
Decline in Processing Speed Tells Only Half the Story: Developmental Delay in Children Living with Sickle Cell Disease

Abstract
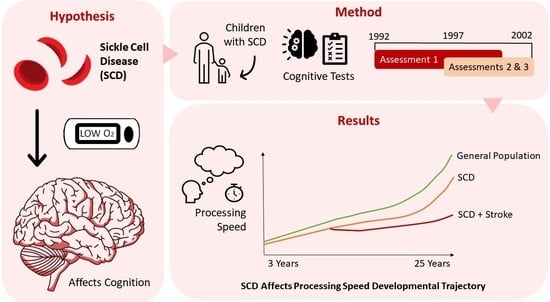
:
1. Introduction
- (1)
- Processing Speed Index (PSI) and individual subtests scores (i.e., Coding and Symbol Search) will be higher for children than for young people living with SCD, providing evidence of decline.
- (2)
- Children with stroke or SCI will have lower PSI and subtest scores than children without infarction detected on MRI.
- (3)
- Children living with SCD will have lower PSI and subtest scores than the normative mean, and there will be an interaction between age and timepoint. Specifically, children will have higher scores at timepoint 1 than at timepoint 3, providing evidence of delay and subsequent decline.
2. Materials and Methods
2.1. The East London Sickle Cell Disease Cohort
2.2. Exclusion Criteria
2.3. Cognitive Assessment
2.4. Magnetic Resonance Imaging
2.5. Statistical Analyses
3. Results
3.1. Longitudinal Change in Processing Speed
3.1.1. Linear Mixed-Effects Regression without Infarct Status Included
3.1.2. Linear Mixed-Effects Regression with Infarct Status Included
4. Discussion
4.1. Limitations
4.2. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-Cell Disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Moser, F.G.; Miller, S.T.; Bello, J.A.; Pegelow, C.H.; Zimmerman, R.A.; Wang, W.C.; Ohene-Frempong, K.; Schwartz, A.; Vichinsky, E.P.; Gallagher, D.; et al. The Spectrum of Brain MR Abnormalities in Sickle-Cell Disease: A Report from the Cooperative Study of Sickle Cell Disease. AJNR Am. J. Neuroradiol. 1996, 17, 965–972. [Google Scholar]
- Powers, W.J.; Grubb, R.L.; Darriet, D.; Raichle, M.E. Cerebral Blood Flow and Cerebral Metabolic Rate of Oxygen Requirements for Cerebral Function and Viability in Humans. J. Cereb. Blood Flow Metab. 1985, 5, 600–608. [Google Scholar] [CrossRef]
- Ford, A.L.; Ragan, D.K.; Fellah, S.; Binkley, M.M.; Fields, M.E.; Guilliams, K.P.; An, H.; Jordan, L.C.; McKinstry, R.C.; Lee, J.-M.; et al. Silent Infarcts in Sickle Cell Disease Occur in the Border Zone Region and Are Associated with Low Cerebral Blood Flow. Blood 2018, 132, 1714–1723. [Google Scholar] [CrossRef]
- Powars, D.; Wilson, B.; Imbus, C.; Pegelow, C.; Allen, J. The Natural History of Stroke in Sickle Cell Disease. Am. J. Med. 1978, 65, 461–471. [Google Scholar] [CrossRef]
- Wang, W.; Enos, L.; Gallagher, D.; Thompson, R.; Guarini, L.; Vichinsky, E.; Wright, E.; Zimmerman, R.; Armstrong, F.D. Neuropsychologic Performance in School-Aged Children with Sickle Cell Disease: A Report from the Cooperative Study of Sickle Cell Disease. J. Pediatr. 2001, 139, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.T.; Davis, P.C.; Lambert, R.; Hsu, L.; Hopkins, K.; Eckman, J. Neurocognitive Functioning and Magnetic Resonance Imaging in Children with Sickle Cell Disease. J. Pediatr. Psychol. 2000, 25, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.J.; Bills, S.E.; Wise, S.M.; Hardy, K.K. Cognitive Abilities Moderate the Effect of Disease Severity on Health-Related Quality of Life in Pediatric Sickle Cell Disease. J. Pediatr. Psychol. 