
Insights into the Pathogenesis of HS and Therapeutical Approaches

 ,
,
Abstract
:1. Introduction
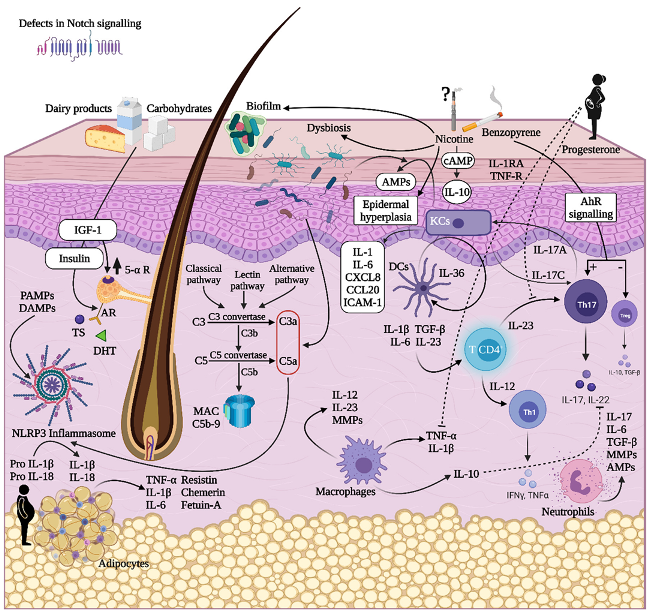
2. Insights into the Pathogenesis of HS
2.1. How Much Does Genetics Play a Role in HS?
2.2. Lifestyle
2.2.1. Obesity
2.2.2. Tobacco Smoking
2.3. The Role of Cutaneous Microbiome in HS
2.4. Sex Hormones
2.5. The Histopathological Findings
2.6. Innate Immunity
2.7. HS Cytokine Milieu
2.7.1. IL-1 Cytokine Family
2.7.2. TNF-α and IFNs
2.7.3. IL-17 Cytokine Family
2.7.4. IL-12 Cytokine Family
2.7.5. IL-10 Cytokine Family
2.7.6. IL-6
2.8. An Overview of the Pathogenetic Mechanisms in HS
3. (New Medical) Therapeutical Approaches in HS
3.1. TNF-α Inhibitors
3.1.1. Adalimumab
3.1.2. Infliximab
3.1.3. Etanercept
3.1.4. Golimumab
3.1.5. Certolizumab Pegol
3.2. IL-1 Inhibitors
3.2.1. Anakinra
3.2.2. Canakinumab
3.2.3. Bermekimab
3.3. IL-17 Inhibitors
3.3.1. Secukinumab
3.3.2. Ixekizumab
3.3.3. Brodalumab
3.3.4. Bimekizumab
3.3.5. CJM112
3.4. IL-12/-23 Inhibitors
Ustekinumab
3.5. IL-23 Inhibitors
3.5.1. Guselkumab
3.5.2. Risankizumab
3.5.3. Tildrakizumab
3.6. Complement 5a Inhibitors
FX-1
3.7. Phosphodiesterase 4 (PDE4) Inhibitors
Apremilast
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Frew, J.W. Hidradenitis suppurativa is an autoinflammatory keratinization disease: A review of the clinical, histologic, and molecular evidence. JAAD Int. 2020, 1, 62–72. [Google Scholar] [CrossRef]
- Sivanand, A.; Alhusayen, R.; Piguet, V.; Alavi, A. “Hidradenitis Suppurativa” Is a Historical Term That Does Not Reflect the Current Understanding of Disease Pathogenesis. J. Cutan. Med. Surg. 2020, 24, 644–645. [Google Scholar] [CrossRef]
- Savage, K.; Brant, E.G.; Flood, K.; Salian, P.; Porter, M.; Kimball, A. Publication trends in hidradenitis suppurativa from 2008 to 2018. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
- Canoui-Poitrine, F.; Le Thuaut, A.; Revuz, J.E.; Viallette, C.; Gabison, G.; Poli, F.; Pouget, F.; Wolkenstein, P.; Bastuji-Garin, S. Identification of Three Hidradenitis Suppurativa Phenotypes: Latent Class Analysis of a Cross-Sectional Study. J. Investig. Dermatol. 2013, 133, 1506–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaysi, G.A.; Cosman, B.; Crikelair, G.F. Hidradenitis Suppurativa. JAMA 1968, 203, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, J.S.; Fitzsimmons, E.M.; Gilbert, G. Familial hidradenitis suppurativa: Evidence in favour of single gene transmission. J. Med. Genet. 1984, 21, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, J.; Guilbert, P.; Fitzsimmons, E. Evidence of genetic factors in hidradenitis suppurativa. Br. J. Dermatol. 1985, 113, 1–8. [Google Scholar] [CrossRef]
- Von Der Werth, J.; Williams, H.; Raeburn, J. The clinical genetics of hidradenitis suppurativa revisited. Br. J. Dermatol. 2000, 142, 947–953. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef]
- Ingram, J.R. The Genetics of Hidradenitis Suppurativa. Dermatol. Clin. 2015, 34, 23–28. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, Y.; Wang, B. γ-Secretase Genetics of Hidradenitis Suppurativa: A Systematic Literature Review. Dermatology 2021, 237, 698–704. [Google Scholar] [CrossRef]
- Melnik, B.C.; Plewig, G. Impaired Notch-MKP-1 signalling in hidradenitis suppurativa: An approach to pathogenesis by evidence from translational biology. Exp. Dermatol. 2013, 22, 172–177. [Google Scholar] [CrossRef]
- Frew, J.; Navrazhina, K. No evidence that impaired Notch signalling differentiates hidradenitis suppurativa from other inflammatory skin diseases. Br. J. Dermatol. 2019, 182, 1042–1043. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Benhadou, F.; Byrd, A.S.; Chandran, N.S.; Giamarellos-Bourboulis, E.J.; Fabbrocini, G.; Frew, J.W.; Fujita, H.; González-López, M.A.; Guillem, P.; et al. What causes hidradenitis suppurativa?—15 years after. Exp. Dermatol. 2020, 29, 1154–1170. [Google Scholar] [CrossRef]
- Duchatelet, S.; Miskinyte, S.; Delage, M.; Ungeheuer, M.-N.; Lam, T.; Benhadou, F.; Del Marmol, V.; Vossen, A.R.V.; Prens, E.P.; Cogrel, O.; et al. Low Prevalence of GSC Gene Mutations in a Large Cohort of Predominantly Caucasian Patients with Hidradenitis Suppurativa. J. Investig. Dermatol. 2020, 140, 2085–2088.e4. [Google Scholar] [CrossRef] [PubMed]
- Jfri, A.H.; O’Brien, E.A.; Litvinov, I.V.; Alavi, A.; Netchiporouk, E. Hidradenitis Suppurativa: Comprehensive Review of Predisposing Genetic Mutations and Changes. J. Cutan. Med. Surg. 2019, 23, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Jemec, G.B.E.; Matusiak, Ł.; Kimball, A.B.; Prens, E.; Wolk, K. Hidradenitis suppurativa. Nat. Rev. Dis. Prim. 2020, 6, 18. [Google Scholar] [CrossRef]
- Macklis, P.C.; Tyler, K.; Kaffenberger, J.; Kwatra, S.; Kaffenberger, B.H. Lifestyle modifications associated with symptom improvement in hidradenitis suppurativa patients. Arch. Dermatol. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Bettoli, V.; Naldi, L.; Cazzaniga, S.; Zauli, S.; Atzori, L.; Borghi, A.; Capezzera, R.; Caproni, M.; Cardinali, C.; DeVita, V.; et al. Overweight, diabetes and disease duration influence clinical severity in hidradenitis suppurativa–acne inversa: Evidence from the national Italian registry. Br. J. Dermatol. 2015, 174, 195–197. [Google Scholar] [CrossRef] [Green Version]
- Kromann, C.; Ibler, K.; Kristiansen, V.; Jemec, G. The Influence of Body Weight on the Prevalence and Severity of Hidradenitis Suppurativa. Acta Derm. Venereol. 2014, 94, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.J.; Mekkes, J.R.; Zouboulis, C.P.D.; Scheinfeld, N.; Kimball, A.; Sundaram, M.; Gu, Y.; Okun, M.M.; Kerdel, F. Association of hidradenitis suppurativa disease severity with increased risk for systemic comorbidities. Br. J. Dermatol. 2014, 171, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, A.; Finelt, N.; Strunk, A.; Garg, A. Association between obesity and hidradenitis suppurativa among children and adolescents: A population-based analysis in the United States. J. Am. Acad. Dermatol. 2020, 82, 502–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riis, P.T.; Saunte, D.M.L.; Benhadou, F.; Del Marmol, V.; Guillem, P.; El-Domyati, M.; Abdel-Wahab, H.; Antoniou, C.; Dessinioti, C.; Gürer, M.; et al. Low and high body mass index in hidradenitis suppurativa patients-different subtypes? J. Eur. Acad. Dermatol. Venereol. 2017, 32, 307–312. [Google Scholar] [CrossRef]
- Phan, K.; Charlton, O.; Smith, S.D. Hidradenitis suppurativa and metabolic syndrome—Systematic review and adjusted meta-analysis. Int. J. Dermatol. 2019, 58, 1112–1117. [Google Scholar] [CrossRef]
- Sabat, R.; Chanwangpong, A.; Schneider-Burrus, S.; Metternich, D.; Kokolakis, G.; Kurek, A.; Philipp, S.; Uribe, D.; Wolk, K.; Sterry, W. Increased Prevalence of Metabolic Syndrome in Patients with Acne Inversa. PLoS ONE 2012, 7, e31810. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.M.; Ellervik, C.; Vinding, G.R.; Zarchi, K.; Ibler, K.S.; Knudsen, K.M.; Jemec, G.B.E. Association of Metabolic Syndrome and Hidradenitis Suppurativa. JAMA Dermatol. 2014, 150, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Vilanova, I.; Hernandez, J.L.; Mata, C.; Durán, C.; García-Unzueta, M.; Portilla, V.; Fuentevilla, P.; Corrales, A.; González-Vela, M.; González-Gay, M.; et al. Insulin resistance in hidradenitis suppurativa: A case-control study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 820–824. [Google Scholar] [CrossRef] [Green Version]
- Van Der Zee, H.H.; Laman, J.D.; Boer, J.; Prens, E. Hidradenitis suppurativa: Viewpoint on clinical phenotyping, pathogenesis and novel treatments. Exp. Dermatol. 2012, 21, 735–739. [Google Scholar] [CrossRef]
- Boer, J. Should Hidradenitis Suppurativa Be Included in Dermatoses Showing Koebnerization? Is It Friction or Fiction? Dermatology 2017, 233, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Boer, J.; Mihajlović, D. Boils at Frictional Locations in a Patient with Hidradenitis Suppurativa. Acta Dermatovenerol. Croat. ADC 2016, 24, 303–304. [Google Scholar]
