression in colorectal cancer. Cell Death Dis. 2016, 7, e2525. [Google Scholar] [CrossRef]
- Chai, H.; Liu, M.; Tian, R.; Li, X.; Tang, H. MiR-20a targets BNIP2 and contributes chemotherapeutic resistance in colorectal adenocarcinoma SW480 and SW620 cell lines. Acta Biochim. Biophys. Sin. 2011, 43, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wan, G.; Spizzo, R.; Ivan, C.; Mathur, R.; Hu, X.; Ye, X.; Lu, J.; Fan, F.; Xia, L.; et al. MiR-203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating ATM kinase. Mol. Oncol. 2014, 8, 83–92. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Qiu, Y.; Yuan, Z.; Li, H.; Yuan, X.; Sun, J.; Xu, J.; Liang, X.; Yin, P. MiR-503-5p confers drug resistance by targeting PUMA in colorectal carcinoma. Oncotarget 2017, 8, 21719–21732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Liang, X.; Cui, D.; Wu, Y.; Shi, W.; Liu, J. MiR-1915 inhibits Bcl-2 to modulate multidrug resistance by increasing drug-sensitivity in human colorectal carcinoma cells. Mol. Carcinog. 2013, 52, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Bitarte, N.; Bandres, E.; Boni, V.; Zarate, R.; Rodriguez, J.; Gonzalez-Huarriz, M.; Lopez, I.; Sola, J.J.; Alonso, M.M.; Fortes, P.; et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells 2011, 29, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Supadmanaba, I.G.P.; Mantini, G.; Randazzo, O.; Capula, M.; Muller, I.B.; Cascioferro, S.; Diana, P.; Peters, G.J.; Giovannetti, E. Interrelationship between miRNA and splicing factors in pancreatic ductal adenocarcinoma. Epigenetics 2021, 1–24. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazan, H.; Ray, D.; Chan, E.T.; Hughes, T.R.; Morris, Q. RNAcontext: A new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol. 2010, 6, e1000832. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passetti, F.; Ferreira, C.G.; Costa, F.F. The impact of microRNAs and alternative splicing in pharmacogenomics. Pharm. J. 2009, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lavorgna, G.; Dahary, D.; Lehner, B.; Sorek, R.; Sanderson, C.M.; Casari, G. In search of antisense. Trends Biochem. Sci. 2004, 29, 88–94. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell. Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, P.; Yang, Z.; Deng, S.; Ni, L.; Zhang, Y.; Jin, L.; Pan, Y. MiR-193a-5p promotes pancreatic cancer cell metastasis through SRSF6-mediated alternative splicing of OGDHL and ECM1. Am. J. Cancer Res. 2020, 10, 38–59. [Google Scholar]
- Rodriguez-Aguayo, C.; Monroig, P.D.C.; Redis, R.S.; Bayraktar, E.; Almeida, M.I.; Ivan, C.; Fuentes-Mattei, E.; Rashed, M.H.; Chavez-Reyes, A.; Ozpolat, B.; et al. Regulation of hnRNPA1 by microRNAs controls the miR-18a-K-RAS axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017, 3, 17029. [Google Scholar] [CrossRef] [Green Version]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem. 2007, 282, 2130–2134. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Aslan, D.; Garde, C.; Nygaard, M.K.; Helbo, A.S.; Dimopoulos, K.; Hansen, J.W.; Severinsen, M.T.; Treppendahl, M.B.; Sjø, L.D.; Grønbæk, K.; et al. Tumor suppressor microRNAs are downregulated in myelodysplastic syndrome with spliceosome mutations. Oncotarget 2016, 7, 9951–9963. [Google Scholar] [CrossRef] [Green Version]
- Braeutigam, C.; Rago, L.; Rolke, A.; Waldmeier, L.; Christofori, G.; Winter, J. The RNA-binding protein Rbfox2: An essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 2014, 33, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Ramya, D.; Siddikuzzaman; Grace, V.M. Effect of all-trans retinoic acid (ATRA) on syndecan-1 expression and its chemoprotective effect in benzo(α)pyrene-induced lung cancer mice model. Immunopharmacol. Immunotoxicol. 2012, 34, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, Ó.; Hernández-Pedro, N.; Fernández-González-Aragón, M.C.; Saavedra-Pérez, D.; Campos-Parra, A.D.; Ríos-Trejo, M.; Cerón-Lizárraga, T.; Martínez-Barrera, L.; Pineda, B.; Ordóñez, G.; et al. Retinoic acid reduces chemotherapy-induced neuropathy in an animal model and patients with lung cancer. Neurology 2011, 77, 987–995. [Google Scholar] [CrossRef]
