
Wiedemann–Steiner Syndrome with a Pathogenic Variant in KMT2A from Taiwan

 ,
,  , and
, and
Abstract
:1. Introduction
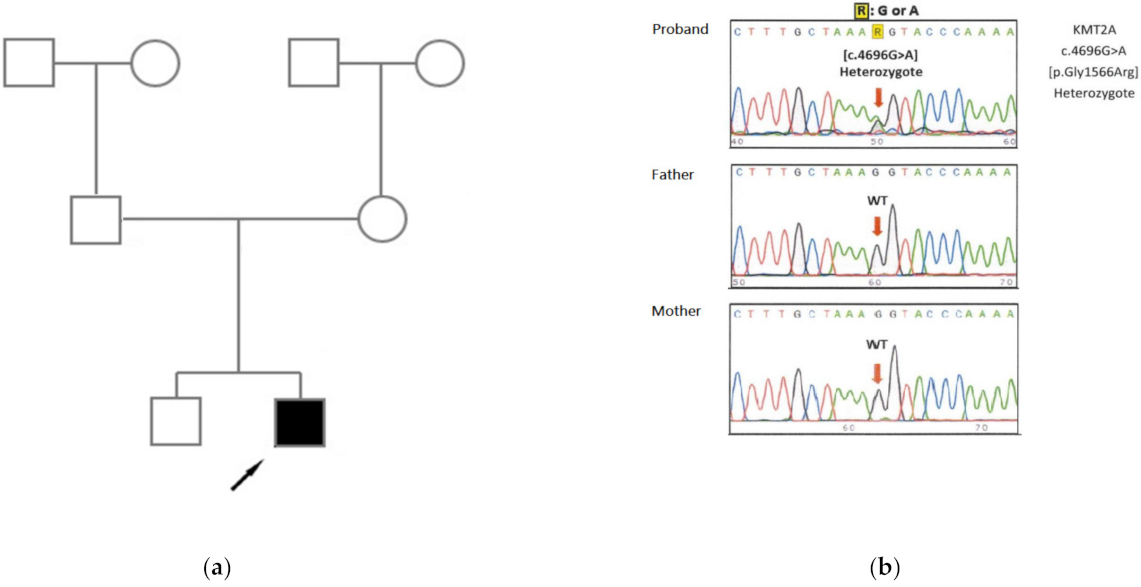
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wiedemann, H.R.; Kunze, J.; Dibbern, H. Atlas of Clinical Syndromes, 2nd ed.; Wolfe Publishing Ltd.: London, UK, 1989; pp. 198–199. [Google Scholar]
- Steiner, C.E.; Marques, A.P. Growth deficiency, mental retardation and unusual facies. Clin. Dysmorphol. 2000, 9, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Baer, S.; Afenjar, A.; Smol, T.; Piton, A.; Gérard, B.; Alembik, Y.; Bienvenu, T.; Boursier, G.; Boute, O.; Colson, C.; et al. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clin. Genet. 2018, 94, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.C.; et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 2012, 91, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, K.I.; Mishra, B.P.; Mandal, S.S. MLL histone methylases in gene expression, hormone signaling and cell cycle. Front. Biosci. 2009, 14, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Puri, R.D.; Bijarnia-Mahay, S.; Verma, I.C. Expanding the phenotypic and genotypic spectrum of Wiedemann-Steiner syndrome: First patient from India. Am. J. Med. Genet. Part A 2020, 182, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.J.S.; Cytrynbaum, C.; Hoang, N.; Ambrozewicz, P.M.; Weksberg, R.; Drmic, I.; Ritzema, A.; Schachar, R.; Walker, S.; Uddin, M.; et al. Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. NPJ Genom. Med. 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, R.; Wu, C.; Li, X.; Wang, Y. A novel de novo mutation (p.Pro1310Glnfs*46) in KMT2A caused Wiedemann-Steiner Syndrome in a Chinese boy with postnatal growth retardation: A case report. Mol. Biol. Rep. 2019, 46, 5555–5559. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.R.; Dlouhy, S.R.; Lah, M.D.; Payne, K.K.; Weaver, D.D. The progression of Wiedemann-Steiner syndrome in adulthood and two novel variants in the KMT2A gene. Am. J. Med. Genet. Part A 2019, 179, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Grangeia, A.; Leao, M.; Moura, C.P. Wiedemann-Steiner syndrome in two patients from Portuga. Am. J. Med. Genet. Part A 2020, 182, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Jinxiu, L.; Shuimei, L.; Ming, X.; Jonathan, L.C.S.; Xiangju, L.; Wenyuan, D. Wiedemann-Steiner syndrome with a de novo mutation in KMT2A: A case report. Medicine 2020, 99, e19813. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Yang, Y.; Wang, P.; Huang, H.; Xiong, S.; Sun, L.; Cheng, M.; Song, C.; Cheng, X.; et al. Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J. Rare Dis. 2018, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Montano, D.; Pachajoa, H. Wiedemann-Steiner syndrome with a novel pathogenic variant in KMT2A: A case report. Colomb. Med. 2019, 50, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyle, G.; Banka, S.; Langley, C.; Jones, E.A.; Banerjee, I. Growth hormone deficiency as a cause for short stature in Wiedemann-Steiner Syndrome. Endocrinol. Diabetes Metab. Case Rep. 2018, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Koenig, R.; Meinecke, P.; Kuechler, A.; Schäfer, D.; Müller, D. Wiedemann-Steiner syndrome: Three further cases. Am. J. Med. Genet. Part A 2010, 152, 2372–2375. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Rodriguez-Buritica, D.F.; Northrup, H. Wiedemann-Steiner syndrome: Novel pathogenic variant and review of literature. Eur. J. Med. Genet. 2017, 60, 285–288. [Google Scholar] [CrossRef]
- Demir, S.; Gürkan, H.; Öz, V.; Yalçıntepe, S.; Atlı, E.I.; Atlı, E. Wiedemann-Steiner Syndrome as a Differential Diagnosis of Cornelia de Lange Syndrome Using Targeted Next-Generation Sequencing: A Case Report. Mol. Syndromol. 2021, 12, 46–51. [Google Scholar]
- Strom, S.P.; Lozano, R.; Lee, H.; Dorrani, N.; Mann, J.; O’Lague, P.F.; Mans, N.; Deignan, J.L.; Vilain, E.; Nelson, S.F.; et al. De Novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med. Genet. 2014, 15, 49. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, G.; Lu, Y.; Luo, X.; Wu, W. Trio-WES reveals a novel de novo missense mutation of KMT2A in a Chinese patient with Wiedemann-Steiner syndrome: A case report. Mol. Genet. Genom. Med. 2021, 9, e1533. [Google Scholar]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Enokizono, T.; Ohto, T.; Tanaka, R.; Tanaka, M.; Suzuki, H.; Sakai, A.; Imagawa, K.; Fukushima, H.; Iwabuti, A.; Fukushima, T.; et al. Preaxial polydactyly in an individual with Wiedemann-Steiner syndrome caused by a novel nonsense mutation in KMT2A. Am. J. Med. Genet. Part A 2017, 173, 2821–2825. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Subspecialist | Considerations/Screening | Evaluation |
|---|---|---|
| Development | Language delay and intellectual disability | Regular speech and occupational therapies |
| Neurology | Hypotonia | Carnitine supplementation |
| Endocrinology | Growth retardation | Pituitary magnetic resonance imaging scan and recombinant human growth hormone treatment |
| Cardiology | Cardiac anomalies | Surgical treatment |
| Geneticist | KMT2A gene analysis and genetic counseling | Diagnostic discussion and family planning |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-L.; Chuang, C.-K.; Chiu, H.-C.; Tu, R.-Y.; Lo, Y.-T.; Chang, Y.-H.; Lin, H.-Y.; Lin, S.-P. Wiedemann–Steiner Syndrome with a Pathogenic Variant in KMT2A from Taiwan. Children 2021, 8, 952. https://0-doi-org.brum.beds.ac.uk/10.3390/children8110952
Lee C-L, Chuang C-K, Chiu H-C, Tu R-Y, Lo Y-T, Chang Y-H, Lin H-Y, Lin S-P. Wiedemann–Steiner Syndrome with a Pathogenic Variant in KMT2A from Taiwan. Children. 2021; 8(11):952. https://0-doi-org.brum.beds.ac.uk/10.3390/children8110952
Chicago/Turabian StyleLee, Chung-Lin, Chih-Kuang Chuang, Huei-Ching Chiu, Ru-Yi Tu, Yun-Ting Lo, Ya-Hui Chang, Hsiang-Yu Lin, and Shuan-Pei Lin. 2021. "Wiedemann–Steiner Syndrome with a Pathogenic Variant in KMT2A from Taiwan" Children 8, no. 11: 952. https://0-doi-org.brum.beds.ac.uk/10.3390/children8110952