
Valine-279 Deletion–Mutation on Arginine Vasopressin Receptor 2 Causes Obstruction in G-Protein Binding Site: A Clinical Nephrogenic Diabetes Insipidus Case and Its Sub-Molecular Pathogenic Analysis

 ,
,  ,
,  and
and
Abstract
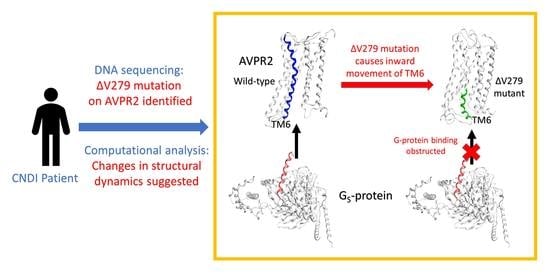
:
1. Introduction
2. Materials and Methods
2.1. Patient Identification and Sample Collection
2.2. Genomic DNA Sequencing
2.3. Plasmid Construction and Immunofluorescence Confocal Microscopy
2.4. Homology Modeling of AVPR2 Structure
2.5. Molecular Dynamics (MD) Simulations
3. Results
3.1. Patient Characteristics
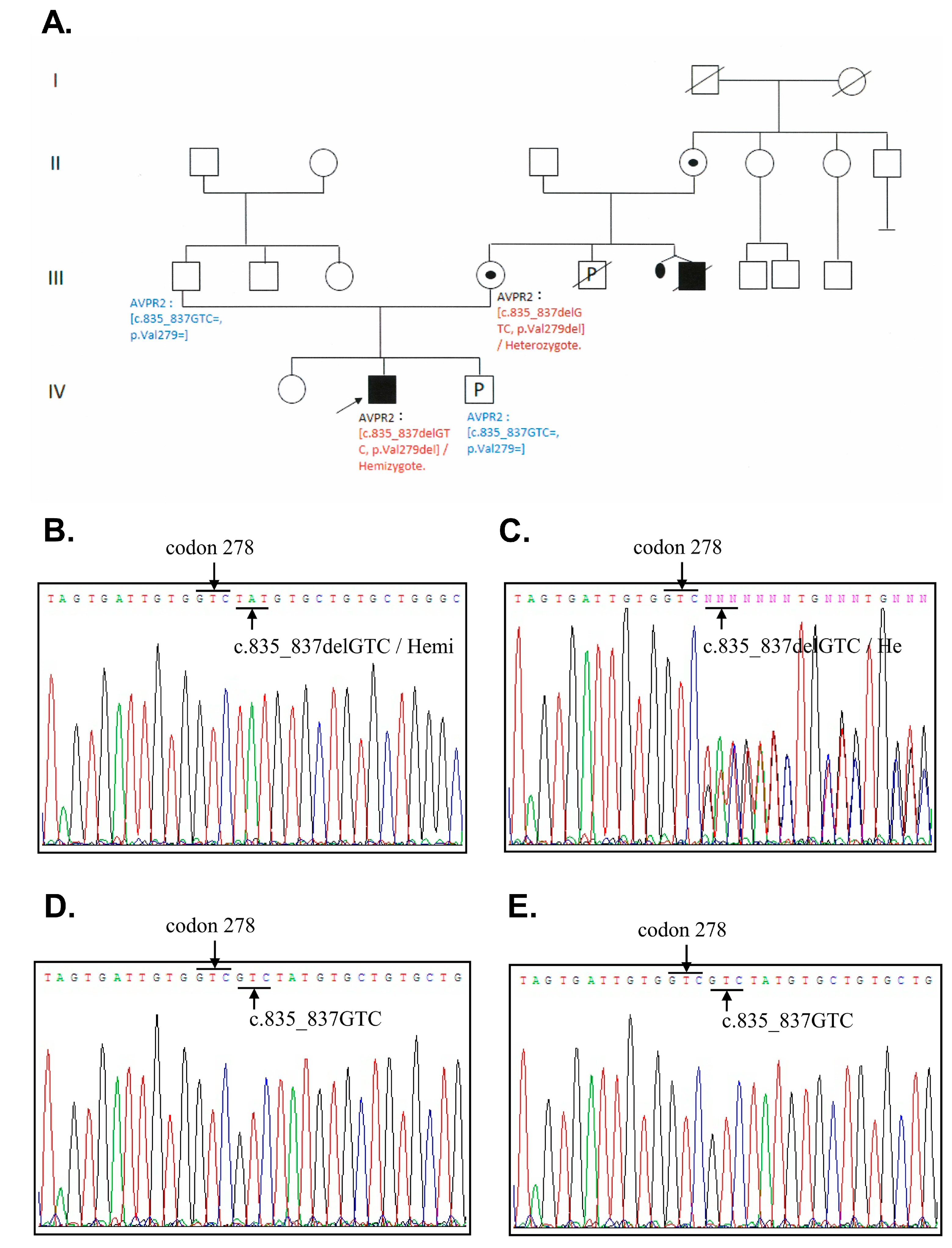
3.2. DNA Sequence Analysis
3.3. Both AVPR2 and AVPR2-∆V279 Can Localize to the Cell Plasma Membrane
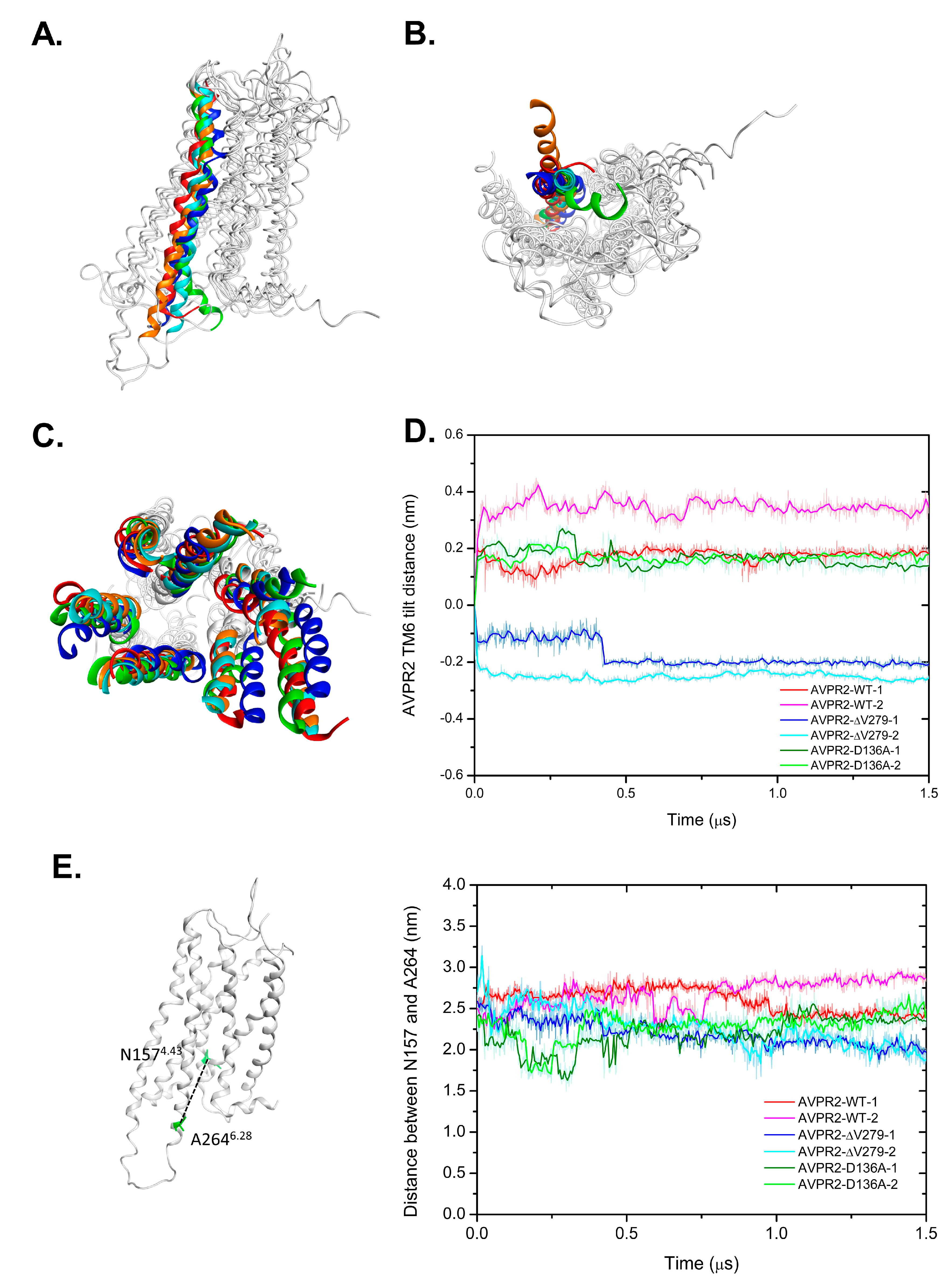
3.4. Conformational Changes in the Distinct Types of AVPR2 during MD Simulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arthus, M.F.; Lonergan, M.; Crumley, M.J.; Naumova, A.K.; Morin, D.; De Marco, L.A.; Kaplan, B.S.; Robertson, G.L.; Sasaki, S.; Morgan, K.; et al. Report of 33 Novel AVPR2 Mutations and Analysis of 117 Families with X-Linked Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2000, 11, 1044–1054. [Google Scholar]
- Celebi, T.A.; Karaduman, T.; Ozcan, T.M.; Sahin, D.; Caltik, Y.A.; Buyukkaragoz, B.; Bulus, A.D.; Mergen, H. A Novel Mutation in the AVPR2 Gene Causing Congenital Nephrogenic Diabetes Insipidus. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 350–356. [Google Scholar]
- Moeller, H.B.; Rittig, S.; Fenton, R.A. Nephrogenic diabetes insipidus: Essential insights into the molecular background and potential therapies for treatment. Endocr. Rev. 2013, 34, 278–301. [Google Scholar] [CrossRef] [Green Version]
- Bichet, D.G. GENETICS IN ENDOCRINOLOGY Pathophysiology, diagnosis and treatment of familial nephrogenic diabetes insipidus. Eur. J. Endocrinol. 2020, 183, R29–R40. [Google Scholar] [CrossRef] [PubMed]
- Guarino, S.; Diplomatico, M.; Marotta, R.; Pecoraro, A.; Furlan, D.; Cerrone, L.; Miraglia Del Giudice, E.; Polito, C.; La Manna, A.; Marzuillo, P. Nephrogenic Diabetes Insipidus in Childhood: Assessment of Volume Status and Appropriate Fluid Replenishment. Pediatr. Emerg. Care 2018. [Google Scholar] [CrossRef] [PubMed]