- Meseguer, S.; Mudduluru, G.; Escamilla, J.M.; Allgayer, H.; Barettino, D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J. Biol. Chem. 2011, 286, 4150–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Zhao, J.; Xie, W.; Sun, Q.; Wang, H.; Qiao, B. LncRNA UCA1 promotes proliferation and cisplatin resistance of oral squamous cell carcinoma by sunppressing miR-184 expression. Cancer Med. 2017, 6, 2897–2908. [Google Scholar] [CrossRef] [PubMed]
- Mukohyama, J.; Isobe, T.; Hu, Q.; Hayashi, T.; Watanabe, T.; Maeda, M.; Yanagi, H.; Qian, X.; Yamashita, K.; Minami, H.; et al. MiR-221 Targets QKI to Enhance the Tumorigenic Capacity of Human Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 5151–5158. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Hamis, S.; Nithiarasu, P.; Powathil, G.G. What does not kill a tumour may make it stronger: In silico insights into chemotherapeutic drug resistance. J. Theor. Biol. 2018, 454, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldie, J.H.; Coldman, A.J. The genetic origin of drug resistance in neoplasms: Implications for systemic therapy. Cancer Res. 1984, 44, 3643–3653. [Google Scholar] [PubMed]
- Kuczynski, E.A.; Sargent, D.J.; Grothey, A.; Kerbel, R.S. Drug rechallenge and treatment beyond progression—Implications for drug resistance. Nat. Rev. Clin. Oncol. 2013, 10, 571–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, D.A. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 2013, 137414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D.; Yu, D.; Lee, D.Y.; Shin, H.S.; Jo, J.H.; Lee, Y.C. Upregulated microRNA-193a-3p is responsible for cisplatin resistance in CD44(+) gastric cancer cells. Cancer Sci. 2019, 110, 662–673. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teplyuk, N.M.; Uhlmann, E.J.; Gabriely, G.; Volfovsky, N.; Wang, Y.; Teng, J.; Karmali, P.; Marcusson, E.; Peter, M.; Mohan, A.; et al. Therapeutic potential of targeting microRNA-10b in established intracranial glioblastoma: First steps toward the clinic. EMBO Mol. Med. 2016, 8, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Zhang, L. Therapeutic nanoparticles to combat cancer drug resistance. Curr. Drug Metab. 2009, 10, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Yuan, R.; Hou, Y.; Sun, W.; Yu, J.; Liu, X.; Niu, Y.; Lu, J.J.; Chen, X. Natural products to prevent drug resistance in cancer chemotherapy: A review. Ann. N. Y. Acad. Sci. 2017, 1401, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Park, N.H.; Cheng, W.; Lai, F.; Yang, C.; de Sessions, P.F.; Periaswamy, B.; Chu, C.W.; Bianco, S.; Liu, S.; Venkataraman, S.; et al. Addressing Drug Resistance in Cancer with Macromolecular Chemotherapeutic Agents. J. Am. Chem. Soc. 2018, 140, 4244–4252. [Google Scholar] [CrossRef] [PubMed]



| Breast | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| HER2 target of splicing factors resulting in increased expression of d16HER2 isoform | Trastuzumab | d16HER2 isoform stops trastuzumab blocking HER2 receptor, increasing proliferation | [52] |
| ECT2 spliced to form ECT2-Ex5+ | Doxorubicin | Levels of splicing factors ZRANB2 and SYF2 is increased in resistant breast cancer. ECT2 is a splicing target of these splicing factors. Increasing levels of the ECT2-Ex5+ variant. This isoform increases resistance to the drug | [50] |
| SRPK1 and Tip60 are alternatively spliced | Cisplatin | Tip60 isoforms change in acetylation of SRPK1, changes in splicing activities. | [49] |
| miRNA | |||
| miRNA | Drug | Effect | Ref |
| miR-22 | Paclitaxel | Low levels pf miR-22 in breast cancer. High levels lead to increased drug sensitivity | [58] |
| miR-155 | PTX | TGF-beta induces miR-155 expression, miR-155 reduced RhoA protein and disrupted tight junction promoting proliferation. | [68] |
| miR-365 | 5-FU | Low levels pf miR-365 in breast cancer. High levels lead to increased drug sensitivity | [56] |
| miR-125b | Expression pattern of miR-125 indicates its involvement with drug resistance through its effect on E2F3 | [69] | |
| mi-R206 | Tamoxifen | Targets WBP2. Decreased expression of this oncogene leads to increasing drug sensitivity | [53] |
| miR-26a | Low levels pf miR-26a in breast cancer. High levels lead to increased drug sensitivity | [55] | |
| Negative feedback loop with E2F7 promotes sensitivity to treatment | [60] | ||
