Role of the Gut–Liver Axis in Driving Parenteral Nutrition-Associated Injury

 ,
, {kind=link}
Abstract
:1. Introduction
2. Evidence Supporting the Importance of Enteral Nutrition and Luminal Content
2.1. Hepatic Injury
2.2. Gut Injury
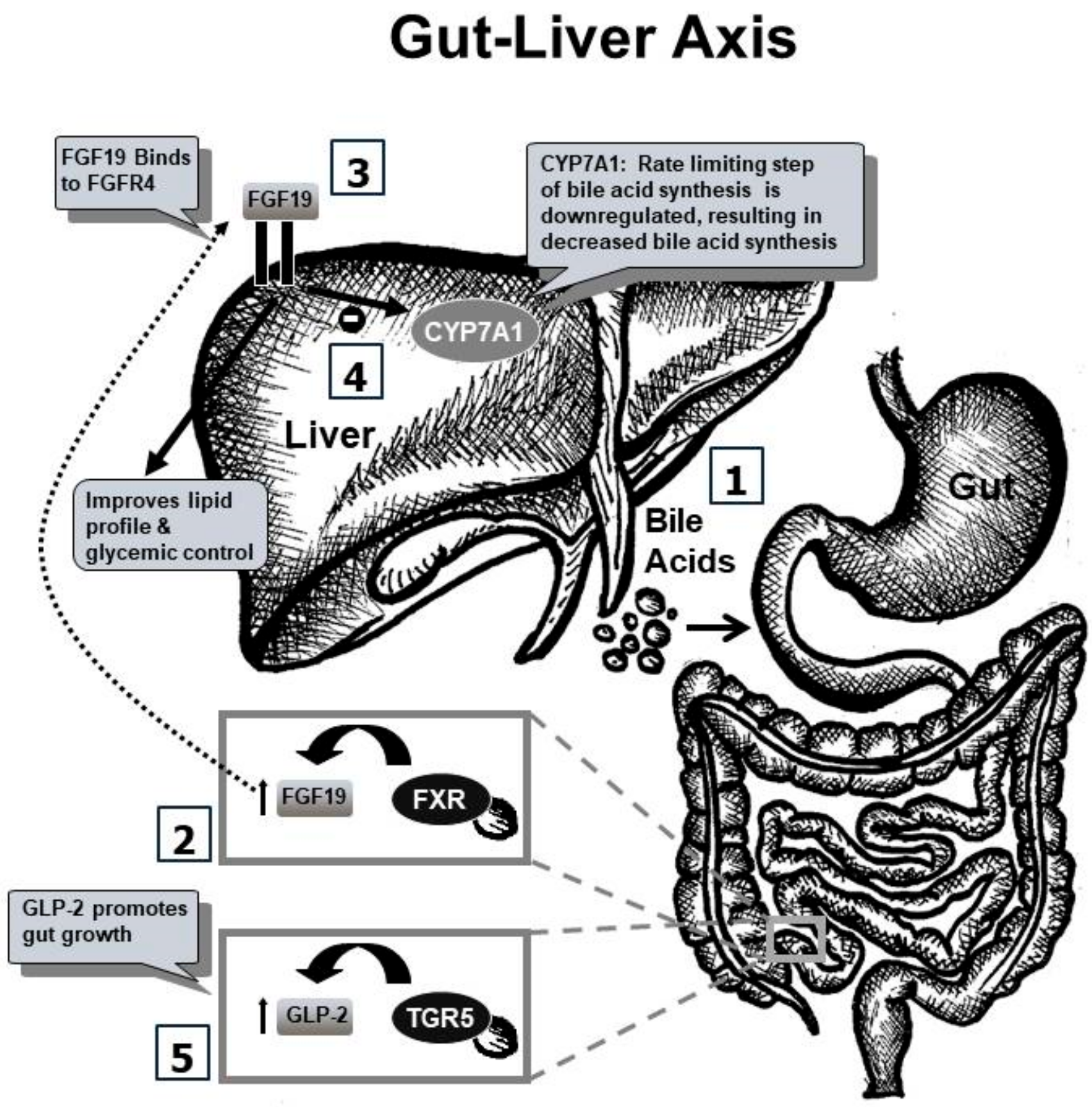
3. Gut–Liver Axis
3.1. Portal System
3.2. Gut Microbiota
3.2.1. Human Gut Microbiota
3.2.2. TPN and Modulation of Gut Microbiota
3.3. Hepatobiliary Receptors and Transporters
3.3.1. FXR-FGF19 Axis
3.3.2. TGR5–GLP Axis
3.4. GLP-2 and EGF
4. Expert Commentary: Five-Year View and Conclusion
5. Key Issues
- Parenteral nutrition (PN) has been a successful method for the intravenous delivery of nutrients and remains an essential therapy for individuals with intolerance of enteral feedings or impaired gut function. PN provides nourishment via the intravenous administration of nutritional components, such as amino acids, glucose, vitamins, minerals, and lipids. When nutritional needs are solely met via such intravenous therapy, the resulting process is called total parenteral nutrition (TPN).