2018, 43, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Hood, A.M.; King, A.A.; Fields, M.E.; Ford, A.L.; Guilliams, K.P.; Hulbert, M.L.; Lee, J.; White, D.A. Higher Executive Abilities Following a Blood Transfusion in Children and Young Adults with Sickle Cell Disease. Pediatr. Blood Cancer 2019, 66, e27899. [Google Scholar] [CrossRef] [PubMed]
- Stotesbury, H.; Kirkham, F.J.; Kölbel, M.; Balfour, P.; Clayden, J.D.; Sahota, S.; Sakaria, S.; Saunders, D.E.; Howard, J.; Kesse-Adu, R.; et al. White Matter Integrity and Processing Speed in Sickle Cell Anemia. Neurology 2018, 90, e2042–e2050. [Google Scholar] [CrossRef] [PubMed]
- Schatz, J.; Brown, R.T.; Pascual, J.M.; Hsu, L.; DeBaun, M.R. Poor School and Cognitive Functioning with Silent Cerebral Infarcts and Sickle Cell Disease. Neurology 2001, 56, 1109–1111. [Google Scholar] [CrossRef]
- Crawford, R.D.; Jonassaint, C.R. Adults with Sickle Cell Disease May Perform Cognitive Tests as Well as Controls When Processing Speed Is Taken into Account: A Preliminary Case-Control Study. J. Adv. Nurs. 2016, 72, 1409–1416. [Google Scholar] [CrossRef]
- Prussien, K.V.; DeBaun, M.R.; Yarboi, J.; Bemis, H.; McNally, C.; Williams, E.; Compas, B.E. Cognitive Function, Coping, and Depressive Symptoms in Children and Adolescents with Sickle Cell Disease. J. Pediatr. Psychol. 2018, 43, 543–551. [Google Scholar] [CrossRef]
- Case, M.; Zhang, H.; Mundahl, J.; Datta, Y.; Nelson, S.; Gupta, K.; He, B. Characterization of Functional Brain Activity and Connectivity Using EEG and FMRI in Patients with Sickle Cell Disease. Neuroimage Clin. 2017, 14, 1–17. [Google Scholar] [CrossRef]
- Hood, A.M.; Kölbel, M.; Stotesbury, H.; Kawadler, J.; Slee, A.; Inusa, B.; Pelidis, M.; Howard, J.; Chakravorty, S.; Height, S.; et al. Biopsychosocial Predictors of Quality of Life in Paediatric Patients with Sickle Cell Disease. Front. Psychol. 2021, 12, 3976. [Google Scholar] [CrossRef]
- Hood, A.M.; Crosby, L.E.; Stotesbury, H.; Kölbel, M.; Kirkham, F.J. Considerations for Selecting Cognitive Endpoints and Psychological Patient-Reported Outcomes for Clinical Trials in Pediatric Patients with Sickle Cell Disease. Front. Neurol. 2022, 13, 835823. [Google Scholar] [CrossRef] [PubMed]
- van der Land, V.; Hijmans, C.T.; de Ruiter, M.; Mutsaerts, H.J.M.M.; Cnossen, M.H.; Engelen, M.; Majoie, C.B.L.M.; Nederveen, A.J.; Grootenhuis, M.A.; Fijnvandraat, K. Volume of White Matter Hyperintensities Is an Independent Predictor of Intelligence Quotient and Processing Speed in Children with Sickle Cell Disease. Br. J. Haematol. 2015, 168, 553–556. [Google Scholar] [CrossRef]
- Kail, R. Developmental Change in Speed of Processing during Childhood and Adolescence. Psychol. Bull. 1991, 109, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, S.; Fize, D.; Marlot, C. Speed of Processing in the Human Visual System. Nature 1996, 381, 520–522. [Google Scholar] [CrossRef]