| miR-15a/16 | E2F7 inhibits miR-15a/16 expression, increasing drug resistance, due to inhibition of Cyclin 1 | [62,64] | |
| miR-210 | Trastuzumab Tamoxifen | Elevated levels of miR-210 is associated with drug resistance | [64] |
| Let-7b | Let-7b downregulates ER-α36 signaling leading to increased drug sensitivity | [70] | |
| miR-200c | Trastuzumab | Targets ZNF217, ZEB1 resulting in suppressed TGFβ signaling, increasing sensitivity to the drug | [71] |
| miR-221 | Targets PTEN for degradation, promoting drug resistance | [72] | |
| miR-155 | VP-16 | TGF-beta induces miR-155 expression, miR-155 reduced FOXO3A and disrupted tight junction promoting proliferation | [68] |
| miR-30c | Doxorubicin | miR-30c targets YWHAZ for degradation, increasing resistance to the drug | [73] |
| miR-155 | TGF-beta induces miR-155 expression, miR-155 reduced Foxo1a and disrupted tight junction promoting proliferation | [68] | |
| miR-34a | miR-34a targets Notch1 leading to decreased drug resistance | [74] | |
| miR130b | miR-130b targets PTEN leading to increased resistance to the drug | [75] | |
| miR-137 | miR-137 targets YB-1 leading to increased drug sensitivity. | [76] | |
| miR-149 | miR-149 targets NDST1 leading to increased sensitivity to the drug | [77] | |
| miR-195 | miR-195 targets RAF-1 leading to increased sensitivity to the drug | [78] | |
| miR-200c | Mir-200c targets and decreases expression of TrKB and BMI1, leading to increased drug sensitivity | [79] | |
| miR-298 | miR-298 targets and decreases levels of MDR1, increasing drug resistance | [80] | |
| miR-17 and miR-20b | Taxol | miR-17 and miR-20b targets and downregulates NCOA3, increasing sensitivity to the drug | [81] |
| miR-181a | Adriamycin | miR-181a targeted BCL-2 leading to enhanced apoptosis and increased sensitivity to this pro-apoptotic drug | [82] |
| miR-34a | Docetaxel | miR-34a targets and decreases expression of BCL-2 and CCND1, increasing drug resistance | [83] |
| miR-96 | Cisplatin | miR-96 targets and downregulates the expression of RAD51 and REV1, resulting in increased drug sensitivity | [84] |
| miR-218 | Taxol | miR-218 targets BIRC5 and decreases SURVIVIN-1 expression, leading to increased drug resistance | [85] |
| miR-20a | Multidrug | miR-20a targets MAPK1 for degradation, inhibiting the MAPK1 signaling pathway downregulating the expression of P-gp and c-Myc, sensitizing cells to the drugs | [8] |
| Cervical Cancer | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| CRKL regulates the splicing of genes related to cancer | Multidrug | Many mRNAs whose splicing is regulated by CRKL are related to malignant transformation, metastases, and chemoresistance | [97] |
| miRNA | |||
| miRNA | Drug | Effects | Ref |
| miR-125a | Paclitaxel | Suppressed miR-125a shows increased resistance to the drug alone but increased sensitivity to paclitaxel and cisplatin combination. miR-125a targets stat3 | [89] |
| miR-30a | Cisplatin | miR-27a suppresses Beclin1-induced autophagy and increases sensitivity to the drug | [98] |
| miR-214 | miR-214 targets and downregulates BCL2L2 expression and increases sensitivity to the drug | [99] | |
| Prostate Cancer | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| Alternative splicing of the androgen receptor (AR) | Enzalutamide | AR-V7 is an AR splice variant that lacks a ligand binding domain, resulting in increased resistance to the drug | [102] |
| miRNA | |||
| miRNA | Drug | Effect | Ref |
| miR-143 | Docetaxel | Downregulation of KRAS inhibits proliferation and migration. Increasing sensitivity to the drug by targeting EGFR/RAS/MAPK signaling | [107] |
| miR-200b | miR-200b enhances sensitivity to the drug by binding to and degrading BMI1 | [111] | |
| miR-148a | Paclitaxel | Increased levels of miR-148 led to increased sensitivity to the drug by targeting and decreasing expression of MSK1 | [108] |
| miR-34a | miRNA-34a expression is decreased in prostate cancer. Decreased expression results in increased drug sensitivity due to upregulated SIRT1 and BCL-2 levels | [106] | |
| Ovarian Cancer | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| ECM1 is spliced to give rise to ECM1a | Cisplatin | ECM1 isoform induces tumorigenesis by activating the AKT/FAK/Rho/cytoskeleton signaling pathway and promotes resistance to the drug by increasing CD326 | [113] |