- TPN remains an essential modality of nutrition delivery worldwide and is commonly used in neonates and pediatric and adult patients with lost or impaired gut function. TPN use has grown exponentially over the last few decades.
- Despite the several benefits of TPN therapy, its use is associated with adverse effects. Parenteral nutrition-associated liver disease (PNALD) is a cholestatic liver disease, which can also present with hepatic inflammation, steatosis, dyslipidemia, glucose intolerance, and/or fibrosis. Significant gut mucosal atrophy has also been shown to occur in association with TPN infusion in several animal studies.
- The pathophysiology and etiology of TPN-associated injury remains largely unknown despite several theories.
- Research into ameliorative therapies and mechanistic pathways continues to be a major focus in the field of gastroenterology and hepatology.
- Recent studies reveal that an alteration in gut-derived signals may occur with administration of TPN in the absence of enteral nutrition. This hypothesis is driven by the observation that hepatic injury is decreased if even a small percentage of nutrition can be provided enterally.
- Based on the concept that gut-derived signals, stimulated by intraluminal nutrients, can maintain liver health, it is hypothesized that in the state of TPN, the enterohepatic axis is disrupted, thus leading to injury.
- Current data, including work from our lab, support the theory that altered gut–liver crosstalk with TPN and bile acids have emerged as key regulators of injury. In fact, treatment with bile acid receptor agonists seems to ameliorate TPN-associated injury in animal models.
- Translating research from bench to clinical targeting of the gut-derived signaling appears to be key in developing novel approaches in mitigating TPN-associated injury and restoring the effectiveness of this lifesaving therapy.
Author Contributions
Funding
Conflicts of Interest
References
- Dudrick, S.J.; Palesty, J.A. Historical highlights of the development of total parenteral nutrition. Surg. Clin. N. Am. 2011, 91, 693–717. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.; Kadam, S. Total parenteral nutrition in neonates. Indian Pediatr. 2006, 43, 953–964. [Google Scholar] [PubMed]
- Dudrick, S.J. Early developments and clinical applications of total parenteral nutrition. J. Parenter. Enter. Nutr. 2003, 27, 291–299. [Google Scholar] [CrossRef] [PubMed]
- DiBaise, J.K.; Scolapio, J.S. Home parenteral and enteral nutrition. Gastroenterol. Clin. N. Am 2007, 36, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.; Malone, M. Clinical outcome of geriatric patients in the United States receiving home parenteral and enteral nutrition. Am. J. Clin. Nutr. 1997, 66, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vantini, I.; Benini, L.; Bonfante, F.; Talamini, G.; Sembenini, C.; Chiarioni, G.; Maragnolli, O.; Benini, F.; Capra, F. Survival rate and prognostic factors in patients with intestinal failure. Dig. Liver Dis. 2004, 36, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Sigalet, D.L. Short bowel syndrome in infants and children: An overview. Semin. Pediatr. Surg. 2001, 10, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ganousse-Mazeron, S.; Lacaille, F.; Colomb-Jung, V.; Talbotec, C.; Ruemmele, F.; Sauvat, F.; Chardot, C.; Canioni, D.; Jan, D.; Revillon, Y.; et al. Assessment and outcome of children with intestinal failure referred for intestinal transplantation. Clin. Nutr. 2015, 34, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Villalona, G.; Price, A.; Blomenkamp, K.S.; Manithody, C.; Ratchford, T.; Saxena, S.; Westrich, M.; Kakarla, V.; Pochampally, S.; Phillips, W.; et al. No gut no gain! Enteral bile acid treatment preserves gut growth but is not protective for parenteral nutrition associated liver injury in a novel extensive short bowel animal model. J. Parenter. Enter. Nutr. 2018, 1–14. [Google Scholar] [CrossRef]