- Sparapani, S.; Millet-Boureima, C.; Oliver, J.; Mu, K.; Hadavi, P.; Kalostian, T.; Ali, N.; Avelar, C.M.; Bardies, M.; Barrow, B.; et al. The Biology of Vasopressin. Biomedicines 2021, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Birnbaumer, M.; Seibold, A.; Gilbert, S.; Ishido, M.; Barberis, C.; Antaramian, A.; Brabet, P.; Rosenthal, W. Molecular cloning of the receptor for human antidiuretic hormone. Nature 1992, 357, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, C.; Uy, N.S. Nephrogenic Diabetes Insipidus. Pediatr. Clin. N. Am. 2019, 66, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Bockenhauer, D. Genetic forms of nephrogenic diabetes insipidus (NDI): Vasopressin receptor defect (X-linked) and aquaporin defect (autosomal recessive and dominant). Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Manaka, K.; Sato, J.; Iiri, T. V2 vasopressin receptor mutations. Vitam. Horm. 2020, 113, 79–99. [Google Scholar] [CrossRef]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef] [Green Version]
- Renault, P.; Louet, M.; Marie, J.; Labesse, G.; Floquet, N. Molecular Dynamics Simulations of the Allosteric Modulation of the Adenosine A2A Receptor by a Mini-G Protein. Sci. Rep. 2019, 9, 5495. [Google Scholar] [CrossRef]
- Bera, A.K.; Akabas, M.H. Spontaneous thermal motion of the GABA(A) receptor M2 channel-lining segments. J. Biol. Chem. 2005, 280, 35506–35512. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; McCammon, J.A. G-protein coupled receptors: Advances in simulation and drug discovery. Curr. Opin. Struct Biol. 2016, 41, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.P.; Karpiak, J.; Kroeze, W.K.; Zhu, H.; Chen, X.; Moy, S.S.; Saddoris, K.A.; Nikolova, V.D.; Farrell, M.S.; Wang, S.; et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Wang, J.; Palczewski, K.; Filipek, S.; Vogel, H.; Liu, Z.J.; Yuan, S. Exploring a new ligand binding site of G protein-coupled receptors. Chem. Sci. 2018, 9, 6480–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Palczewski, K.; Peng, Q.; Kolinski, M.; Vogel, H.; Filipek, S. The mechanism of ligand-induced activation or inhibition of mu- and kappa-opioid receptors. Angew. Chem. Int. Ed. Engl. 2015, 54, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Peces, R.; Mena, R.; Peces, C.; Santos-Simarro, F.; Fernández, L.; Afonso, S.; Lapunzina, P.; Selgas, R.; Nevado, J. Severe congenital nephrogenic diabetes insipidus in a compound heterozygote with a new large deletion of the AQP2 gene. A case report. Mol. Genet Genom. Med. 2019, 7, e00568. [Google Scholar] [CrossRef]
