
Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism

 , , , ,
, , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Samples
2.2. DNA Extraction and Bisulfite Conversion
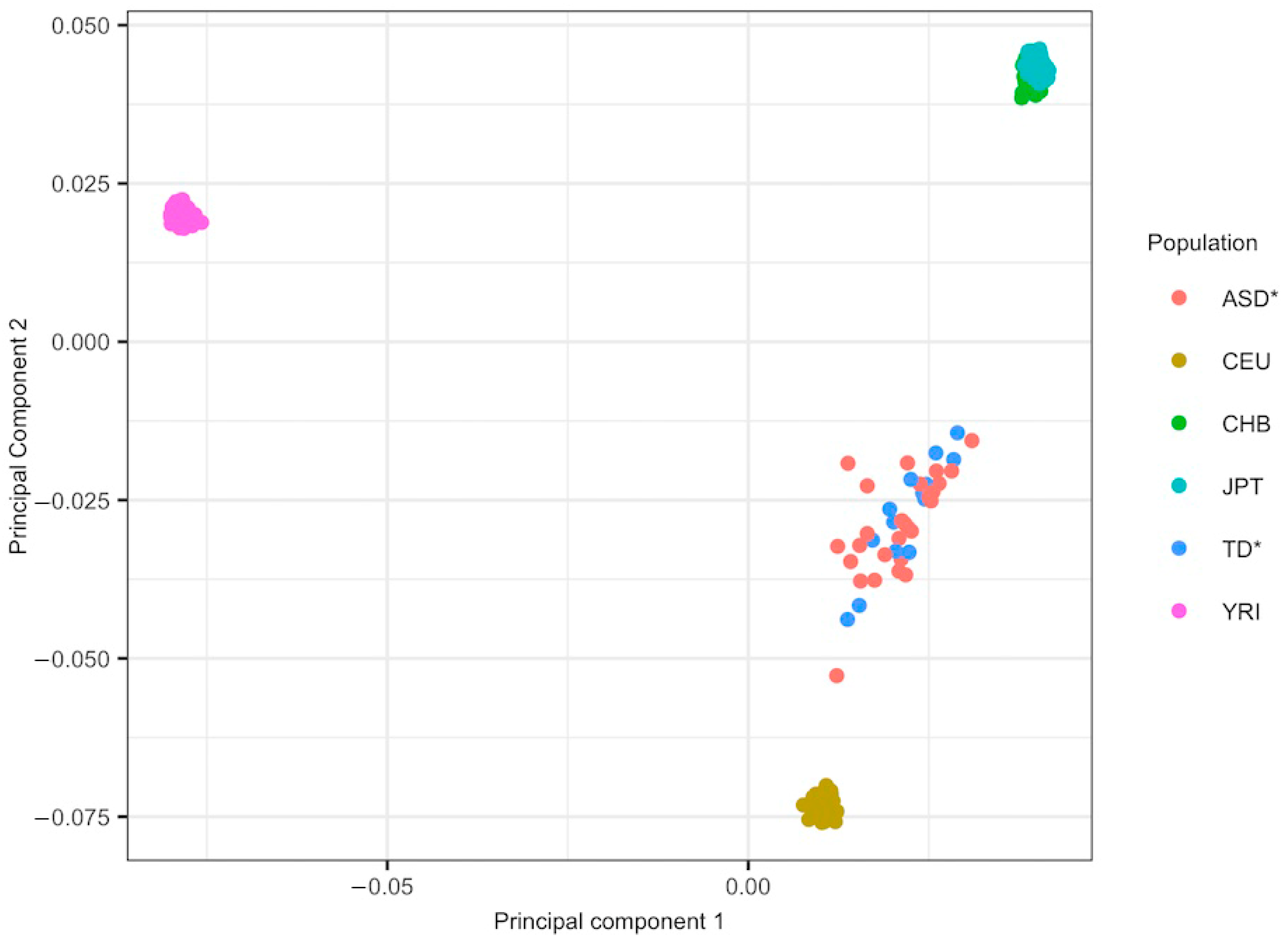
2.3. Ancestry Analysis
2.4. Genome-Wide Analysis of Differential DNA Methylation
3. Results
3.1. Clinical and Demographic Data
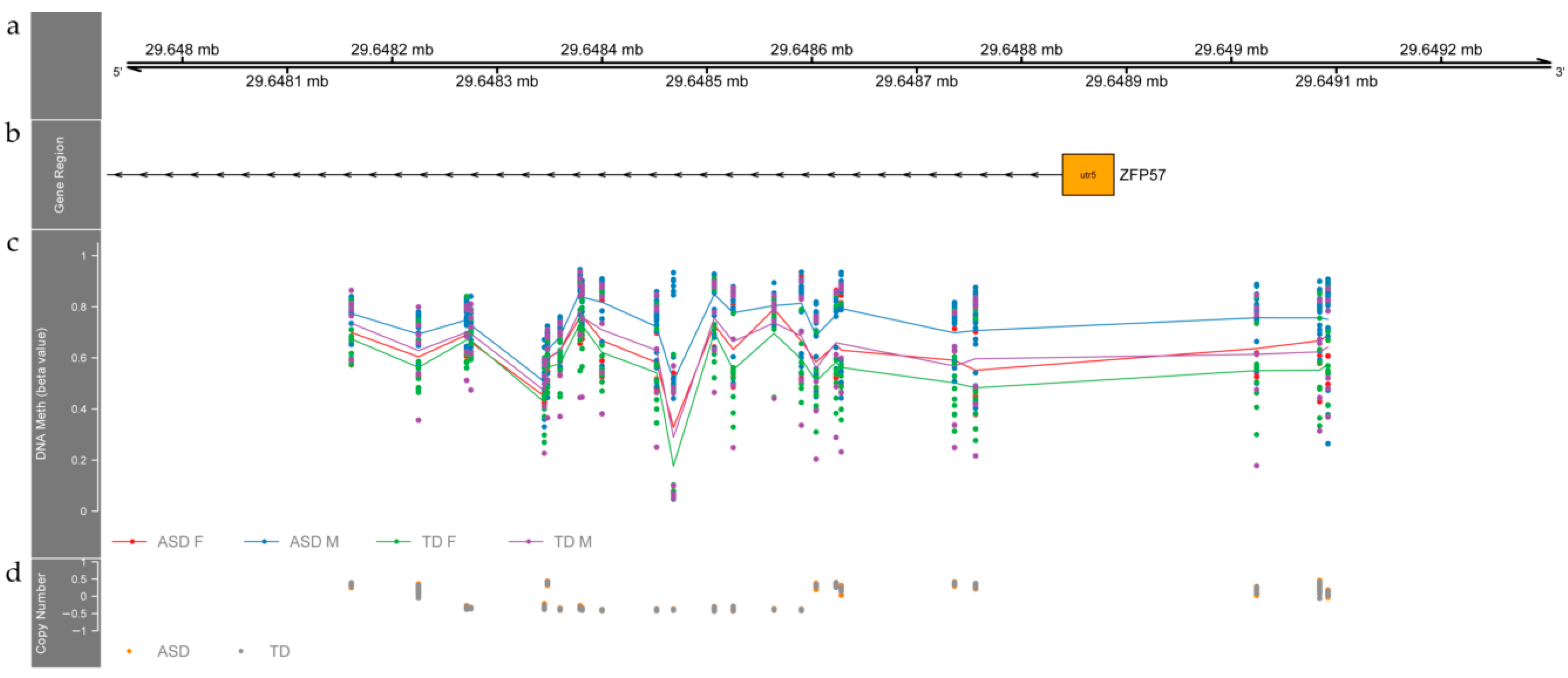
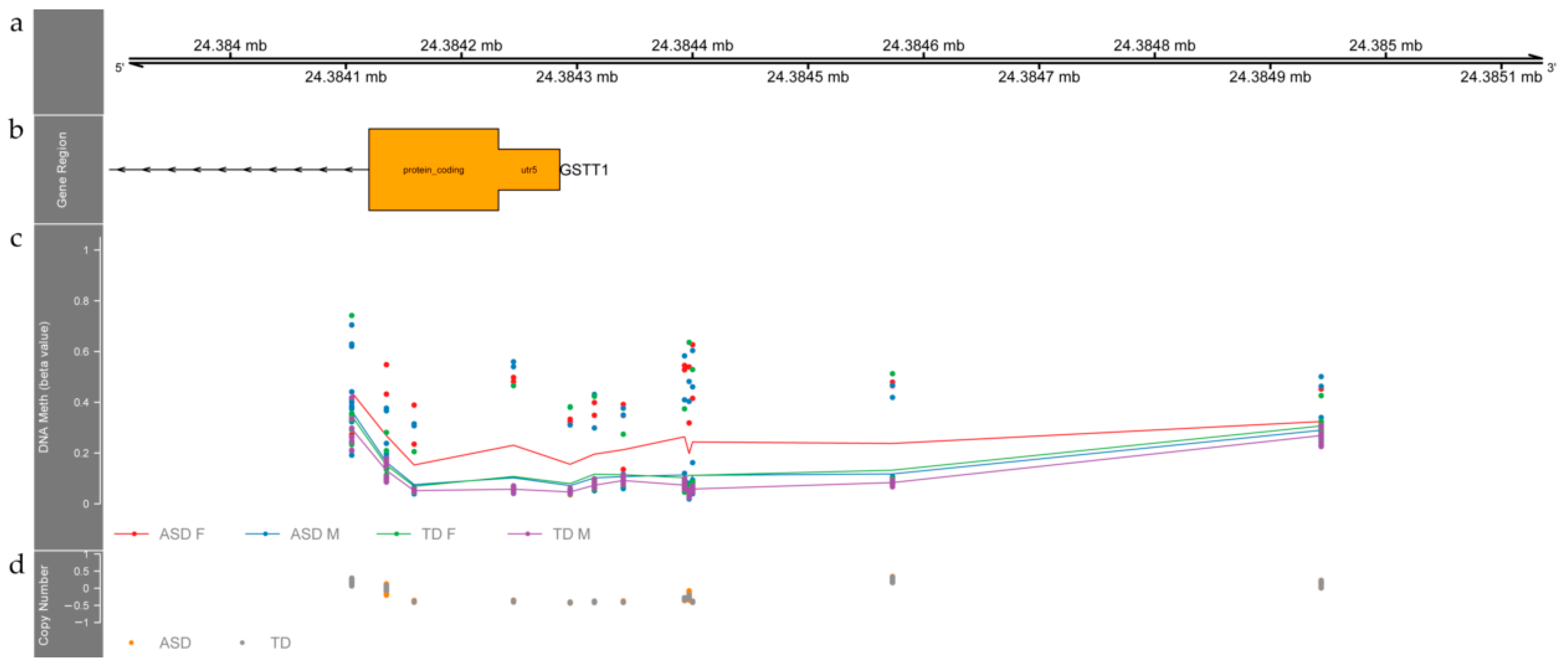
3.2. Differentially Methylated Regions between ASD and TD Individuals
3.3. Differentially Methylated Regions Affected by Sex and ASD Diagnosis
4. Discussion
4.1. Differentially Methylated Regions between ASD and TD Children Relate to Genes Involved in Neurodevelopment
4.2. Differentially Methylated Regions in ASD Interact with Sex
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Autism Spectrum Disorders. Available online: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders (accessed on 5 February 2022).
- Elsabbagh, M.; Divan, G.; Koh, Y.-J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global Prevalence of Autism and Other Pervasive Developmental Disorders: Global Epidemiology of Autism. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Christensen, D.L.; Maenner, M.J.; Bilder, D.; Constantino, J.N.; Daniels, J.; Durkin, M.S.; Fitzgerald, R.T.; Kurzius-Spencer, M.; Pettygrove, S.D.; Robinson, C.; et al. Prevalence and Characteristics of Autism Spectrum Disorder among Children Aged 4 Years—Early Autism and Developmental Disabilities Monitoring Network, Seven Sites, United States, 2010, 2012, and 2014. MMWR Surveill. Summ. 2019, 68, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hallmayer, J. Genetic Heritability and Shared Environmental Factors among Twin Pairs with Autism. Arch. Gen. Psychiatry 2011, 68, 1095. [Google Scholar] [CrossRef]