- Kumpf, V.J. Parenteral nutrition-associated liver disease in adult and pediatric patients. Nutr. Clin. Pract. 2006, 21, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tomar, B.S. Hepatobiliary abnormalities and parenteral nutrition. Indian J. Pediatr. 2000, 67, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Yonekura, T.; Hoki, M.; Oyanagi, H.; Kawahara, H.; Yagi, M.; Imura, K.; Iiboshi, Y.; Wasa, K.; Kamata, S.; et al. Total parenteral nutrition-associated intrahepatic cholestasis in infants: 25 years’ experience. J. Pediatr. Surg. 2000, 35, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Wen, J.X.; Blomenkamp, K.S.; Arora, S.; Blaufuss, T.A.; Rodrigues, J.; Long, J.P.; Neuschwander-Tetri, B.A.; Teckman, J.H. Oleanolic Acid Improves Gut Atrophy Induced by Parenteral Nutrition. J. Parenter. Enter. Nutr. 2016, 40, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Niinikoski, H.; Stoll, B.; Guan, X.; Kansagra, K.; Lambert, B.D.; Stephens, J.; Hartmann, B.; Holst, J.J.; Burrin, D.G. Onset of small intestinal atrophy is associated with reduced intestinal blood flow in TPN-fed neonatal piglets. J. Nutr. 2004, 134, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Ekelund, M.; Kristensson, E.; Ekelund, M.; Ekblad, E. Total parenteral nutrition causes circumferential intestinal atrophy, remodeling of the intestinal wall, and redistribution of eosinophils in the rat gastrointestinal tract. Dig. Dis. Sci. 2007, 52, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Javid, P.J.; Collier, S.; Richardson, D.; Iglesias, J.; Gura, K.; Lo, C.; Kim, H.B.; Duggan, C.P.; Jaksic, T. The role of enteral nutrition in the reversal of parenteral nutrition-associated liver dysfunction in infants. J. Pediatr. Surg. 2005, 40, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Stoll, B.; Burrin, D.G.; Holst, J.J.; Moore, D.D. Enteral bile acid treatment improves parenteral nutrition-related liver disease and intestinal mucosal atrophy in neonatal pigs. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G218–G224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.A.; Taylor, O.A.; Prendergast, D.R.; Zimmerman, T.L.; Von Furstenberg, R.; Moore, D.D.; Karpen, S.J. Stigmasterol, a soy lipid-derived phytosterol, is an antagonist of the bile acid nuclear receptor FXR. Pediatr. Res. 2007, 62, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Gohel, T.; Deen, O.J.; DeChicco, R.; Shatnawei, A. Fish oil-based lipid emulsion: Current updates on a promising novel therapy for the management of parenteral nutrition-associated liver disease. Gastroenterol. Rep. 2015, 3, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.A.; Shulman, R.J. Mechanisms of disease: Update on the molecular etiology and fundamentals of parenteral nutrition associated cholestasis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. 2011, 205–259. [Google Scholar] [CrossRef] [Green Version]
- Drongowski, R.A.; Coran, A.G. An analysis of factors contributing to the development of total parenteral nutrition-induced cholestasis. J. Parenter. Enter. Nutr. 1989, 13, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Repa, A.; Lochmann, R.; Unterasinger, L.; Weber, M.; Berger, A.; Haiden, N. Aggressive nutrition in extremely low birth weight infants: Impact on parenteral nutrition associated cholestasis and growth. PeerJ 2016, 4, e2483. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A. Total parenteral nutrition-associated liver disease. J. Parenter. Enter. Nutr. 2002, 26 (Suppl. 5), S43–S48. [Google Scholar] [CrossRef] [PubMed]