- Wolk, K.; Sabat, R. Adipokines in psoriasis: An important link between skin inflammation and metabolic alterations. Rev. Endocr. Metab. Disord. 2016, 17, 305–317. [Google Scholar] [CrossRef]
- Danby, F.W. Diet in the prevention of hidradenitis suppurativa (acne inversa). J. Am. Acad. Dermatol. 2015, 73, S52–S54. [Google Scholar] [CrossRef] [PubMed]
- Saric-Bosanac, S.; Clark, A.K.; Sivamani, R.K.; Shi, V.Y. The role of hypothalamus-pituitary-adrenal (HPA)-like axis in inflammatory pilosebaceous disorders. Dermatol. Online J. 2020, 26. [Google Scholar] [CrossRef]
- Sivanand, A.; Gulliver, W.P.; Josan, C.K.; Alhusayen, R.; Fleming, P.J. Weight Loss and Dietary Interventions for Hidradenitis Suppurativa: A Systematic Review. J. Cutan. Med. Surg. 2019, 24, 64–72. [Google Scholar] [CrossRef]
- König, A.; Lehmann, C.; Rompel, R.; Happle, R. Cigarette smoking as a triggering factor of hidradenitis suppurativa. Dermatology 1999, 198, 261–264. [Google Scholar] [CrossRef]
- Bukvić Mokos, Z.; Miše, J.; Balić, A.; Marinović, B. Understanding the Relationship Between Smoking and Hidradenitis Suppurativa. Acta Dermatovenerol. Croat. 2020, 28, 9–13. [Google Scholar]
- Happle, R.; König, A. A lesson to be learned from Karl Marx: Smoking triggers hidradenitis suppurativa. Br. J. Dermatol. 2008, 159, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Shuster, S. A lesson to be learned from Karl Marx: Smoking triggers hidradenitis suppurativa: Reply from author. Br. J. Dermatol. 2008, 159, 256–257. [Google Scholar] [CrossRef]
- Garg, A.; Papagermanos, V.; Midura, M.; Strunk, A. Incidence of hidradenitis suppurativa among tobacco smokers: A population-based retrospective analysis in the U.S.A. Br. J. Dermatol. 2018, 178, 709–714. [Google Scholar] [CrossRef] [Green Version]
- Saleem, M.D.; Arnold, D.L.; Feldman, S.R. Hidradenitis and smoking. Br. J. Dermatol. 2018, 178, 810–811. [Google Scholar] [CrossRef]
- Micheletti, R. Tobacco smoking and hidradenitis suppurativa: Associated disease and an important modifiable risk factor. Br. J. Dermatol. 2018, 178, 587–588. [Google Scholar] [CrossRef]
- Dessinioti, C.; Zisimou, C.; Tzanetakou, V.; Ntritsos, G.; Kontochristopoulos, G.; Antoniou, C. A retrospective institutional study of the association of smoking with the severity of hidradenitis suppurativa. J. Dermatol. Sci. 2017, 87, 206–207. [Google Scholar] [CrossRef] [Green Version]
- Matusiak, L.; Bieniek, A.; Szepietowski, J. Hidradenitis suppurativa and associated factors: Still unsolved problems. J. Am. Acad. Dermatol. 2009, 61, 362–365. [Google Scholar] [CrossRef]
- Kromann, C.; Deckers, I.; Esmann, S.; Boer, J.; Prens, E.; Jemec, G. Risk factors, clinical course and long-term prognosis in hidradenitis suppurativa: A cross-sectional study. Br. J. Dermatol. 2014, 171, 819–824. [Google Scholar] [CrossRef]
- Denny, G.; Anadkat, M.J. The effect of smoking and age on the response to first-line therapy of hidradenitis suppurativa: An institutional retrospective cohort study. J. Am. Acad. Dermatol. 2016, 76, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Melnik, B.; John, S.; Chen, W.; Plewig, G. T helper 17 cell/regulatory T-cell imbalance in hidradenitis suppurativa/acne inversa: The link to hair follicle dissection, obesity, smoking and autoimmune comorbidities. Br. J. Dermatol. 2018, 179, 260–272. [Google Scholar] [CrossRef]
- Wolk, K.; Join-Lambert, O.; Sabat, R. Aetiology and pathogenesis of hidradenitis suppurativa. Br. J. Dermatol. 2020, 183, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Hana, A.; Booken, D.; Henrich, C.; Gratchev, A.; Maas-Szabowski, N.; Goerdt, S.; Kurzen, H. Functional significance of non-neuronal acetylcholine in skin epithelia. Life Sci. 2007, 80, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Y.; Xu, T.; Zhang, Q.-Z.; Bai, J.; Wang, J.; Zhu, T.; Lou, Q.; Götz, F.; Qu, D.; et al. Nicotine Enhances Staphylococcus epidermidis Biofilm Formation by Altering the Bacterial Autolysis, Extracellular DNA Releasing, and Polysaccharide Intercellular Adhesin Production. Front. Microbiol. 2018, 9, 2575. [Google Scholar] [CrossRef] [PubMed]
- Mitri, A.; Lin, G.; Waldman, R.A.; Grant-Kels, J.M. Effects of tobacco and vaping on the skin. Clin. Dermatol. 2021. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenei, A.; Dajnoki, Z.; Medgyesi, B.; Gáspár, K.; Béke, G.; Kinyó, Á.; Méhes, G.; Hendrik, Z.; Dinya, T.; Törőcsik, D.; et al. Apocrine Gland–Rich Skin Has a Non-Inflammatory IL-17–Related Immune Milieu, that Turns to Inflammatory IL-17–Mediated Disease in Hidradenitis Suppurativa. J. Investig. Dermatol. 2019, 139, 964–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, S.L.; Schneider, A.M.; Nelson, A.M. Yin and Yang: A disrupted skin microbiome and an aberrant host immune response in hidradenitis suppurativa. Exp. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wark, K.J.L.; Cains, G.D. The Microbiome in Hidradenitis Suppurativa: A Review. Dermatol. Ther. 2020, 11, 39–52. [Google Scholar] [CrossRef]
- Dréno, B.; Pécastaings, S.; Corvec, S.; Veraldi, S.; Khammari, A.; Roques, C. Cutibacterium acnes (Propionibacterium acnes) and acne vulgaris: A brief look at the latest updates. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Ring, H.C.; Thorsen, J.; Saunte, D.M.L.; Lilje, B.; Bay, L.; Riis, P.T.; Larsen, N.; Andersen, L.O.; Nielsen, H.V.; Miller, I.M.; et al. The Follicular Skin Microbiome in Patients with Hidradenitis Suppurativa and Healthy Controls. JAMA Dermatol. 2017, 153, 897–905. [Google Scholar] [CrossRef]
- Ring, H.; Sigsgaard, V.; Thorsen, J.; Fuursted, K.; Fabricius, S.; Saunte, D.; Jemec, G. The microbiome of tunnels in hidradenitis suppurativa patients. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1775–1780. [Google Scholar] [CrossRef]
- Grand, D.; Navrazhina, K.; Frew, J.W. Integrating complement into the molecular pathogenesis of Hidradenitis Suppurativa. Exp. Dermatol. 2019, 29, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Segre, J.A. Dialogue between skin microbiota and immunity. Science 2014, 346, 954–959. [Google Scholar] [CrossRef]
- Gallo, R.L.; Hooper, L.V. Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 2012, 12, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Bouladoux, N.; Wilhelm, C.; Molloy, M.J.; Salcedo, R.; Kastenmuller, W.; Deming, C.; Quinones, M.; Koo, L.; Conlan, S.; et al. Compartmentalized Control of Skin Immunity by Resident Commensals. Science 2012, 337, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Lousada, M.; Lachnit, T.; Edelkamp, J.; Rouillé, T.; Ajdic, D.; Uchida, Y.; Di Nardo, A.; Bosch, T.; Paus, R. Exploring the human hair follicle microbiome. Br. J. Dermatol. 2020, 184, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The immune response toPrevotellabacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzecry, V.; Grancini, A.; Guanziroli, E.; Nazzaro, G.; Barbareschi, M.; Marzano, A.V.; Muratori, S.; Veraldi, S. Hidradenitis suppurativa/acne inversa: A prospective bacteriological study and review of the literature. G. Ital. Dermatol. Venereol. 2020, 155, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, G.; Passoni, E.; Veraldi, S.; Marzano, A.V. Lymph node involvement in hidradenitis suppurativa: Ultrasound and color Doppler study of 85 patients. Ski. Res. Technol. 2020, 26, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Yidana, D.B. Hidradenitis suppurativa—The role of interleukin-17, the aryl hydrocarbon receptor and the link to a possible fungal aetiology. Med. Hypotheses 2021, 149, 110530. [Google Scholar] [CrossRef]
- Mortimer, P.S.; Dawber, R.P.; A Gales, M.; A Moore, R. Mediation of hidradenitis suppurativa by androgens. BMJ 1986, 292, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Riis, P.T.; Ring, H.C.; Themstrup, L.; Jemec, G.B. The Role of Androgens and Estrogens in Hidradenitis Suppurativa—A Systematic Review. Acta Dermatovenerol. Croat. ADC 2016, 24, 239–249. [Google Scholar]