- Werling, D.M.; Geschwind, D.H. Sex Differences in Autism Spectrum Disorders. Curr. Opin. Neurol. 2013, 26, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Hull, L.; Petrides, K.V.; Mandy, W. The Female Autism Phenotype and Camouflaging: A Narrative Review. Rev. J. Autism Dev. Disord. 2020, 7, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Cheroni, C.; Caporale, N.; Testa, G. Autism Spectrum Disorder at the Crossroad between Genes and Environment: Contributions, Convergences, and Interactions in ASD Developmental Pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a Strongly Genetic Disorder: Evidence from a British Twin Study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare De Novo Variants Associated with Autism Implicate a Large Functional Network of Genes Involved in Formation and Function of Synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [Green Version]
- Bacchelli, E.; Loi, E.; Cameli, C.; Moi, L.; Vega Benedetti, A.; Blois, S.; Fadda, A.; Bonora, E.; Mattu, S.; Fadda, R.; et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. J. Clin. Med. 2019, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Grabrucker, A.M. Environmental Factors in Autism. Front. Psychiatry 2012, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Karimi, P.; Kamali, E.; Mousavi, S.M.; Karahmadi, M. Environmental Factors Influencing the Risk of Autism. J. Res. Med. Sci. 2017, 22, 27. [Google Scholar] [CrossRef]
- Kubota, T.; Mochizuki, K. Epigenetic Effect of Environmental Factors on Autism Spectrum Disorders. Int. J. Environ. Res. Public Health 2016, 13, 504. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, A.A.; Liu, G.; Kay, S.-I.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA Methylation Alterations in Multiple Brain Regions in Autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [Green Version]
- Nardone, S.; Sams, D.S.; Zito, A.; Reuveni, E.; Elliott, E. Dysregulation of Cortical Neuron DNA Methylation Profile in Autism Spectrum Disorder. Cereb. Cortex 2017, 27, 5739–5754. [Google Scholar] [CrossRef] [Green Version]