| miRNA | |||
| miRNA | Drug | Effects | Ref |
| Let-7b | Tamoxifen | Targets and downregulates ER-α36, increasing sensitivity to the drug | [70] |
| Let-7e | Cisplatin | Let-7e increased sensitivity to the drug by reducing the expression of proteins related to the increased resistance to the drug, namely BRCA1, EZH2, CCND1 | [132] |
| miR-199b-5p | miR-199b-5p downregulated JAG1 leading to increased sensitivity to the drug | [118] | |
| miR-93 | Regulates PTEN/AKT signaling resulting in increased drug sensitivity | [121] | |
| miR-106a | Increased resistance to the drug by targeting and decreasing PDCD4 expression | [133] | |
| miR-130b | Increased sensitivity to the drug by targeting and downregulating CSF-1 expression | [134] | |
| miR-214 | Increases resistance to the drug by targeting PTEN | [135] | |
| miR-370 | miR-370 decreases the expression of ENG leading to increased drug sensitivity | [117] | |
| miR-489 | miR-489 increased sensitivity to the drug by targeting Akt3 | [119] | |
| miR-31 | Paclitaxel | Increases MET expression leading to increased drug resistance | [128] |
| miR-136 | miR-136 targets Notch3 leading to increased sensitivity to the drug | [136] | |
| miR-130b | Increased sensitivity to the drug by targeting and decreasing CSF-1 expression | [134] | |
| miR-200c | miR-200c targets TuBB3, TrKB resulting in increased sensitivity to the drug | [126] | |
| miR-145 | miR-145 targets and downregulates expression of SP1 and CDK6, increasing the sensitivity to the drug | [137] | |
| miR-591 | miR-591 increases resistance to the drug by targeting Bcl-10, Caspase7 and Zeb1. | [129] | |
| miR-100 | Doxorubicin | Re-sensitizes resistant cells to the drug by targeting PLK1 | [138] |
| miR-197 | Taxol | miR-197 regulates NLK expression to increase resistance to the drug | [139] |
| let-7 | This miRNA family targets IMP-1, destabilizing MDR1 and sensitizing cells to the drug | [115] | |
| miRNA-200c | Taxane | miRNA-200c inhibits class III β-tubulin by targeting ZEB1 and ZEB2, increasing drug sensitivity | [124] |
| miRNA-200c | Targets TUBB3 resulting in increased sensitivity to the drug | [127] | |
| Leukemia | |||
|---|---|---|---|
| Alternative Splicing | |||
| Splicing Event | Drug | Effect | Ref |
| Cip2a alternatively spliced to form nociva | Dasatinib and Nilotinib | High levels of NOCIVA are also associated with dasatinib and nilotinib resistance | [141] |
| DCK splicing | Cytarabine | Some isoforms of DCK lack the ability to process cytarabine into its active metabolite, contributing to resistant AML | [142] |
| BRD4 | SPHINX31 | Short to long BRD4 isoform switch leads to reduced AML cell survival/proliferation and increased drug sensitivity | [143] |
| miRNA | |||
| miRNA | Drug | Effects | Ref |
| miR-30e | Imatinib | Downregulated in CML. Increased miR-30e led to increased drug sensitivity via BCR-ABL expression | [144] |
| miR-203 | Increased sensitivity to the drug by downregulating BCR-ABL expression | [145] | |
| miR-486 | Promotes resistance to the drug by targeting PTEN and FOXO1 | [147] | |
| miR-125b | Doxorubicin | Represses BAK1 protein expression leading to increased drug resistance | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marima, R.; Francies, F.Z.; Hull, R.; Molefi, T.; Oyomno, M.; Khanyile, R.; Mbatha, S.; Mabongo, M.; Owen Bates, D.; Dlamini, Z. MicroRNA and Alternative mRNA Splicing Events in Cancer Drug Response/Resistance: Potent Therapeutic Targets. Biomedicines 2021, 9, 1818. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121818
Marima R, Francies FZ, Hull R, Molefi T, Oyomno M, Khanyile R, Mbatha S, Mabongo M, Owen Bates D, Dlamini Z. MicroRNA and Alternative mRNA Splicing Events in Cancer Drug Response/Resistance: Potent Therapeutic Targets. Biomedicines. 2021; 9(12):1818. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121818
Chicago/Turabian StyleMarima, Rahaba, Flavia Zita Francies, Rodney Hull, Thulo Molefi, Meryl Oyomno, Richard Khanyile, Sikhumbuzo Mbatha, Mzubanzi Mabongo, David Owen Bates, and Zodwa Dlamini. 2021. "MicroRNA and Alternative mRNA Splicing Events in Cancer Drug Response/Resistance: Potent Therapeutic Targets" Biomedicines 9, no. 12: 1818. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121818