- Liu, C.D.; Lee, H.L.; Peng, C.W. B cell specific transcription activator PAX5 recruits p300 to support EBNA1-driven transcription. J. Virol. 2020, 94, e02028-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, J.; Cao, S.; Sun, D.; Liu, H.; Lu, Q.; Liu, Z.; Du, Y.; Zhang, C. Cryo-EM structure of the AVP-vasopressin receptor 2-Gs signaling complex. Cell Res. 2021. [Google Scholar] [CrossRef]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.E.; Zhang, Y.; Hu, H.; Suomivuori, C.M.; Kadji, F.M.N.; Aoki, J.; Krishna Kumar, K.; Fonseca, R.; Hilger, D.; Huang, W.; et al. Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 2019, 572, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.W.; Chang, F.T.; Chung, Y.; Chen, W.Y.; Fischer, W.B.; Hsu, H.J. In silico analysis reveals sequential interactions and protein conformational changes during the binding of chemokine CXCL-8 to its receptor CXCR1. PLoS ONE 2014, 9, e94178. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.H.; Li, Y.T.; Hsu, H.J. Activation-Induced Conformational Changes of Dopamine D3 Receptor Promote the Formation of the Internal Water Channel. Sci. Rep. 2017, 7, 12792. [Google Scholar] [CrossRef]
- Chang, C.C.; Liou, J.W.; Primus Dass, T.K.; Li, Y.T.; Jiang, S.J.; Pan, S.F.; Yeh, Y.C.; Hsu, H.J. Internal water channel formation in CXCR4 is crucial for Gi-protein coupling upon activation by CXCL12. Comm. Chem. 2020, 3, s42004–s42020. [Google Scholar] [CrossRef]
- Morin, D.; Cotte, N.; Balestre, M.N.; Mouillac, B.; Manning, M.; Breton, C.; Barberis, C. The D136A mutation of the V2 vasopressin receptor induces a constitutive activity which permits discrimination between antagonists with partial agonist and inverse agonist activities. FEBS Lett. 1998, 441, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Yin, J.; Chen, K.M.; Clark, M.J.; Hijazi, M.; Kumari, P.; Bai, X.C.; Sunahara, R.K.; Barth, P.; Rosenbaum, D.M. Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 2020, 584, 125–129. [Google Scholar] [CrossRef]
- Sharma, S.; Ashton, E.; Iancu, D.; Arthus, M.F.; Hayes, W.; Van’t Hoff, W.; Kleta, R.; Bichet, D.G.; Bockenhauer, D. Long-term outcome in inherited nephrogenic diabetes insipidus. Clin. Kidney J. 2019, 12, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Fenske, W.; Refardt, J.; Chifu, I.; Schnyder, I.; Winzeler, B.; Drummond, J.; Ribeiro-Oliveira, A., Jr.; Drescher, T.; Bilz, S.; Vogt, D.R.; et al. A Copeptin-Based Approach in the Diagnosis of Diabetes Insipidus. N. Engl. J. Med. 2018, 379, 428–439. [Google Scholar] [CrossRef]