- Karagiannidis, I.; Nikolakis, G.; Sabat, R.; Zouboulis, C.C. Hidradenitis suppurativa/Acne inversa: An endocrine skin disorder? Rev. Endocr. Metab. Disord. 2016, 17, 335–341. [Google Scholar] [CrossRef]
- Collier, E.K.; Price, K.N.; Grogan, T.R.; Naik, H.B.; Shi, V.Y.; Hsiao, J.L. Characterizing perimenstrual flares of hidradenitis suppurativa. Int. J. Women’s Dermatol. 2020, 6, 372–376. [Google Scholar] [CrossRef]
- Vossen, A.R.; van Straalen, K.; Prens, E.P.; van der Zee, H.H. Menses and pregnancy affect symptoms in hidradenitis suppurativa: A cross-sectional study. J. Am. Acad. Dermatol. 2017, 76, 155–156. [Google Scholar] [CrossRef] [Green Version]
- Perng, P.; Zampella, J.; Okoye, G. Considering the impact of pregnancy on the natural history of hidradenitis suppurativa. Br. J. Dermatol. 2017, 178, e13–e14. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.J.; Kumar, S.; Read, G.F.; Edwards, C.A.; Scanlon, M.F.; Hughes, L.E. Hidradenitis suppurativa: Evidence for an endocrine abnormality. BJS 1985, 72, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Jemec, G. The symptomatology of hidradenitis suppurativa in women. Br. J. Dermatol. 1988, 119, 345–350. [Google Scholar] [CrossRef]
- Harrison, B.J.; Read, G.F.; Hughes, L.E. Endocrine basis for the clinical presentation of hidradenitis suppurativa. BJS 1988, 75, 972–975. [Google Scholar] [CrossRef]
- Barth, J.H.; Kealey, T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br. J. Dermatol. 1991, 125, 304–308. [Google Scholar] [CrossRef]
- Buimer, M.G.; Wobbes, T.; Klinkenbijl, J.H.G.; Reijnen, M.M.P.J.; Blokx, W.A.M. Immunohistochemical Analysis of Steroid Hormone Receptors in Hidradenitis Suppurativa. Am. J. Dermatopathol. 2015, 37, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Gauntner, T.D. Hormonal, stem cell and Notch signalling as possible mechanisms of disease in hidradenitis suppurativa: A systems-level transcriptomic analysis. Br. J. Dermatol. 2018, 180, 203–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouboulis, C.C.; Da Costa, A.N.; Fimmel, S.; Zouboulis, K.C. Apocrine glands are bystanders in hidradenitis suppurativa and their involvement is gender specific. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Nikolakis, G.; Kyrgidis, A.; Zouboulis, C.C. Is There a Role for Antiandrogen Therapy for Hidradenitis Suppurativa? A Systematic Review of Published Data. Am. J. Clin. Dermatol. 2019, 20, 503–513. [Google Scholar] [CrossRef]
- C.-W.Yu, C.; Cook, M.G. Hidradenitis suppurativa: A disease of follicular epithelium, rather than apocrine glands. Br. J. Dermatol. 1990, 122, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Jemec, G.; Hansen, U. Histology of hidradenitis suppurativa. J. Am. Acad. Dermatol. 1996, 34, 994–999. [Google Scholar] [CrossRef]
- Boer, J.; Weltevreden, E.F. Hidradenitis suppurativa or acne inversa. A clinicopathological study of early lesions. Br. J. Dermatol. 1996, 135, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Von Laffert, M.; Helmbold, P.; Wohlrab, J.; Fiedler, E.; Stadie, V.; Marsch, W.C. Hidradenitis suppurativa (acne inversa): Early inflammatory events at terminal follicles and at interfollicular epidermis. Exp. Dermatol. 2009, 19, 533–537. [Google Scholar] [CrossRef]
- Von Laffert, M.; Stadie, V.; Wohlrab, J.; Marsch, W. Hidradenitis suppurativa/acne inversa: Bilocated epithelial hyperplasia with very different sequelae. Br. J. Dermatol. 2010, 164, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Danby, F.; Jemec, G.; Marsch, W.; Von Laffert, M. Preliminary findings suggest hidradenitis suppurativa may be due to defective follicular support. Br. J. Dermatol. 2013, 168, 1034–1039. [Google Scholar] [CrossRef]
- Rongioletti, F. Histopathology. In Hidradenitis Suppurativa; Micali, G., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Hunger, R.; Surovy, A.; Hassan, A.; Braathen, L.; Yawalkar, N. Toll-like receptor 2 is highly expressed in lesions of acne inversa and colocalizes with C-type lectin receptor. Br. J. Dermatol. 2008, 158, 691–697. [Google Scholar] [CrossRef]
- Shah, A.; Alhusayen, R.; Amini-Nik, S. The critical role of macrophages in the pathogenesis of hidradenitis suppurativa. Inflamm. Res. 2017, 66, 931–945. [Google Scholar] [CrossRef]
- Frew, J.W.; Grand, D.; Navrazhina, K.; Krueger, J.G. Beyond antibodies: B cells in Hidradenitis Suppurativa: Bystanders, contributors or therapeutic targets? Exp. Dermatol. 2020, 29, 509–515. [Google Scholar] [CrossRef]
- Frew, J.W.; Navrazhina, K.; Marohn, M.; Lu, C.; Krueger, J.G. Contribution of fibroblasts to tunnel formation and inflammation in hidradenitis suppurativa/ acne inversa. Exp. Dermatol. 2019, 28, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.S.; Carmona-Rivera, C.; O’Neil, L.J.; Carlucci, P.M.; Cisar, C.; Rosenberg, A.Z.; Kerns, M.L.; Caffrey, J.A.; Milner, S.M.; Sacks, J.M.; et al. Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci. Transl. Med. 2019, 11, eaav5908. [Google Scholar] [CrossRef]
- Wolk, K.; Brembach, T.; Šimaitė, D.; Bartnik, E.; Cucinotta, S.; Pokrywka, A.; Irmer, M.; Triebus, J.; Witte-Händel, E.; Salinas, G.; et al. Activity and components of the granulocyte colony-stimulating factor pathway in hidradenitis suppurativa. Br. J. Dermatol. 2021, 185, 164–176. [Google Scholar] [CrossRef]
- Zouboulis, C.; Da Costa, A.N.; Makrantonaki, E.; Hou, X.; Almansouri, D.; Dudley, J.; Edwards, H.; Readhead, B.; Balthasar, O.; Jemec, G.; et al. Alterations in innate immunity and epithelial cell differentiation are the molecular pillars of hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 846–861. [Google Scholar] [CrossRef]
- Herman, A.; Herman, A.P. Antimicrobial peptides activity in the skin. Ski. Res. Technol. 2018, 25, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Warszawska, K.; Hoeflich, C.; Witte, E.; Schneider-Burrus, S.; Witte, K.; Kunz, S.; Buss, A.; Roewert, H.J.; Krause, M.; et al. Deficiency of IL-22 Contributes to a Chronic Inflammatory Disease: Pathogenetic Mechanisms in Acne Inversa. J. Immunol. 2010, 186, 1228–1239. [Google Scholar] [CrossRef] [Green Version]
- Scala, E.; Di Caprio, R.; Cacciapuoti, S.; Caiazzo, G.; Fusco, A.; Tortorella, E.; Fabbrocini, G.; Balato, A. A new T helper 17 cytokine in hidradenitis suppurativa: Antimicrobial and proinflammatory role of interleukin-26. Br. J. Dermatol. 2019, 181, 1038–1045. [Google Scholar] [CrossRef]
- Hofmann, S.C.; Saborowski, V.; Lange, S.; Kern, W.V.; Bruckner-Tuderman, L.; Rieg, S. Expression of innate defense antimicrobial peptides in hidradenitis suppurativa. J. Am. Acad. Dermatol. 2012, 66, 966–974. [Google Scholar] [CrossRef]
- Hotz, C.; Boniotto, M.; Guguin, A.; Surenaud, M.; Jean-Louis, F.; Tisserand, P.; Ortonne, N.; Hersant, B.; Bosc, R.; Poli, F.; et al. Intrinsic Defect in Keratinocyte Function Leads to Inflammation in Hidradenitis Suppurativa. J. Investig. Dermatol. 2016, 136, 1768–1780. [Google Scholar] [CrossRef] [Green Version]
- Bechara, F.G.; Sand, M.; Skrygan, M.; Kreuter, A.; Altmeyer, P.; Gambichler, T. Acne Inversa: Evaluating Antimicrobial Peptides and Proteins. Ann. Dermatol. 2012, 24, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Thomi, R.; Schlapbach, C.; Yawalkar, N.; Simon, D.; Yerly, D.; Hunger, R.E. Elevated levels of the antimicrobial peptide LL-37 in hidradenitis suppurativa are associated with a Th1/Th17 immune response. Exp. Dermatol. 2017, 27, 172–177. [Google Scholar] [CrossRef]
- Shanmugam, V.K.; Jones, D.; McNish, S.; Bendall, M.L.; Crandall, K.A. Transcriptome patterns in hidradenitis suppurativa: Support for the role of antimicrobial peptides and interferon pathways in disease pathogenesis. Clin. Exp. Dermatol. 2019, 44, 882–892. [Google Scholar] [CrossRef]