- Burrin, D.; Stoll, B.; Moore, D. Digestive physiology of the pig symposium: Intestinal bile acid sensing is linked to key endocrine and metabolic signaling pathways. J. Anim. Sci. 2013, 91, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Wen, J.X.; Arora, S.; Blomenkamp, K.S.; Rodrigues, J.; Blaufuss, T.A.; Liou, V.; Burrin, D.G.; Long, J.P.; Teckman, J.H. Validating hyperbilirubinemia and gut mucosal atrophy with a novel ultramobile ambulatory total parenteral nutrition piglet model. Nutr. Res. 2015, 35, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Moukarzel, A.A.; Bhuta, S.; Belle, M.; Ament, M.E.; Eckhert, C.D.; Hollander, D.; Gornbein, J.; Kopple, J.D.; Vijayaroghavan, S.R. Parenteral nutrition is associated with intestinal morphological and functional changes in humans. J. Parenter. Enter. Nutr. 1995, 19, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Omata, J.; Fukatsu, K.; Murakoshi, S.; Noguchi, M.; Miyazaki, H.; Moriya, T.; Okamoto, K.; Fukazawa, S.; Akase, T.; Saitoh, D. Enteral refeeding rapidly restores PN-induced reduction of hepatic mononuclear cell number through recovery of small intestine and portal vein blood flows. J. Parenter. Enter. Nutr. 2009, 33, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Sharma, A.; Arora, S.; Blomenkamp, K.S.; Jun, I.; Luong, R.; Westrich, D.J.; Mittal, A.; Buchanan, P.M.; Guzman, M.A.; et al. Preserved gut microbial diversity accompanies upregulation of TGR5 and hepatobiliary transporters in bile acid treated animals on Parenteral Nutrition. J. Parenter. Enter. Nutr. 2017, 41, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Bull, M.J.; Plummer, N.T. Part 1: The Human Gut Microbiome in Health and Disease. Integr. Med. 2014, 13, 17–22. [Google Scholar]
- Morowitz, M.J.; Carlisle, E.M.; Alverdy, J.C. Contributions of intestinal bacteria to nutrition and metabolism in the critically ill. Surg. Clin. N. Am. 2011, 91, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Angenent, L.T.; Gordon, J.I. Getting a grip on things: How do communities of bacterial symbionts become established in our intestine? Nat. Immunol. 2004, 5, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.J.; Lorenz, R.G. Gut Microbiota and Obesity. Curr. Obes. Rep. 2012, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roychowdhury, S.; Selvakumar, P.C.; Cresci, G.A.M. The Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease. Med. Sci. 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Hodin, C.M.; Visschers, R.G.; Rensen, S.S.; Boonen, B.; Olde Damink, S.W.; Lenaerts, K.; Buurman, W.A. Total parenteral nutrition induces a shift in the Firmicutes to Bacteroidetes ratio in association with Paneth cell activation in rats. J. Nutr. 2012, 142, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, A.F.; Pierre, J.F.; Tandee, K.; Shanmuganayagam, D.; Wang, X.; Reed, J.D.; Steele, J.L.; Kudsk, K.A. Parenteral Nutrition Decreases Paneth cell function and Intestinal Bactericidal Activity while Increasing Susceptibility to Bacterial Enteroinvasion. J. Parenter. Enter. Nutr. 2014, 38, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, F.; Li, Y.; Wang, J.; Li, J. Fecal microbiota signatures of adult patients with different types of short bowel syndrome. J. Gastroenterol. Hepatol. 2017, 32, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Joly, F.; Mayeur, C.; Bruneau, A.; Noordine, M.L.; Meylheuc, T.; Langella, P.; Messing, B.; Duée, P.H.; Cherbuy, C.; Thomas, M. Drastic changes in fecal and mucosa-associated microbiota in adult patients with short bowel syndrome. Biochimie 2010, 92, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Engelstad, H.; Barron, L.; Moen, J.; Wylie, T.; Wylie, K.; Rubin, D.; Davidson, N.; Cade, W.; Warner, B.; Warner, B. Remnant small bowel length in pediatric short bowel syndrome and the correlation with intestinal dysbiosis and linear growth. J. Am. Coll. Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powell, J.; Mathioudakis, N.; Kane, S.; Fernandez, E.; Sears, C.L. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-κB pathway. Infect. Immun. 2004, 72, 5832–5839. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.Y.; Yoo, D.; Sim, Y.S.; Kim, Y.J.; Oh, Y.K.; Kang, J.S.; Kim, S.; Kim, J.S.; Kim, J.M. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I κB kinase/NF-κB-dependent pathway. Infect. Immun. 2010, 78, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Shiryaev, S.A.; Strongin, A.Y. Distinct interactions with cellular E-cadherin of the two virulent metalloproteinases encoded by a Bacteroides fragilis pathogenicity island. PLoS ONE 2014, 9, e113896. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lim, K.C.; Huang, J.; Saidi, R.F.; Sears, C.L. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E.-cadherin. Proc. Natl. Acad. Sci. USA 1998, 95, 14979–14984. [Google Scholar] [CrossRef] [PubMed]
- Green, R.M.; Beier, D.; Gollan, J.L. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology 1996, 111, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Alrefai, W.A.; Gill, R.K. Bile acid transporters: Structure, function, regulation and pathophysiological implications. Pharm. Res. 2007, 24, 1803–1823. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Arrese, M.; Lee, H.; Boyer, J.L.; Karpen, S.J. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J. Clin. Investig. 1998, 101, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Whiting, J.F.; Green, R.M.; Rosenbluth, A.B.; Gollan, J.L. Tumor necrosis factor-alpha decreases hepatocyte bile salt uptake and mediates endotoxin-induced cholestasis. Hepatology 1995, 22 Pt 1, 1273–1278. [Google Scholar]
- Zheng, Y.J.; Tam, Y.K.; Coutts, R.T. Endotoxin and cytokine released during parenteral nutrition. J. Parenter. Enter. Nutr. 2004, 28, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, G.L.; Carreras, F.I.; Soria, L.R.; Gradilone, S.A.; Marinelli, R.A. LPS induces the TNF-alpha-mediated downregulation of rat liver aquaporin-8: Role in sepsis-associated cholestasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G567–G575. [Google Scholar] [CrossRef] [PubMed]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R.B. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Freund, H.R.; Muggia-Sullam, M.; LaFrance, R.; Enrione, E.B.; Popp, M.B.; Bjornson, H.S. A possible beneficial effect of metronidazole in reducing TPN-associated liver function derangements. J. Surg. Res. 1985, 38, 356–363. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Yoshitake, M.; Kumemura, H.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Kumashiro, R.; Sata, M. Abnormal accumulation in lipopolysaccharide in biliary epithelial cells of rats with self-filling blind loop. Int. J. Mol. Med. 2002, 9, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, M.N.; Widlak, M.; Bhala, N.; Moore, D.; Price, M.; Sharma, N.; Iqbal, T.H. Systematic review with meta-analysis: The efficacy of faecal microbiota transplantation for the treatment of recurrent and refractory Clostridium difficile infection. Aliment. Pharmacol. Ther. 2017, 46, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Boesjes, M.; Brufau, G. Metabolic effects of bile acids in the gut in health and disease. Curr. Med. Chem. 2014, 21, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, Y.K.; Bundman, D.; Han, Y.; Thevananther, S.; Kim, C.-S.; Chua, S.S.; Wei, P.; Heyman, R.A.; Karin, M.; et al. Redundant pathways for negative feedback regulation of bile acid production. Dev. Cell 2002, 2, 721–731. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutanen, A.; Lohi, J.; Heikkila, P.; Jalanko, H.; Pakarinen, M.P. Loss of ileum decreases serum fibroblast growth factor 19 in relation to liver inflammation and fibrosis in pediatric onset intestinal failure. J. Hepatol. 