- Ellis, S.E.; Gupta, S.; Moes, A.; West, A.B.; Arking, D.E. Exaggerated CpH Methylation in the Autism-Affected Brain. Mol. Autism 2017, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain Transcriptional and Epigenetic Associations with Autism. PLoS ONE 2012, 7, e44736. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Chen, J.; Perrone-Bizzozero, N.; Bustillo, J.R.; Du, Y.; Calhoun, V.D.; Liu, J. Characterization of Cross-Tissue Genetic-Epigenetic Effects and Their Patterns in Schizophrenia. Genome Med. 2018, 10, 13. [Google Scholar] [CrossRef]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional Annotation of the Human Brain Methylome Identifies Tissue-Specific Epigenetic Variation across Brain and Blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA Extracted from Saliva for Methylation Studies of Psychiatric Traits: Evidence Tissue Specificity and Relatedness to Brain. Am. J. Med. Genet. 2015, 168, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-Control Meta-Analysis of Blood DNA Methylation and Autism Spectrum Disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic Epigenetic Dysregulation of Ectodermal Cells in Autism Spectrum Disorder. PLoS Genet 2014, 10, e1004402. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.-H.; Chuang, L.-C.; Su, M.-H.; Chen, C.-H.; Chen, C.-H.; Wu, J.-Y.; Yen, C.-J.; Wu, Y.-Y.; Liu, S.-K.; Chou, M.-C.; et al. Genome-Wide Association Study for Autism Spectrum Disorder in Taiwanese Han Population. PLoS ONE 2015, 10, e0138695. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A Revised Version of a Diagnostic Interview for Caregivers of Individuals with Possible Pervasive Developmental Disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Márquez, C.; Nicolini, H.; Crowley, M.J.; Solís-Vivanco, R. Early Processing (N170) of Infant Faces in Mothers of Children with Autism Spectrum Disorder and Its Association with Maternal Sensitivity. Autism Res. 2019, 12, 744–758. [Google Scholar] [CrossRef]