- Wechsler, D. Manual for the Wechsler Intelligence Scale for Children—Third UK Edition (WISC-III UK); Psychological Corporation: San Antonio, TX, USA, 1991. [Google Scholar]
- Wechsler, D. WAIS-III: Wechsler Adult Intelligence Scale—Third Edition; Pearson: London, UK, 1997. [Google Scholar]
- Wechsler, D. WAIS-R: Wechsler Adult Intelligence Scale—Revised; Psychological Corporation Harcourt Brace Jovanovich: San Antonio, TX, USA, 1981. [Google Scholar]
- Wechsler, D. WPPSI-R: Wechsler Preschool and Primary Scale of Intelligence—Revised; Psychological Corporation: San Antonio, TX, USA, 1989. [Google Scholar]
- Akshoomoff, N.; Brown, T.T.; Bakeman, R.; Hagler, D.J. Developmental Differentiation of Executive Functions on the NIH Toolbox Cognition Battery. Neuropsychology 2018, 32, 777–783. [Google Scholar] [CrossRef] [PubMed]
- McAuley, T.; White, D.A. A Latent Variables Examination of Processing Speed, Response Inhibition, and Working Memory during Typical Development. J. Exp. Child Psychol. 2011, 108, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P. Assessment and Development of Executive Function (EF) During Childhood. Child Neuropsychol. 2002, 8, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Crowe, L.M.; Dooley, J.; Collie, A.; Davis, G.; McCrory, P.; Clausen, H.; Maddocks, D.; Anderson, V. Developmental Trajectory of Information-Processing Skills in Children: Computer-Based Assessment. Appl. Neuropsychol. Child 2016, 5, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Koelbel, M.; Hamdule, S.; Kirkham, F.J.; Stotesbury, H.; Hood, A.M.; Dimitriou, D. Mind the Gap: Trajectory of Cognitive Development in Young Individuals with Sickle Cell Disease: A Cross-Sectional Study. Front. Neurol. 2023, 14, 1087054. [Google Scholar] [CrossRef] [PubMed]
- Kawadler, J.M.; Clayden, J.D.; Clark, C.A.; Kirkham, F.J. Intelligence Quotient in Paediatric Sickle Cell Disease: A Systematic Review and Meta-Analysis. Dev. Med. Child Neurol. 2016, 58, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Anderson, L.M.; Rothman, J.A.; Bonner, M.J. Executive Functioning and Health-Related Quality of Life in Pediatric Sickle Cell Disease. Child Neuropsychol. 2017, 23, 889–906. [Google Scholar] [CrossRef]
- Prussien, K.V.; Jordan, L.C.; DeBaun, M.R.; Compas, B.E. Cognitive Function in Sickle Cell Disease Across Domains, Cerebral Infarct Status, and the Lifespan: A Meta-Analysis. J. Pediatr. Psychol. 2019, 44, 948–958. [Google Scholar] [CrossRef]
- Downes, M.; Kirkham, F.J.; Telfer, P.T.; De Haan, M. Assessment of Executive Functions in Preschool Children with Sickle Cell Anemia. J. Int. Neuropsychol. Soc. 2018, 24, 949–954. [Google Scholar] [CrossRef]
- Brugulat-Serrat, A.; Salvadó, G.; Operto, G.; Cacciaglia, R.; Sudre, C.H.; Grau-Rivera, O.; Suárez-Calvet, M.; Falcon, C.; Sánchez-Benavides, G.; Gramunt, N.; et al. White Matter Hyperintensities Mediate Gray Matter Volume and Processing Speed Relationship in Cognitively Unimpaired Participants. Hum. Brain Mapp. 2020, 41, 1309–1322. [Google Scholar] [CrossRef]