- Coates, M.; Mariottoni, P.; Corcoran, D.L.; Kirshner, H.F.; Jaleel, T.; Brown, D.A.; Brooks, S.R.; Murray, J.; Morasso, M.I.; MacLeod, A.S. The skin transcriptome in hidradenitis suppurativa uncovers an antimicrobial and sweat gland gene signature which has distinct overlap with wounded skin. PLoS ONE 2019, 14, e0216249. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Wenzel, J.; Tsaousi, A.; Witte-Händel, E.; Babel, N.; Zelenak, C.; Volk, H.-D.; Sterry, W.; Schneider-Burrus, S.; Sabat, R. Lipocalin-2 is expressed by activated granulocytes and keratinocytes in affected skin and reflects disease activity in acne inversa/hidradenitis suppurativa. Br. J. Dermatol. 2017, 177, 1385–1393. [Google Scholar] [CrossRef]
- Ghias, M.H.; Hyde, M.J.; Tomalin, L.E.; Morgan, B.P.; Alavi, A.; Lowes, M.A.; Piguet, V. Role of the Complement Pathway in Inflammatory Skin Diseases: A Focus on Hidradenitis Suppurativa. J. Investig. Dermatol. 2020, 140, 531–536.e1. [Google Scholar] [CrossRef]
- Kanni, T.; Zenker, O.; Habel, M.; Riedemann, N.; Giamarellos-Bourboulis, E.J. Complement activation in hidradenitis suppurativa: A new pathway of pathogenesis? Br. J. Dermatol. 2018, 179, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Del Duca, E.; Morelli, P.; Bennardo, L.; Di Raimondo, C.; Nisticò, S.P. Cytokine Pathways and Investigational Target Therapies in Hidradenitis Suppurativa. Int. J. Mol. Sci. 2020, 21, 8436. [Google Scholar] [CrossRef]
- Ardon, C.; Wang, C.; Prens, E.; van Straalen, K. Noninvasive assessment of cytokine and antimicrobial peptide levels in hidradenitis suppurativa using transdermal analysis patches. Br. J. Dermatol. 2020, 184, 343–345. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Kelly, G.; Hughes, R.; McGarry, T.; Born, M.V.D.; Adamzik, K.; Fitzgerald, R.; Lawlor, C.; Tobin, A.-M.; Sweeney, C.; Kirby, B. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br. J. Dermatol. 2015, 173, 1431–1439. [Google Scholar] [CrossRef]
- McGovern, N.; Schlitzer, A.; Gunawan, M.; Jardine, L.; Shin, A.; Poyner, E.; Green, K.; Dickinson, R.; Wang, X.-N.; Low, D.; et al. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages. Immunity 2014, 41, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Kanni, T.; Tzanetakou, V.; Savva, A.; Kersten, B.; Pistiki, A.; Van De Veerdonk, F.L.; Netea, M.G.; Van Der Meer, J.W.; Giamarellos-Bourboulis, E.J. Compartmentalized Cytokine Responses in Hidradenitis Suppurativa. PLoS ONE 2015, 10, e0130522. [Google Scholar] [CrossRef] [Green Version]
- Van Der Zee, H.; De Ruiter, L.; Broecke, D.V.D.; Dik, W.; Laman, J.; Prens, E. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: A rationale for targeting TNF-α and IL-1β. Br. J. Dermatol. 2011, 164, 1292–1298. [Google Scholar] [CrossRef]
- Witte-Händel, E.; Wolk, K.; Tsaousi, A.; Irmer, M.L.; Mößner, R.; Shomroni, O.; Lingner, T.; Witte, K.; Kunkel, D.; Salinas, G.; et al. The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction. J. Investig. Dermatol. 2019, 139, 1294–1305. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Lima, A.; Karl, I.; Giner, T.; Poppe, H.; Schmidt, M.; Presser, D.; Goebeler, M.; Bauer, B.L. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br. J. Dermatol. 2015, 174, 514–521. [Google Scholar] [CrossRef]
- Frings, V.G.; Sennefelder, H.; Presser, D.; Goebeler, M.; Schmidt, M. Altered NOX expression does not seem to account for epidermal NLRP 3 inflammasome activation in hidradenitis suppurativa. Br. J. Dermatol. 2019, 181, 391–392. [Google Scholar] [CrossRef]
- Di Caprio, R.; Balato, A.; Caiazzo, G.; Lembo, S.; Raimondo, A.; Fabbrocini, G.; Monfrecola, G. IL-36 cytokines are increased in acne and hidradenitis suppurativa. Arch. Dermatol. Res. 2017, 309, 673–678. [Google Scholar] [CrossRef]
- Thomi, R.; Kakeda, M.; Yawalkar, N.; Schlapbach, C.; Hunger, R.E. Increased expression of the interleukin-36 cytokines in lesions of hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2091–2096. [Google Scholar] [CrossRef]
- Hessam, S.; Sand, M.; Gambichler, T.; Skrygan, M.; Rüddel, I.; Bechara, F. Interleukin-36 in hidradenitis suppurativa: Evidence for a distinctive proinflammatory role and a key factor in the development of an inflammatory loop. Br. J. Dermatol. 2018, 178, 761–767. [Google Scholar] [CrossRef]
- Hayran, Y.; Allı, N.; Yücel, Ç.; Akdoğan, N.; Turhan, T. Serum IL-36α, IL-36β, and IL-36γ levels in patients with hidradenitis suppurativa: Association with disease characteristics, smoking, obesity, and metabolic syndrome. Arch. Dermatol. Res. 2019, 312, 187–196. [Google Scholar] [CrossRef]
- Savage, K.T.; Flood, K.S.; Porter, M.L.; Kimball, A.B. TNF-α inhibitors in the treatment of hidradenitis suppurativa. Ther. Adv. Chronic Dis. 2019, 10, 2040622319851640. [Google Scholar] [CrossRef]
- Moran, B.; Sweeney, C.M.; Hughes, R.; Malara, A.; Kirthi, S.; Tobin, A.-M.; Kirby, B.; Fletcher, J.M. Hidradenitis Suppurativa Is Characterized by Dysregulation of the Th17:Treg Cell Axis, Which Is Corrected by Anti-TNF Therapy. J. Investig. Dermatol. 2017, 137, 2389–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Gallo, D.; de la Varga-Martínez, R.; Ossorio-García, L.; Collantes-Rodríguez, C.; Linares-Barrios, M. Effects of adalimumab on T-helper-17 lymphocyte- and neutrophil-related inflammatory serum markers in patients with moderate-to-severe hidradenitis suppurativa. Cytokine 2017, 103, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Balato, A.; Caiazzo, G.; Annunziata, M.; Marasca, C.; Scala, E.; Cacciapuoti, S.; Fabbrocini, G. Anti- TNF -α therapy modulates mTORC 1 signalling in hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2018, 33, e43–e45. [Google Scholar] [CrossRef] [PubMed]
- Bettuzzi, T.; Frumholtz, L.; Jachiet, M.; Lepelletier, C.; Djermane, M.; Cordoliani, F.; Saussine, A.; Bouaziz, J.-D.; Bachelez, H. Sirolimus as combination rescue therapy with tumor necrosis alpha inhibitors for severe, refractory hidradenitis suppurativa. J. Am. Acad. Dermatol. 2020, 83, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Mozeika, E.; Pilmane, M.; Nürnberg, B.; Jemec, G. Tumour Necrosis Factor-alpha and Matrix Metalloproteinase-2 are Expressed Strongly in Hidradenitis Suppurativa. Acta Derm. Venereol. 2013, 93, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Éno, B.D.; Khammari, A.; Brocard, A.; Moyse, D.; Blouin, E.; Guillet, G.; Éonard, F.L.; Knol, A.-C. Hidradenitis Suppurativa. Arch. Dermatol. 2012, 148, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Cardoso, A.; Goncalves, R.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; McNish, S.; Shanmugam, V.K. Interferon-gamma (IFN-γ) is Elevated in Wound Exudate from Hidradenitis Suppurativa. Immunol. Investig. 2016, 46, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Gudjonsson, J.E.; Tsoi, L.C.; Ma, F.; Billi, A.C.; van Straalen, K.; Vossen, A.; Van Der Zee, H.; Harms, P.W.; Wasikowski, R.; Yee, C.M.; et al. Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis. JCI Insight 2020, 5, e139930. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Moran, B.; Petrasca, A.; Smith, C.M. IL-17 in inflammatory skin diseases psoriasis and hidradenitis suppurativa. Clin. Exp. Immunol. 2020, 201, 121–134. [Google Scholar] [CrossRef]
- Yao, Y.; Thomsen, S.F. The role of interleukin-17 in the pathogenesis of hidradenitis suppurativa. Dermatol. Online J. 2017, 23. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Krueger, J.G. IL-17C: A Unique Epithelial Cytokine with Potential for Targeting across the Spectrum of Atopic Dermatitis and Psoriasis. J. Investig. Dermatol. 2018, 138, 1467–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.-A.; Suh, J.W.; Lee, K.H.; Kang, J.L.; Woo, S.-Y. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1β by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int. Immunol. 2011, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Navrazhina, K.; Frew, J.; Krueger, J. Interleukin 17C is elevated in lesional tissue of hidradenitis suppurativa. Br. J. Dermatol. 2019, 182, 1045–1047. [Google Scholar] [CrossRef]