- Morales-Marín, M.E.; Aguilar, M.; Albores, L.; Ballesteros, A.; Castro, X.; Chicalote, C.; Gómez, A.; Gutiérrez, N.; Lanzagorta, N.; López, F.; et al. Effect of the polymorphism BDNF rs6265 G/A in Mexican outpatient children with autism spectrum disorders. Salud Ment. 2018, 41, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Constantino, J.N. Social Responsiveness Scale. In Encyclopedia of Autism Spectrum Disorders; Volkmar, F.R., Ed.; Springer: New York, NY, USA, 2013; pp. 2919–2929. ISBN 9781441916976. [Google Scholar]
- Bölte, S.; Poustka, F.; Constantino, J.N. Assessing autistic traits: Cross-cultural validation of the social responsiveness scale (SRS). Autism Res. 2008, 1, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Data Quality Control in Genetic Case-Control Association Studies. Nat. Protoc. 2010, 5, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- The International HapMap 3 Consortium. Integrating Common and Rare Genetic Variation in Diverse Human Populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of Cross-Reactive Probes and Polymorphic CpGs in the Illumina Infinium HumanMethylation450 Microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Touleimat, N.; Tost, J. Complete Pipeline for Infinium® Human Methylation 450K BeadChip Data Processing Using Subset Quantile Normalization for Accurate DNA Methylation Estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Irimia, A.; Lei, X.; Torgerson, C.M.; Jacokes, Z.J.; Abe, S.; Van Horn, J.D. Support Vector Machines, Multidimensional Scaling and Magnetic Resonance Imaging Reveal Structural Brain Abnormalities Associated With the Interaction Between Autism Spectrum Disorder and Sex. Front. Comput. Neurosci. 2018, 12, 93. [Google Scholar] [CrossRef]
- Hu, V.W.; Hong, Y.; Xu, M.; Shu, H.T. Altered DNA Methylation in a Severe Subtype of Idiopathic Autism: Evidence for Sex Differences in Affected Metabolic Pathways. Autism 2021, 25, 887–910. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De Novo Identification of Differentially Methylated Regions in the Human Genome. Epigenet. Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart—Biological Queries Made Easy. BMC Genomics 2009, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 11 February 2022).