- Lejarraga, H.; Caletti, M.G.; Caino, S.; Jimenez, A. Long-term growth of children with nephrogenic diabetes insipidus. Pediatr. Nephrol. 2008, 23, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, E.; Milord, E.; Gragnoli, C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: Review and missense mutation significance. J. Cell. Physiol. 2008, 217, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Kvistgaard, H.; Kamperis, K.; Færch, M.; Hagstrøm, S.; Gregersen, N.; Rittig, S.; Christensen, J.H. Novel and recurrent variants in AVPR2 in 19 families with X-linked congenital nephrogenic diabetes insipidus. Eur. J. Pediatr. 2018, 177, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Matsubara, H.; Aritaki, S.; Kimura, T.; Abe, S.; Inada, M. Two Novel Mutations in the Vasopressin V2 Receptor Gene in Unrelated Japanese Kindreds With Nephrogenic Diabetes Insipidus. Biochem. Biophys. Res. Commun. 1993, 15, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Faa, V.; Ventruto, M.L.; Loche, S.; Bozzola, M.; Podda, R.; Cao, A.; Rosatelli, M.C. Mutations in the vasopressin V2-receptor gene in three families of Italian descent with nephrogenic diabetes insipidus. Hum. Mol. Genet 1994, 3, 1685–1686. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Takahashi, T.; Suzuki, Y.; Suzuki, T.; Komatsu, K.; Hirono, H.; Shoji, Y.; Yokoyama, T.; Kito, H.; Takada, G. Mutational analyses of AVPR2 gene in three Japanese families with X-linked nephrogenic diabetes insipidus: Two recurrent mutations, R137H and deltaV278, caused by the hypermutability at CpG dinucleotides. Hum. Mutat. 1998, 11 (Suppl. 1), S278–S283. [Google Scholar] [CrossRef] [PubMed]
- Wildin, R.S.; Cogdell, D.E.; Valadez, V. AVPR2 variants and V2 vasopressin receptor function in nephrogenic diabetes insipidus. Kidney Int. 1998, 54, 1909–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robben, J.H.; Knoers, N.V.; Deen, P.M. Characterization of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus in a polarized cell model. Am. J. Physiol. Renal. Physiol. 2005, 289, F265–F272. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Metzenberg, A.; Das, S.; Jing, B.; Gitschier, J. Mutations in the V2 vasopressin receptor gene are associated with X-linked nephrogenic diabetes insipidus. Nat. Genet 1992, 2, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Lolait, S.J.; O’Carroll, A.M.; McBride, O.W.; Konig, M.; Morel, A.; Brownstein, M.J. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 1992, 357, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Nehme, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Wescott, M.P.; Kufareva, I.; Paes, C.; Goodman, J.R.; Thaker, Y.; Puffer, B.A.; Berdougo, E.; Rucker, J.B.; Handel, T.M.; Doranz, B.J. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc. Natl. Acad. Sci. USA 2016, 113, 9928–9933. [Google Scholar] [CrossRef] [Green Version]