- Adams, R.J. Stroke Prevention and Treatment in Sickle Cell Disease. Arch. Neurol. 2001, 58, 565–568. [Google Scholar] [CrossRef]
- Scantlebury, N.; Mabbott, D.; Janzen, L.; Rockel, C.; Widjaja, E.; Jones, G.; Kirby, M.; Odame, I. White Matter Integrity and Core Cognitive Function in Children Diagnosed with Sickle Cell Disease. J. Pediatr. Hematol. Oncol. 2011, 33, 163–171. [Google Scholar] [CrossRef]
- Watkins, K.E.; Hewes, D.K.M.; Connelly, A.; Kendall, B.E.; Kingsley, D.P.E.; Evans, J.E.P.; Gadian, D.G.; Vargha-Khadem, F.; Kirkham, F.J. Cognitive Deficits Associated with Frontal-Lobe Infarction in Children with Sickle Cell Disease. Dev. Med. Child Neurol. 1998, 40, 536–543. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Statistics for Windows 2020; IBM Corp.: Rochester, MN, USA, 2020. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Weiler, M.D.; Forbes, P.; Kirkwood, M.; Waber, D. The Developmental Course of Processing Speed in Children with and without Learning Disabilities. J. Exp. Child Psychol. 2003, 85, 178–194. [Google Scholar] [CrossRef]
- Martin, R.; Sawrie, S.; Gilliam, F.; Mackey, M.; Faught, E.; Knowlton, R.; Kuzniekcy, R. Determining Reliable Cognitive Change after Epilepsy Surgery: Development of Reliable Change Indices and Standardized Regression-based Change Norms for the WMS-III and WAIS-III. Epilepsia 2002, 43, 1551–1558. [Google Scholar] [CrossRef]
- Duff, K. Evidence-Based Indicators of Neuropsychological Change in the Individual Patient: Relevant Concepts and Methods. Arch. Clin. Neuropsychol. 2012, 27, 248–261. [Google Scholar] [CrossRef]
- Groth-Marnat, G.; Wright, J. Context of Clinical Assessment. In Handbook of Psychological Assessment; Wiley: Hoboken, NJ, USA, 2016; pp. 37–68. [Google Scholar]
- Kail, R. Speed of Information Processing: Developmental Change and Links to Intelligence. J. Sch. Psychol. 2000, 38, 51–61. [Google Scholar] [CrossRef]
- Prussien, K.V.; Siciliano, R.E.; Ciriegio, A.E.; Anderson, A.S.; Sathanayagam, R.; DeBaun, M.R.; Jordan, L.C.; Compas, B.E. Correlates of Cognitive Function in Sickle Cell Disease: A Meta-Analysis. J. Pediatr. Psychol. 2020, 45, 145–155. [Google Scholar] [CrossRef]
- Smith, K.E.; Schatz, J. Working Memory in Children with Neurocognitive Effects from Sickle Cell Disease: Contributions of the Central Executive and Processing Speed. Dev. Neuropsychol. 2016, 41, 231–244. [Google Scholar] [CrossRef]
- Milne, S.; McDonald, J.; Comino, E.J. The Use of the Bayley Scales of Infant and Toddler Development III with Clinical Populations: A Preliminary Exploration. Phys. Occup. Ther. Pediatr. 2012, 32, 24–33. [Google Scholar] [CrossRef]
- Kavšek, M. Predicting Later IQ from Infant Visual Habituation and Dishabituation: A Meta-Analysis. J. Appl. Dev. Psychol. 2004, 25, 369–393. [Google Scholar] [CrossRef]
- Schatz, J. Cognitive Functioning in Children with Sickle Cell Disease: A Meta-Analysis. J. Pediatr. Psychol. 2002, 27, 739–748. [Google Scholar] [CrossRef] [PubMed]
- McMorris, T.; Hale, B.J.; Barwood, M.; Costello, J.; Corbett, J. Effect of Acute Hypoxia on Cognition: A Systematic Review and Meta-Regression Analysis. Neurosci. Biobehav. Rev. 2017, 74, 225–232. [Google Scholar] [CrossRef]