- Matusiak, Ł.; Szczęch, J.; Bieniek, A.; Nowicka-Suszko, D.; Szepietowski, J. Increased interleukin (IL)-17 serum levels in patients with hidradenitis suppurativa: Implications for treatment with anti-IL-17 agents. J. Am. Acad. Dermatol. 2017, 76, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Gallo, D.; Martínez, R.D.L.V.; García, L.O.; Albarrán-Planelles, C.; Rodríguez, C.; Linares-Barrios, M. The Clinical Significance of Increased Serum Proinflammatory Cytokines, C-Reactive Protein, and Erythrocyte Sedimentation Rate in Patients with Hidradenitis Suppurativa. Mediat. Inflamm. 2017, 2017, 2450401. [Google Scholar] [CrossRef]
- Öztürk, G.S.; Ergun, T.; Eyüboğlu, I.P.; Akkiprik, M. Serum high-sensitivity C-reactive protein, tumor necrosis factor-α, interleukin (IL)-1β, IL-17A and IL-23 levels in patients with hidradenitis suppurativa. Cytokine 2021, 144, 155585. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Thirunavukkarasu, S. The Tale of IL-12 and IL-23: A Paradigm Shift. J. Immunol. 2019, 202, 629–630. [Google Scholar] [CrossRef] [Green Version]
- Schlapbach, C.; Hänni, T.; Yawalkar, N.; Hunger, R.E. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2011, 65, 790–798. [Google Scholar] [CrossRef]
- Vossen, A.R.J.V.; Van Der Zee, H.H.; Tsoi, L.C.; Xing, X.; Devalaraja, M.; Gudjonsson, J.E.; Prens, E.P. Novel cytokine and chemokine markers of hidradenitis suppurativa reflect chronic inflammation and itch. Allergy 2018, 74, 631–634. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Banerjee, A.; Berger, P.Z.; Gross, A.; McNish, S.; Amdur, R.; Shanmugam, V.K. Inherent differences in keratinocyte function in hidradenitis suppurativa: Evidence for the role of IL-22 in disease pathogenesis. Immunol. Investig. 2017, 47, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zee, H.; Laman, J.; De Ruiter, L.; Dik, W.; Prens, E. Adalimumab (antitumour necrosis factor-α) treatment of hidradenitis suppurativa ameliorates skin inflammation: An in situ and ex vivo study. Br. J. Dermatol. 2012, 166, 298–305. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorganic Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, X.; He, Y.; Zhang, X.; Li, C.; Mao, Q.; Wu, X.; Wang, B. Increased serum interleukin-6 levels in patients with hidradenitis suppurativa. Adv. Dermatol. Allergol. 2017, 1, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, A.J.; Hsiao, J.L.; Lowes, M.A.; Shi, V.Y. A Comparison of International Management Guidelines for Hidradenitis Suppurativa. Dermatology 2019, 237, 81–96. [Google Scholar] [CrossRef]
- Zouboulis, C.; Bechara, F.; Dickinson-Blok, J.; Gulliver, W.; Horváth, B.; Hughes, R.; Kimball, A.; Kirby, B.; Martorell, A.; Podda, M.; et al. Hidradenitis suppurativa/acne inversa: A practical framework for treatment optimization—Systematic review and recommendations from the HS ALLIANCE working group. J. Eur. Acad. Dermatol. Venereol. 2018, 33, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Frew, J.W.; Giamarellos-Bourboulis, E.J.; Jemec, G.B.; del Marmol, V.; Marzano, A.V.; Nikolakis, G.; Sayed, C.J.; Tzellos, T.; Wolk, K.; et al. Target molecules for future hidradenitis suppurativa treatment. Exp. Dermatol. 2021, 30, 8–17. [Google Scholar] [CrossRef]
- Narla, S.; Azzam, M.; Townsend, S.; Vellaichamy, G.; Marzano, A.; Alavi, A.; Lowes, M.; Hamzavi, I. Identifying key components and therapeutic targets of the immune system in hidradenitis suppurativa with an emphasis on neutrophils. Br. J. Dermatol. 2020, 184, 1004–1013. [Google Scholar] [CrossRef]
- Study of Efficacy and Safety of Investigational Treatments in Patients with Moderate to Severe Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT03827798 (accessed on 1 August 2021).
- A Global Study Comparing Risankizumab to Placebo in Adult Participants with Moderate to Severe Hidradenitis Suppura-Tiva (Determined 1). Available online: https://clinicaltrials.gov/ct2/show/NCT03926169 (accessed on 1 August 2021).
- Molecular Characteristics of Brodalumab in Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04979520 (accessed on 1 August 2021).
- A Study to Evaluate the Efficacy and Safety of Bimekizumab in Study Participants with Moderate to Severe Hidradenitis Suppurativa (Be Heard I). Available online: https://clinicaltrials.gov/ct2/show/NCT04242446 (accessed on 1 August 2021).
- A Study to Test the Efficacy and Safety of Bimekizumab in Study Participants with Moderate to Severe Hidradenitis Suppurativa (Be heard II). Available online: https://clinicaltrials.gov/ct2/show/NCT04242498 (accessed on 1 August 2021).
- A Study to Test the Long-Term Treatment of Bimekizumab in Study Participants with Moderate to Severe Hidradenitis Suppurativa (Be Heard EXT). Available online: https://clinicaltrials.gov/ct2/show/NCT04901195 (accessed on 1 August 2021).
- A Study of Oral Upadacitinib Tablet Compared to Placebo in Adult Participants with Moderate to Severe Hidradenitis Suppurativa to Assess Change in Disease Symptoms. Available online: https://clinicaltrials.gov/ct2/show/NCT04430855 (accessed on 1 August 2021).
- To Assess the Efficacy and Safety of INCB054707 in Participants with Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04476043 (accessed on 1 August 2021).
- Orismilast for the Treatment of Mild to Severe Hidradenitis Suppurativa (OSIRIS). Available online: https://clinicaltrials.gov/ct2/show/NCT04982432 (accessed on 1 August 2021).
- Extension Study to Assess Effects of Non-Interrupted Versus Interrupted and Long-Term Treatment of Two Dose Regimes of Secukinumab in Subjects with Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04179175 (accessed on 1 August 2021).
- This Is a Study of Efficacy and Safety of Two Secukinumab Dose Regimens in Subjects with Moderate to Severe Hidradenitis Suppurativa (HS). (SUNSHINE). Available online: https://clinicaltrials.gov/ct2/show/NCT03713619 (accessed on 1 August 2021).
- Study of Efficacy and Safety of Two Secukinumab Dose Regimens in Subjects with Moderate to Severe Hidradenitis Suppurativa (HS) (SUNRISE). Available online: https://clinicaltrials.gov/ct2/show/NCT03713632 (accessed on 1 August 2021).
- A Study to Test Whether Spesolimab Helps People with A Skin Disease Called Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04762277 (accessed on 1 August 2021).
- A Study Investigating Long-Term Treatment with Spesolimab in People with A Skin Disease Called Hidradenitis Suppurativa Who Completed A Previous Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT04876391 (accessed on 1 August 2021).
- A Study to Evaluate the Efficacy and Safety of Imsidolimab (ANB019) in The Treatment of Subjects with Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04856930 (accessed on 1 August 2021).
- A Study of LY3041658 in Adults with Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04493502 (accessed on 1 August 2021).
- A Study to Evaluate the Safety and Efficacy of PF-06650833, PF-06700841, and PF 06826647 in Adults with Hidradenitis Suppurativa. Available online: https://clinicaltrials.gov/ct2/show/NCT04092452 (accessed on 1 August 2021).
- A Single and Multiple Ascending Dose Trial of KT-474 in Healthy Adult Volunteers and Patients with Atopic Dermatitis (AD) or Hidradenitis Suppurativa (HS). Available online: https://clinicaltrials.gov/ct2/show/NCT04772885 (accessed on 1 August 2021).
- Guselkumab for Hidradenitis Suppurativa, a Mode of Action Study. (HiGUS). Available online: https://clinicaltrials.gov/ct2/show/NCT04061395 (accessed on 1 August 2021).
- Evaluation of Safety and Efficacy of Avacopan in Subjects with Moderate to Severe Hidradenitis Suppurativa (AURORA). Available online: https://clinicaltrials.gov/ct2/show/NCT03852472 (accessed on 1 August 2021).
- Safety and Pharmacokinetics of Repeat Doses of CSL324 in Subjects with Hidradenitis Suppurativa and Palmoplantar Pustulosis. Available online: https://clinicaltrials.gov/ct2/show/NCT03972280 (accessed on 1 August 2021).
- Cost-Effectiveness of Adalimumab and Surgery vs. Adalimumab in HS (HS-COST). Available online: https://clinicaltrials.gov/ct2/show/NCT03221621 (accessed on 1 August 2021).
- Tofacitinib for Immune Skin Conditions in Down Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT04246372 (accessed on 1 August 2021).