2015, 62, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Lundasen, T.; Galman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, P.B.; Pertkiewicz, M.; Messing, B.; Iyer, K.; Seidner, D.L.; O’keefe, S.J.D.; Forbes, A.; Heinze, H.; Joelsson, B. Teduglutide reduces need for parenteral support among patients with short bowel syndrome with intestinal failure. Gastroenterology 2012, 143, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Mutanen, A.; Pakarinen, M.P. Serum fasting GLP-1 and GLP-2 associate with intestinal adaptation in pediatric onset intestinal failure. Clin. Nutr. 2017, 36, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Augustin, O.; Sanchez de Medina, F. Intestinal bile acid physiology and pathophysiology. World J. Gastroenterol. 2008, 14, 5630–5640. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Hirata, S.; Inoue, K.; Mashima, H.; Ohnishi, H.; Yoshiba, M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem. Biophys. Res. Commun. 2007, 354, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Sato, H.; Gioiello, A.; Costantino, G.; Macchiarulo, A.; Sadeghpour, B.M.; Giorgi, G.; Schoonjans, K.; Auwerx, J. Nongenomic actions of bile acids. Synthesis and preliminary characterization of 23- and 6,23-alkyl-substituted bile acid derivatives as selective modulators for the G-protein coupled receptor TGR5. J. Med. Chem. 2007, 50, 4265–4268. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Bradley, D.; Setchell, K.D.; Eagon, J.C.; Abumrad, N.; Klein, S. Weight loss induced by Roux-en-Y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J. Clin. Endocrinol. Metab. 2013, 98, E708–E712. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The role of gut hormones in glucose homeostasis. J. Clin. Investig. 2007, 117, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, P.A.; Goodlad, R.A.; FitzGerald, A.J.; Mandir, N.; Ghatei, M.A.; Bloom, S.R.; Berlanga-Acosta, J.; Playford, R.J.; Forbes, A.; Walters, J.R.F. Intestinal growth in parenterally-fed rats induced by the combined effects of glucagon-like peptide 2 and epidermal growth factor. J. Parenter. Enter. Nutr. 2005, 29, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Demehri, F.R.; Xiao, W.; Tsai, Y.-H.; Jones, J.C.; Brindley, C.D.; Threadgill, D.W.; Holst, J.J.; Hartmann, B.; Barrett, T.A.; et al. Interdependency of EGF and GLP-2 Signaling in Attenuating Mucosal Atrophy in a Mouse Model of Parenteral Nutrition. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, H.; Yang, S.; Li, Z.; Zhong, J.; Fang, R. Epidermal Growth Factor and Intestinal Barrier Function. Mediat. Inflamm. 2016, 2016, 1927348. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denton, C.; Price, A.; Friend, J.; Manithody, C.; Blomenkamp, K.; Westrich, M.; Kakarla, V.; Phillips, W.; Krebs, J.; Munoz Abraham, A.S.; et al. Role of the Gut–Liver Axis in Driving Parenteral Nutrition-Associated Injury. Children 2018, 5, 136. https://0-doi-org.brum.beds.ac.uk/10.3390/children5100136
Denton C, Price A, Friend J, Manithody C, Blomenkamp K, Westrich M, Kakarla V, Phillips W, Krebs J, Munoz Abraham AS, et al. Role of the Gut–Liver Axis in Driving Parenteral Nutrition-Associated Injury. Children. 2018; 5(10):136. https://0-doi-org.brum.beds.ac.uk/10.3390/children5100136
Chicago/Turabian StyleDenton, Christine, Amber Price, Julie Friend, Chandrashekhara Manithody, Keith Blomenkamp, Matthew Westrich, Vindhya Kakarla, William Phillips, Joseph Krebs, Armando Salim Munoz Abraham, and et al. 2018. "Role of the Gut–Liver Axis in Driving Parenteral Nutrition-Associated Injury" Children 5, no. 10: 136. https://0-doi-org.brum.beds.ac.uk/10.3390/children5100136