- Bush, A.M.; Borzage, M.T.; Choi, S.; Václavů, L.; Tamrazi, B.; Nederveen, A.J.; Coates, T.D.; Wood, J.C. Determinants of Resting Cerebral Blood Flow in Sickle Cell Disease. Am. J. Hematol. 2016, 91, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Schatz, J.; Finke, R.; Roberts, C.W. Interactions of Biomedical and Environmental Risk Factors for Cognitive Development. J. Dev. Behav. Pediatr. 2004, 25, 303–310. [Google Scholar] [CrossRef]
- Bills, S.E.; Katz, T.; McNeil, J.; Schatz, J. Does Obstructive Sleep Apnea Increase Cognitive Deficits in Pediatric Sickle Cell Disease? J. Int. Neuropsychol. Soc. 2019, 25, 922–930. [Google Scholar] [CrossRef]
- Fields, M.E.; Mirro, A.E.; Guilliams, K.P.; Binkley, M.M.; Gil Diaz, L.; Tan, J.; Fellah, S.; Eldeniz, C.; Chen, Y.; Ford, A.L.; et al. Functional Connectivity Decreases with Metabolic Stress in Sickle Cell Disease. Ann. Neurol. 2020, 88, 995–1008. [Google Scholar] [CrossRef]
- Choi, S.; Bush, A.M.; Borzage, M.T.; Joshi, A.A.; Mack, W.J.; Coates, T.D.; Leahy, R.M.; Wood, J.C. Hemoglobin and Mean Platelet Volume Predicts Diffuse T1-MRI White Matter Volume Decrease in Sickle Cell Disease Patients. Neuroimage Clin. 2017, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Puffer, E.; Schatz, J.; Roberts, C.W. The Association of Oral Hydroxyurea Therapy with Improved Cognitive Functioning in Sickle Cell Disease. Child Neuropsychol. 2007, 13, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Oluwole, O.; Owusu-Ansah, A.; Nouraie, S.M.; Novelli, E.M. The Impact of Hydroxyurea Use on Cognitive Functioning of Children with Sickle Cell Disease in Ghana. Blood 2020, 136, 22. [Google Scholar] [CrossRef]
- Tarazi, R.A.; Patrick, K.E.; Iampietro, M.; Apollonsky, N. Hydroxyurea Use Associated with Nonverbal and Executive Skills in Sickle Cell Anemia. J. Pediatr. Psychol. 2021, 46, 710–718. [Google Scholar] [CrossRef]
- Heitzer, A.M.; Longoria, J.; Okhomina, V.; Wang, W.C.; Raches, D.; Potter, B.; Jacola, L.M.; Porter, J.; Schreiber, J.E.; King, A.A.; et al. Hydroxyurea Treatment and Neurocognitive Functioning in Sickle Cell Disease from School Age to Young Adulthood. Br. J. Haematol. 2021, 195, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Wailoo, K. Sickle Cell Disease—A History of Progress and Peril. N. Engl. J. Med. 2017, 376, 805–807. [Google Scholar] [CrossRef] [PubMed]
- Prussien, K.V.; Patel, D.A.; Wilkerson, K.; Armstrong, B.; Karnik, L.; la Fuente, J.; Kassim, A.A. Improvement in Processing Speed Following Haploidentical Bone Marrow Transplant with Posttransplant Cytoxan in Children and Adolescents with Sickle Cell Disease. Pediatr. Blood Cancer 2020, 67, e28001. [Google Scholar] [CrossRef]
- Wang, W.C.; Zou, P.; Hwang, S.N.; Kang, G.; Ding, J.; Heitzer, A.M.; Schreiber, J.E.; Helton, K.; Hankins, J.S. Effects of Hydroxyurea on Brain Function in Children with Sickle Cell Anemia. Pediatr. Blood Cancer 2021, 68, e29254. [Google Scholar] [CrossRef]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P. Methodological Issues in Group-Matching Designs: Alpha Levels for Control Variable Comparisons and Measurement Characteristics of Control and Target Variables. J. Autism Dev. Disord. 2004, 34, 7–17. [Google Scholar] [CrossRef]