- Włodarek, K.; Ponikowska, M.; Matusiak, Ł.; Szepietowski, J.C. Biologics for hidradenitis suppurativa: An update. Immunotherapy 2019, 11, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Kimball, A.; Okun, M.M.; Williams, D.A.; Gottlieb, A.B.; Papp, K.A.; Zouboulis, C.C.; Armstrong, A.W.; Kerdel, F.; Gold, M.H.; Forman, S.B.; et al. Two Phase 3 Trials of Adalimumab for Hidradenitis Suppurativa. N. Engl. J. Med. 2016, 375, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-W.; Huang, Y.-W.; Chen, T.-L. Efficacy and safety of adalimumab in hidradenitis suppurativa: A systematic review and meta-analysis of randomized controlled trials. Medicine 2021, 100, e26190. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Okun, M.M.; Prens, E.P.; Gniadecki, R.; Foley, P.A.; Lynde, C.; Weisman, J.; Gu, Y.; Williams, D.A.; Jemec, G.B. Long-term adalimumab efficacy in patients with moderate-to-severe hidradenitis suppurativa/acne inversa: 3-year results of a phase 3 open-label extension study. J. Am. Acad. Dermatol. 2019, 80, 60–69.e2. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Hansen, H.; Caro, R.D.C.; Damiani, G.; Delorme, I.; Pascual, J.C.; Reguiai, Z.; Trigoni, A.; Vilarrasa, E.; Roldán, F.A. Adalimumab Dose Intensification in Recalcitrant Hidradenitis Suppurativa/Acne Inversa. Dermatology 2019, 236, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Sobell, J.M.; Leonardi, C.L.; Lynde, C.W.; Karunaratne, M.; Valdecantos, W.C.; Hendrickson, B.A. Safety of Adalimumab Dosed Every Week and Every Other Week: Focus on Patients with Hidradenitis Suppurativa or Psoriasis. Am. J. Clin. Dermatol. 2018, 19, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Frew, J.W.; Jiang, C.S.; Singh, N.; Grand, D.; Navrazhina, K.; Vaughan, R.; Krueger, J.G. Malignancy and infection risk during adalimumab therapy in hidradenitis suppurativa. Clin. Exp. Dermatol. 2020, 45, 859–865. [Google Scholar] [CrossRef]
- Cao, Y.; Hong, F.; Conlon, D.; Sidur, L.; Smith, K.; Fang, Y.; Cuff, C.; Kaymakcalan, Z.; Ruzek, M. Potential predictive biomarkers of adalimumab response in patients with hidradenitis suppurativa. Br. J. Dermatol. 2021. [Google Scholar] [CrossRef]
- Martínez, F.; Nos, P.; Benlloch, S.; Ponce, J. Hidradenitis suppurativa and Crohn’s disease: Response to treatment with infliximab. Inflamm. Bowel Dis. 2001, 7, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Christodoulou, D.K.; Tsianos, E.V. Axillary hidradenitis suppurativa successfully treated with infliximab in a Crohn’s disease patient. Am. J. Gastroenterol. 2002, 97, 2155–2156. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Gonzalez, T.; Montgomery, M.O.; Cardenas, V.; Kerdel, F.A. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: A randomized, double-blind, placebo-controlled crossover trial. J. Am. Acad. Dermatol. 2010, 62, 205–217. [Google Scholar] [CrossRef]
- Van Rappard, D.C.; Leenarts, M.F.E.; Oost, L.M.-V.; Mekkes, J.R. Comparing treatment outcome of infliximab and adalimumab in patients with severe hidradenitis suppurativa. J. Dermatol. Treat. 2011, 23, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskardmay, A.N.; Miles, J.A.; Sayed, C.J. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2019, 81, 702–708. [Google Scholar] [CrossRef]
- Ghias, M.; Johnston, A.; Kutner, A.J.; Micheletti, R.G.; Hosgood, H.D.; Cohen, S.R. High-dose, high-frequency infliximab: A novel treatment paradigm for hidradenitis suppurativa. J. Am. Acad. Dermatol. 2019, 82, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Prens, L.; Bouwman, K.; Aarts, P.; Arends, S.; van Straalen, K.; Dudink, K.; Horváth, B.; Prens, E. Adalimumab and infliximab survival in patients with hidradenitis suppurativa: A daily practice cohort study. Br. J. Dermatol. 2021, 185, 177–184. [Google Scholar] [CrossRef] [PubMed]
- DeFazio, M.V.; Economides, J.M.; King, K.S.; Han, K.D.; Shanmugam, V.K.; Attinger, C.E.; Evans, K.K. Outcomes After Combined Radical Resection and Targeted Biologic Therapy for the Management of Recalcitrant Hidradenitis Suppurativa. Ann. Plast. Surg. 2016, 77, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.S.; Mysler, E.; Moots, R.J. Etanercept for the treatment of rheumatoid arthritis. Immunotherapy 2018, 10, 433–445. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.; Pelekanou, E.; Antonopoulou, A.; Petropoulou, H.; Baziaka, F.; Karagianni, V.; Stavrianeas, N.; Giamarellou, H. An open-label phase II study of the safety and efficacy of etanercept for the therapy of hidradenitis suppurativa. Br. J. Dermatol. 2007, 158, 567–572. [Google Scholar] [CrossRef]
- Lee, R.A.; Dommasch, E.; Treat, J.; Sciacca-Kirby, J.; Chachkin, S.; Williams, J.; Shin, D.B.; Leyden, J.J.; Vittorio, C.; Gelfand, J.M. A prospective clinical trial of open-label etanercept for the treatment of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2009, 60, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.R.; Yankura, J.A.; Fogelberg, A.C.; Anderson, B.E. Treatment of Hidradenitis Suppurativa With Etanercept Injection. Arch. Dermatol. 2010, 146, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zee, H.; Prens, E. Failure of Anti-Interleukin-1 Therapy in Severe Hidradenitis Suppurativa: A Case Report. Dermatology 2013, 226, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A. Concomitant hidradenitis suppurativa and pyostomatitis vegetans in silent ulcerative colitis successfully treated with golimumab. Dig. Liver Dis. 2016, 48, 1511–1512. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Stephens, S. Certolizumab Pegol. mAbs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, J.G.; Jørgensen, A.R.; Yao, Y.; Thomsen, S.F. Certolizumab pegol for hidradenitis suppurativa: Case report and literature review. Dermatol. Ther. 2020, 33, e14494. [Google Scholar] [CrossRef]
- Esme, P.; Akoglu, G.; Caliskan, E. Rapid Response to Certolizumab Pegol in Hidradenitis Suppurativa: A Case Report. Ski. Appendage Disord. 2020, 7, 58–61. [Google Scholar] [CrossRef]
- Tampouratzi, E.; Kanni, T.; Katsantonis, J.; Douvali, T. Case report: Treating a co-existence of hidradenitis suppurativa and psoriasis with different therapeutic approaches. F1000Research 2020, 8, 2002. [Google Scholar] [CrossRef]
- Porter, M.L.; Golbari, N.M.; Lockwood, S.J.; Kimball, A.B. Overview and update on biologic therapy for moderate-to-severe hidradenitis suppurativa. Semin. Cutan. Med. Surg. 2018, 37, 182–189. [Google Scholar] [CrossRef]
- Tegtmeyer, K.; Atassi, G.; Zhao, J.; Maloney, N.J.; Lio, P.A. Off-Label studies on anakinra in dermatology: A review. J. Dermatol. Treat. 2020, 1–14. [Google Scholar] [CrossRef]
- Leslie, K.S.; Tripathi, S.V.; Nguyen, T.V.; Pauli, M.; Rosenblum, M.D. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J. Am. Acad. Dermatol. 2014, 70, 243–251. [Google Scholar] [CrossRef]
- Tzanetakou, V.; Kanni, T.; Giatrakou, S.; Katoulis, A.; Papadavid, E.; Netea, M.G.; Dinarello, C.A.; Van Der Meer, J.W.M.; Rigopoulos, D.; Giamarellos-Bourboulis, E.J. Safety and Efficacy of Anakinra in Severe Hidradenitis Suppurativa: A Randomized Clinical Trial. JAMA Dermatol. 2016, 152, 52–59. [Google Scholar] [CrossRef]
- André, R.; Marescassier, H.; Gabay, C.; Pittet, B.; Laffitte, E. Long-term therapy with anakinra in hidradenitis suppurativa in three patients. Int. J. Dermatol. 2019, 58, e208–e209. [Google Scholar] [CrossRef]
- Russo, V.; Alikhan, A. Failure of Anakinra in a Case of Severe Hidradenitis Suppurativa. J. Drugs Dermatol. JDD 2016, 15, 772–774. [Google Scholar]
- Gram, H. The long and winding road in pharmaceutical development of canakinumab from rare genetic autoinflammatory syndromes to myocardial infarction and cancer. Pharmacol. Res. 2019, 154, 104139. [Google Scholar] [CrossRef]
- Houriet, C.; Jafari, S.M.S.; Thomi, R.; Schlapbach, C.; Borradori, L.; Yawalkar, N.; Hunger, R.E. Canakinumab for Severe Hidradenitis Suppurativa. JAMA Dermatol. 2017, 153, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, T.; Andres, C.; Grosber, M.; Zirbs, M.; Hein, R.; Ring, J.; Traidl-Hoffmann, C. Pyoderma gangrenosum and concomitant hidradenitis suppurativa—Rapid response to canakinumab (anti-IL-1β). Eur. J. Dermatol. EJD 2013, 23, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.Z.; Ro, T.; Jolly, P.; Sayed, C.J. Non-response to Interleukin-1 Antagonist Canakinumab in Two Patients with Refractory Pyoderma Gangrenosum and Hidradenitis Suppurativa. J. Clin. Aesthetic Dermatol. 2017, 10, 36–38. [Google Scholar]
- Tekin, B.; Salman, A.; Ergun, T. Hidradenitis suppurativa unresponsive to canakinumab treatment: A case report. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 615–617. [Google Scholar] [CrossRef]