- Deupi, X.; Standfuss, J. Structural insights into agonist-induced activation of G-protein- coupled receptors. Curr. Opin. Struct Biol. 2011, 21, 514–551. [Google Scholar] [CrossRef]
- Milano, S.; Carmosino, M.; Gerbino, A.; Svelto, M.; Procino, G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update. Int. J. Mol. Sci. 2017, 18, 2385. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Duration (h) | Body Weight (g) | Body Weight Loss (%) | Serum Sodium (mmol/L) | Serum Osmolarity (mOsm/kg) | Urine Specific Gravity | Urine Sodium (mmol/L) | Urine Osmolarity (mOsm/kg) |
|---|---|---|---|---|---|---|---|
| 0 | 6424 | 0 | 156 | 327 | 1.010 | 14 | 152 |
| 1 | 6321 | 1.6 | 156 | 322 | 1.007 | ND | 193 |
| 2 * | 6224 | 3.1 | 160 | 328 | 1.005 | 8 | 128 |
| 3 | 6294 | 2.0 | 155 | ND | 1.006 | 18 | 168 |
| 4 | 6156 | 4.2 | 160 | 330 | 1.008 | 21 | 210 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-C.; Hsiao, Y.-C.; Chang, C.-C.; Pan, S.-F.; Peng, C.-W.; Li, Y.-T.; Liu, C.-D.; Liou, J.-W.; Hsu, H.-J. Valine-279 Deletion–Mutation on Arginine Vasopressin Receptor 2 Causes Obstruction in G-Protein Binding Site: A Clinical Nephrogenic Diabetes Insipidus Case and Its Sub-Molecular Pathogenic Analysis. Biomedicines 2021, 9, 301. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030301
Chen M-C, Hsiao Y-C, Chang C-C, Pan S-F, Peng C-W, Li Y-T, Liu C-D, Liou J-W, Hsu H-J. Valine-279 Deletion–Mutation on Arginine Vasopressin Receptor 2 Causes Obstruction in G-Protein Binding Site: A Clinical Nephrogenic Diabetes Insipidus Case and Its Sub-Molecular Pathogenic Analysis. Biomedicines. 2021; 9(3):301. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030301
Chicago/Turabian StyleChen, Ming-Chun, Yu-Chao Hsiao, Chun-Chun Chang, Sheng-Feng Pan, Chih-Wen Peng, Ya-Tzu Li, Cheng-Der Liu, Je-Wen Liou, and Hao-Jen Hsu. 2021. "Valine-279 Deletion–Mutation on Arginine Vasopressin Receptor 2 Causes Obstruction in G-Protein Binding Site: A Clinical Nephrogenic Diabetes Insipidus Case and Its Sub-Molecular Pathogenic Analysis" Biomedicines 9, no. 3: 301. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030301