- Hood, A.M.; Reife, I.; King, A.A.; White, D.A. Brief Screening Measures Identify Risk for Psychological Difficulties Among Children with Sickle Cell Disease. J. Clin. Psychol. Med. Settings 2020, 27, 651–661. [Google Scholar] [CrossRef]
- Lorig, K.R.; Holman, H.R. Self-Management Education: History, Definition, Outcomes, and Mechanisms. Ann. Behav. Med. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Jones, K.E.; Nyman, T.M.; Daly, B.P.; Jacobson, L.A.; Tarazi, R.A. Executive Functioning Predicts Adaptive Functioning and Self-Care Independence in Pediatric Sickle Cell Disease. J. Pediatr. Psychol. 2022, 47, 206–214. [Google Scholar] [CrossRef]
- Heitzer, A.M.; Hamilton, L.; Stafford, C.; Gossett, J.; Ouellette, L.; Trpchevska, A.; King, A.A.; Kang, G.; Hankins, J.S. Academic Performance of Children with Sickle Cell Disease in the United States: A Meta-Analysis. Front. Neurol. 2021, 12, 786065. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| ≤8.99 Years (n = 58) | >9 Years (n = 45) | Total (n = 103) | p Value | |
|---|---|---|---|---|
| Age | ||||
| M (SD) | 6.2 (1.5) | 12.6 (2.2) | 9.01 (3.7) | |
| Baseline Intelligence Quotient | 0.03 | |||
| M (SD) | 85.67 (12.54) | 80.11 (13.01) | 83.18 (12.99) | |
| Range | 49–111 | 55–107 | 49–111 | |
| Baseline Processing speed Index | ||||
| M (SD) | 88.55 (17.09) | 81.57 (15.78) | 85.47 (16.77) | 0.08 |
| Range | 50-130 | 54-132 | 50-132 | |
| Sex | 0.73 | |||
| Female | 25 (43.10%) | 17 (37.77%) | 42 (40.78%) | |
| Male | 33 (56.90%) | 28 (62.22%) | 61 (59.22%) | |
| Genotype | 0.68 | |||
| HbSC | 9 (15.52%) | 5 (11.11%) | 14 (13.59%) | |
| HbSS | 45 (77.59%) | 38 (84.44%) | 83 (80.58%) | |
| HbS Thalassaemia | 4 (6.90%) | 2 (4.44%) | 6 (5.83%) | |
| MRI Infarct Status | 0.40 | |||
| No Infarct | 20 (34.50%) | 20 (44.44%) | 40 (38.83%) | |
| SCI | 11 (18.97%) | 10 (22.22%) | 21 (20.39%) | |
| Stroke | 13 (22.41%) | 6 (13.33%) | 19 (18.45%) | |
| No MRI available | 14 (24.14%) | 9 (20.00%) | 23 (22.33%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, E.J.; Kirkham, F.J.; Hood, A.M. Decline in Processing Speed Tells Only Half the Story: Developmental Delay in Children Living with Sickle Cell Disease. Children 2024, 11, 277. https://0-doi-org.brum.beds.ac.uk/10.3390/children11030277
Walker EJ, Kirkham FJ, Hood AM. Decline in Processing Speed Tells Only Half the Story: Developmental Delay in Children Living with Sickle Cell Disease. Children. 2024; 11(3):277. https://0-doi-org.brum.beds.ac.uk/10.3390/children11030277
Chicago/Turabian StyleWalker, Elise Jade, Fenella Jane Kirkham, and Anna Marie Hood. 2024. "Decline in Processing Speed Tells Only Half the Story: Developmental Delay in Children Living with Sickle Cell Disease" Children 11, no. 3: 277. https://0-doi-org.brum.beds.ac.uk/10.3390/children11030277