- Kanni, T.; Argyropoulou, M.; Spyridopoulos, T.; Pistiki, A.; Stecher, M.; Dinarello, C.A.; Simard, J.; Giamarellos-Bourboulis, E.J. MABp1 Targeting IL-1α for Moderate to Severe Hidradenitis Suppurativa Not Eligible for Adalimumab: A Randomized Study. J. Investig. Dermatol. 2018, 138, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Kanni, T.; Argyropoulou, M.; Dinarello, C.A.; Simard, J.; Giamarellos-Bourboulis, E.J. MABp1 targeting interleukin-1α in hidradenitis suppurativa ineligible for adalimumab treatment: Results of the open-label extension period. Clin. Exp. Dermatol. 2020, 46, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Natsis, N.E.; Kerdel, F.; Forman, S.; Gonzalez, E.; Jimenez, G.; Hernandez, L.; Kaffenberger, J.; Guido, G.; Lucas, K.; et al. A Phase II Open-Label Study of Bermekimab in Patients with Hidradenitis Suppurativa Shows Resolution of Inflammatory Lesions and Pain. J. Investig. Dermatol. 2020, 140, 1538–1545.e2. [Google Scholar] [CrossRef] [PubMed]
- Giuseppe, P.; Nicola, P.; Valentina, C.; Elena, C.; Salvatrice, C.; Rosario, G.; Rita, B.M. A Case of Moderate Hidradenitis Suppurativa and Psoriasis Treated with Secukinumab. Ann. Dermatol. 2018, 30, 462–464. [Google Scholar] [CrossRef]
- Schuch, A.; Fischer, T.; Boehner, A.; Biedermann, T.; Volz, T. Successful Treatment of Severe Recalcitrant Hidradenitis Suppurativa with the Interleukin-17A Antibody Secukinumab. Acta Derm. Venereol. 2018, 98, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.-H.R.; Yao, Y.; Thomsen, S.F. Therapeutic Response to Secukinumab in a 36-Year-Old Woman with Hidradenitis Suppurativa. Case Rep. Dermatol. Med. 2018, 2018, 8685136. [Google Scholar] [CrossRef] [Green Version]
- Thorlacius, L.; Riis, P.T.; Jemec, G. Severe hidradenitis suppurativa responding to treatment with secukinumab: A case report. Br. J. Dermatol. 2018, 179, 182–185. [Google Scholar] [CrossRef]
- Głowaczewska, A.; Szepietowski, J.C.; Matusiak, Ł. Severe hidradenitis suppurativa successfully treated with secukinumab. Dermatol. Ther. 2020, 33, e13845. [Google Scholar] [CrossRef]
- Prussick, L.; Rothstein, B.; Joshipura, D.; Saraiya, A.; Turkowski, Y.; Abdat, R.; Alomran, A.; Zancanaro, P.; Kachuk, C.; Dumont, N.; et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitis suppurativa. Br. J. Dermatol. 2019, 181, 609–611. [Google Scholar] [CrossRef]
- Reguiaï, Z.; Fougerousse, A.; Maccari, F.; Bécherel, P. Effectiveness of secukinumab in hidradenitis suppurativa: An open study (20 cases). J. Eur. Acad. Dermatol. Venereol. 2020, 34, e750–e751. [Google Scholar] [CrossRef]
- Casseres, R.G.; Prussick, L.; Zancanaro, P.; Rothstein, B.; Joshipura, D.; Saraiya, A.; Turkowski, Y.; Au, S.C.; Alomran, A.; Abdat, R.; et al. Secukinumab in the treatment of moderate to severe hidradenitis suppurativa: Results of an open-label trial. J. Am. Acad. Dermatol. 2020, 82, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Ribero, S.; Ramondetta, A.; Fabbrocini, G.; Bettoli, V.; Potenza, C.; Chiricozzi, A.; Licciardello, M.; Marzano, A.; Bianchi, L.; Rozzo, G.; et al. Effectiveness of Secukinumab in the treatment of moderate–severe hidradenitis suppurativa: Results from an Italian multicentric retrospective study in a real-life setting. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e441–e442. [Google Scholar] [CrossRef] [PubMed]
- Marasca, C.; Megna, M.; Balato, A.; Balato, N.; Napolitano, M.; Fabbrocini, G. Secukinumab and hidradenitis suppurativa: Friends or foes? JAAD Case Rep. 2019, 5, 184–187. [Google Scholar] [CrossRef] [Green Version]
- Odorici, G.; Pellacani, G.; Conti, A. Ixekizumab in hidradenitis suppurativa in a psoriatic patient. G. Ital. Dermatol. Venereol. 2021, 155, 788–789. [Google Scholar] [CrossRef] [PubMed]
- Megna, M.; Ruggiero, A.; Di Guida, A.; Patrì, A.; Fabbrocini, G.; Marasca, C. Ixekizumab: An efficacious treatment for both psoriasis and hidradenitis suppurativa. Dermatol. Ther. 2020, 33, e13756. [Google Scholar] [CrossRef] [PubMed]
- Reardon, K.; Levin, J.; Levin, C. Severe hidradenitis suppurativa with herpes simplex virus 1 superinfection and clinical responsiveness to ixekizumab. JAAD Case Rep. 2021, 9, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Cotter, C.; Tobin, A.M.; O’Connor, R.; Gallagher, C.; Connolly, M. Severe refractory hidradenitis suppurativa: Treatment with ixekizumab, two case reports. Br. J. Dermatol. 2018, 179, 70. [Google Scholar]
- Frew, J.W.; Navrazhina, K.; Grand, D.; Sullivan-Whalen, M.; Gilleaudeau, P.; Garcet, S.; Ungar, J.; Krueger, J.G. The effect of subcutaneous brodalumab on clinical disease activity in hidradenitis suppurativa: An open-label cohort study. J. Am. Acad. Dermatol. 2020, 83, 1341–1348. [Google Scholar] [CrossRef]
- Arenbergerova, M.; Arenberger, P.; Marques, E.; Gkalpakiotis, S. Successful treatment of recalcitrant gluteal hidradenitis suppurativa with brodalumab after anti-TNF failure. Int. J. Dermatol. 2020, 59, 733–735. [Google Scholar] [CrossRef]
- Yoshida, Y.; Oyama, N.; Iino, S.; Shimizu, C.; Hasegawa, M. Long-standing refractory hidradenitis suppurativa responded to a brodalumab monotherapy in a patient with psoriasis: A possible involvement of Th17 across the spectrum of both diseases. J. Dermatol. 2021, 48, 916–920. [Google Scholar] [CrossRef]
- Frew, J.; Navrazhina, K.; Sullivan-Whalen, M.; Gilleaudeau, P.; Garcet, S.; Krueger, J. Weekly administration of brodalumab in hidradenitis suppurativa: An open-label cohort study. Br. J. Dermatol. 2020, 184, 350–352. [Google Scholar] [CrossRef]
- Oliveira, D.G.; Faria, R.; Torres, T. An Overview of Bimekizumab for the Treatment of Psoriatic Arthritis: The Evidence so Far. Drug Des. Dev. Ther. 2021, 15, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Efficacy, Safety, and Pharmacokinetics Study of cjm112 in Hidradenitis Suppurativa Patients. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02421172 (accessed on 1 August 2021).
- Gulliver, W.; Jemec, G.; Baker, K. Experience with ustekinumab for the treatment of moderate to severe Hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2011, 26, 911–914. [Google Scholar] [CrossRef]
- Blok, J.L.; Li, K.; Brodmerkel, C.; Horvatovich, P.; Jonkman, M.F.; Horváth, B. Ustekinumab in hidradenitis suppurativa: Clinical results and a search for potential biomarkers in serum. Br. J. Dermatol. 2016, 174, 839–846. [Google Scholar] [CrossRef]
- Montero-Vilchez, T.; Pozo-Román, T.; Sánchez-Velicia, L.; Vega-Gutiérrez, J.; Arias-Santiago, S.; Molina-Leyva, A. Ustekinumab in the treatment of patients with hidradenitis suppurativa: Multicenter case series and systematic review. J. Dermatol. Treat. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Romaní, J.; Vilarrasa, E.; Martorell, A.; Fuertes, I.; Ciudad, C.; Molina-Leyva, A. Ustekinumab with Intravenous Infusion: Results in Hidradenitis Suppurativa. Dermatology 2019, 236, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, E.M.; García-Ruiz, R.; Moneva-Léniz, L.M.; Mateu-Puchades, A. Effectiveness and safety of ustekinumab in patients with hidradenitis suppurativa using intravenous induction. Dermatol. Ther. 2020, 33, e14054. [Google Scholar] [CrossRef]
- Scholl, L.; Hessam, S.; Garcovich, S.; Bechara, F.G. High-dosage ustekinumab for the treatment of severe hidradenitis suppurativa. Eur. J. Dermatol. EJD 2019, 29, 659–661. [Google Scholar] [CrossRef]
- Takeda, K.; Kikuchi, K.; Kanazawa, Y.; Yamasaki, K.; Aiba, S. Ustekinumab treatment for hidradenitis suppurativa. J. Dermatol. 2019, 46, 1215–1218. [Google Scholar] [CrossRef]
- Valenzuela-Ubiña, S.; Jiménez-Gallo, D.; Villegas-Romero, I.; Rodríguez-Mateos, M.E.; Linares-Barrios, M. Effectiveness of ustekinumab for moderate-to-severe hidradenitis suppurativa: A case series. J. Dermatol. Treat. 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Provini, L.E.; Stellar, J.J.; Stetzer, M.N.; Nguyen, P.D.; Jen, M. Combination hyperbaric oxygen therapy and ustekinumab for severe hidradenitis suppurativa. Pediatr. Dermatol. 2019, 36, 381–383. [Google Scholar] [CrossRef]
- Berman, H.S.; Villa, N.M.; Shi, V.Y.; Hsiao, J.L. Guselkumab in the treatment of concomitant hidradenitis suppurativa, psoriasis, and Crohn’s disease. J. Dermatol. Treat. 2019, 32, 261–263. [Google Scholar] [CrossRef]
- Montero-Vilchez, T.; Martinez-Lopez, A.; Salvador-Rodriguez, L.; Arias-Santiago, S.; Molina-Leyva, A. The use of guselkumab 100 mg every 4 weeks on patients with hidradenitis suppurativa and a literature review. Dermatol. Ther. 2020, 33, e13456. [Google Scholar] [CrossRef] [PubMed]
- Casseres, R.G.; Kahn, J.S.; Her, M.J.; Rosmarin, D. Guselkumab in the treatment of hidradenitis suppurativa: A retrospective chart review. J. Am. Acad. Dermatol. 2019, 81, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Kearney, N.; Byrne, N.; Kirby, B.; Hughes, R. Successful use of guselkumab in the treatment of severe hidradenitis suppurativa. Clin. Exp. Dermatol. 2020, 45, 618–619. [Google Scholar] [CrossRef]
- Kovacs, M.; Podda, M. Guselkumab in the treatment of severe hidradenitis suppurativa. J. Eur. Acad. Dermatol. Venereol. 2018, 33, e140–e141. [Google Scholar] [CrossRef]
- Jørgensen, A.R.; Holm, J.G.; Thomsen, S.F. Guselkumab for hidradenitis suppurativa in a patient with concomitant Crohn’s disease: Report and systematic literature review of effectiveness and safety. Clin. Case Rep. 2020, 8, 2874–2877. [Google Scholar] [CrossRef]
- Burzi, L.; Repetto, F.; Ramondetta, A.; Rozzo, G.; Licciardello, M.; Ribero, S.; Quaglino, P.; Dapavo, P. Guselkumab in the treatment of severe hidradenitis suppurativa, a promising role? Dermatol. Ther. 2021, 34, e14930. [Google Scholar] [CrossRef]
- Marques, E.; Arenberger, P.; Smetanová, A.; Gkalpakiotis, S.; Zimová, D.; Arenbergerová, M. Successful treatment of recalcitrant hidradenitis suppurativa with risankizumab after failure of anti-tumour necrosis factor alpha. Br. J. Dermatol. 2020, 184, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Licata, G.; Gambardella, A.; Buononato, D.; De Rosa, A.; Calabrese, G.; Pellerone, S.; Argenziano, G. A case of moderate hidradenitis suppurativa and psoriasis successfully treated with risankizumab. Int. J. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kok, Y.; Nicolopoulos, J.; Howard, A.; Varigos, G.; Kern, J.; Dolianitis, C. Tildrakizumab in the treatment of moderate-to-severe hidradenitis suppurativa. Australas. J. Dermatol. 2020, 61, e488–e490. [Google Scholar] [CrossRef]
- Kok, Y.; Nicolopoulos, J.; Dolianitis, C. Tildrakizumab as a potential long-term therapeutic agent for severe Hidradenitis Suppurativa: A 15 months experience of an Australian institution. Australas. J. Dermatol. 2021, 62, e313–e316. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.; Argyropoulou, M.; Kanni, T.; Spyridopoulos, T.; Otto, I.; Zenker, O.; Guo, R.; Riedemann, N. Clinical efficacy of complement C5a inhibition by IFX-1 in hidradenitis suppurativa: An open-label single-arm trial in patients not eligible for adalimumab. Br. J. Dermatol. 2020, 183, 176–178. [Google Scholar] [CrossRef]
- Schafer, P.H.; Parton, A.; Gandhi, A.K.; Capone, L.; Adams, M.; Wu, L.; Bartlett, J.B.; A Loveland, M.; Gilhar, A.; Cheung, Y.-F.; et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br. J. Pharmacol. 2009, 159, 842–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossen, A.R.; van Doorn, M.B.; van der Zee, H.H.; Prens, E.P. Apremilast for moderate hidradenitis suppurativa: Results of a randomized controlled trial. J. Am. Acad. Dermatol. 2018, 80, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Aarts, P.; Vossen, A.R.; van der Zee, H.H.; Prens, E.P.; van Straalen, K.R. Long-term treatment with apremilast in hidradenitis suppurativa: A 2-year follow-up of initial responders. J. Am. Acad. Dermatol. 2021, 85, 258–260. [Google Scholar] [CrossRef]
- Weber, P.; Jafari, S.M.S.; Yawalkar, N.; Hunger, R.E. Apremilast in the treatment of moderate to severe hidradenitis suppurativa: A case series of 9 patients. J. Am. Acad. Dermatol. 2017, 76, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Garcovich, S.; Giovanardi, G.; Malvaso, D.; De Simone, C.; Peris, K. Apremilast for the treatment of hidradenitis suppurativa associated with psoriatic arthritis in multimorbid patients: Case report and review of literature. Medicine 2020, 99, e18991. [Google Scholar] [CrossRef]
- Lanna, C.; Mazzilli, S.; Zangrilli, A.; Bianchi, L.; Campione, E. One drug and two diseases: A case of multidrug-resistant hidradenitis suppurativa and psoriasis treated with apremilast. Dermatol. Ther. 2019, 32, e13089. [Google Scholar] [CrossRef]
- McKay, C.; Kuraitis, D.; Murina, A. Serum cytokine levels in patients with hidradenitis suppurativa vary with race. J. Am. Acad. Dermatol. 2020, 84, 1405–1406. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Boniotto, M.; Genovese, G.; Zouboulis, C.C.; Marzano, A.V.; Crovella, S. An Integrated Approach to Unravel Hidradenitis Suppurativa Etiopathogenesis. Front. Immunol. 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- Walss, M.; Anzengruber, F.; Arafa, A.; Djamei, V.; Navarini, A.A. Implementing Medical Chatbots: An Application on Hidradenitis Suppurativa. Dermatology 2021, 237, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Gomolin, A.; Netchiporouk, E.; Gniadecki, R.; Litvinov, I.V. Artificial Intelligence Applications in Dermatology: Where Do We Stand? Front. Med. 2020, 7, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Intervention | Target | Primary Outcome | NCT Number | Phase | Status |
|---|---|---|---|---|---|
| CFZ533 (MA) LYS006 (SM) Placebo | CD40 LTA4H | HiSCR at 16 weeks | NCT03827798 [155] | Phase 2 | Recruiting |
| Risankizumab (MA) Placebo | IL-23 | HiSCR at 16 weeks | NCT03926169 [156] | Phase 2 | Active, not recruiting |
| Brodalumab (MA) | IL-17RA | IL-17RA saturation at week 12 | NCT04979520 [157] | Early Phase 1 | Recruiting |
| Bimekizumab (MA) Placebo | IL-17A IL-17F | HiSCR at 16 weeks | NCT04242446 [158] | Phase 3 | Recruiting |
| Bimekizumab (MA) Placebo | IL-17A IL-17F | HiSCR at 16 weeks | NCT04242498 [159] | Phase 3 | Recruiting |
| Bimekizumab (MA) | IL-17A IL-17F | Percentage of participants with TEAEs up to week 120 | NCT04901195 [160] | Phase 3 | Enrolling by invitation |
| Upadacitinib (SM) Placebo | JAK1/2 | HiSCR at 12 weeks | NCT04430855 [161] | Phase 2 | Active, not recruiting |
| INCB054707 (SM) Placebo | JAK1 | Mean change in total AN count at 16 weeks | NCT04476043 [162] | Phase 2 | Recruiting |
| Orismilast (SM) | PDE4 | Percent change in AN count at 16 weeks | NCT04982432 [163] | Phase 2 | Not yet recruiting |
| Secukinumab (MA) | IL-17A | Time to LOR in HiSCR responders at weeks 52–104 | NCT04179175 [164] | Phase 3 | Recruiting |
| Secukinumab (MA) Placebo | IL-17A | HiSCR at 16 weeks | NCT03713619 [165] | Phase 3 | Active, not recruiting |
| Secukinumab (MA) Placebo | IL-17A | HiSCR at 16 weeks | NCT03713632 [166] | Phase 3 | Active, not recruiting |
| Spesolimab (MA) Placebo | IL-36R | Percent change in total AN count at 12 weeks | NCT04762277 [167] | Phase 2 | Recruiting |
| Spesolimab (MA) | IL-36R | Occurrence TEAEs up to week 120 | NCT04876391 [168] | Phase 2 | Not yet recruiting |
| Imsidolimab (MA) Placebo solution | IL-36R | Change in AN count at 16 weeks | NCT04856930 [169] | Phase 2 | Not yet recruiting |
| LY3041658 (MA) Placebo | ELR+CXC chemokine family | HiSCR at 16 weeks | NCT04493502 [170] | Phase 2 | Recruiting |
| PF-06650833 (SM) PF-06700841 (SM) PF-06826647 (SM) Placebo | IRAK4 JAK1 TYK2 TYK2 | HiSCR at 16 weeks | NCT04092452 [171] | Phase 2 | Recruiting |
| KT-474 (SM) | IRAK4 | Incidence and severity of TEAEs up to 28 days | NCT04772885 [172] | Phase 1 | Recruiting |
| Guselkumab | IL-23 | Changes in levels of cytokines in the skin at week 0 and week 16 | NCT04061395 [173] | Phase 2 | Not yet recruiting |
| Avacopan (SM) Placebo | C5aR | HiSCR at 12 weeks | NCT03852472 [174] | Phase 2 | Active, not recruiting |
| Recombinant anti G-CSF receptor (MA) | G-CSF receptor | Incidence of TEAEs (up to 24 weeks) Incidence of AESIs | NCT03972280 [175] | Phase 1 | Recruiting |
| Adalimumab (MA) Monotherapy Adalimumab + surgery | TNF-α | Cost-utility, 2 years | NCT03221621 [176] | Phase 4 | Recruiting |
| Tofacitinib (SM) | JAK1 JAK3 | Number of SAEs up to week 18 Change in IFN scores (Baseline and 16 weeks) | NCT04246372 [177] | Phase 2 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosi, E.; Fastame, M.T.; Scandagli, I.; Di Cesare, A.; Ricceri, F.; Pimpinelli, N.; Prignano, F. Insights into the Pathogenesis of HS and Therapeutical Approaches. Biomedicines 2021, 9, 1168. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091168
Rosi E, Fastame MT, Scandagli I, Di Cesare A, Ricceri F, Pimpinelli N, Prignano F. Insights into the Pathogenesis of HS and Therapeutical Approaches. Biomedicines. 2021; 9(9):1168. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091168
Chicago/Turabian StyleRosi, Elia, Maria Thais Fastame, Ilaria Scandagli, Antonella Di Cesare, Federica Ricceri, Nicola Pimpinelli, and Francesca Prignano. 2021. "Insights into the Pathogenesis of HS and Therapeutical Approaches" Biomedicines 9, no. 9: 1168. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091168