- Theda, C.; Hwang, S.H.; Czajko, A.; Loke, Y.J.; Leong, P.; Craig, J.M. Quantitation of the Cellular Content of Saliva and Buccal Swab Samples. Sci. Rep. 2018, 8, 6944. [Google Scholar] [CrossRef]
- Cousins, S.; Blencowe, N.S.; Blazeby, J.M. What Is an Invasive Procedure? A Definition to Inform Study Design, Evidence Synthesis and Research Tracking. BMJ Open 2019, 9, e028576. [Google Scholar] [CrossRef] [Green Version]
- Selvey, P.; Stypulkowski, K.; Waisbren, S. Surgical Management of the Patient Living with Autism. Surg. Open Sci. 2019, 1, 90–96. [Google Scholar] [CrossRef]
- Jones, K.B.; Klein, O.D. Oral Epithelial Stem Cells in Tissue Maintenance and Disease: The First Steps in a Long Journey. Int. J. Oral Sci. 2013, 5, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Li, M. Human Brain Transcriptome. Available online: http://hbatlas.org/ (accessed on 1 August 2019).
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-Temporal Transcriptome of the Human Brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, B.; Taylor, V.; Welcher, A.A.; McClelland, M.; Suter, U. Neural Membrane Protein 35 (NMP35): A Novel Member of a Gene Family Which Is Highly Expressed in the Adult Nervous System. Mol. Cell Neurosci. 1998, 11, 260–273. [Google Scholar] [CrossRef]
- Hurtado de Mendoza, T.; Perez-Garcia, C.G.; Kroll, T.T.; Hoong, N.H.; O’Leary, D.D.M.; Verma, I.M. Antiapoptotic Protein Lifeguard Is Required for Survival and Maintenance of Purkinje and Granular Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17189–17194. [Google Scholar] [CrossRef] [Green Version]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of Cerebellar Basket and Stellate Cells in Autism: Evidence for a Late Developmental Loss of Purkinje Cells. J. Neurosci. Res. 2009, 87, 2245–2254. [Google Scholar] [CrossRef] [Green Version]
- Gil-Varea, E.; Urcelay, E.; Vilariño-Güell, C.; Costa, C.; Midaglia, L.; Matesanz, F.; Rodríguez-Antigüedad, A.; Oksenberg, J.; Espino-Paisan, L.; Dessa Sadovnick, A.; et al. Exome Sequencing Study in Patients with Multiple Sclerosis Reveals Variants Associated with Disease Course. J. Neuroinflamm. 2018, 15, 265. [Google Scholar] [CrossRef] [Green Version]
- Edmonson, C.A.; Ziats, M.N.; Rennert, O.M. A Non-Inflammatory Role for Microglia in Autism Spectrum Disorders. Front. Neurol. 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Qin, L.; Min, Z.; Zhao, Y.; Zhu, L.; Zhu, J.; Yu, S. SOX7 Interferes with β-Catenin Activity to Promote Neuronal Apoptosis. Eur. J. Neurosci. 2015, 41, 1430–1437. [Google Scholar] [CrossRef]
- Takash, W. SOX7 Transcription Factor: Sequence, Chromosomal Localisation, Expression, Transactivation and Interference with Wnt Signalling. Nucleic Acids Res. 2001, 29, 4274–4283. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Pan, T.; Yang, S.; Liu, J.; Tao, H.; Zhao, Y.; Xu, D.; Shao, W.; Wu, J.; Liu, X.; et al. Up-Regulated NRIP2 in Colorectal Cancer Initiating Cells Modulates the Wnt Pathway by Targeting RORβ. Mol. Cancer 2017, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Riso, V.; Cammisa, M.; Kukreja, H.; Anvar, Z.; Verde, G.; Sparago, A.; Acurzio, B.; Lad, S.; Lonardo, E.; Sankar, A.; et al. ZFP57 Maintains the Parent-of-Origin-Specific Expression of the Imprinted Genes and Differentially Affects Non-Imprinted Targets in Mouse Embryonic Stem Cells. Nucleic Acids Res. 2016, 44, 8165–8178. [Google Scholar] [CrossRef]
- Connolly, S.; Anney, R.; Gallagher, L.; Heron, E.A. A Genome-Wide Investigation into Parent-of-Origin Effects in Autism Spectrum Disorder Identifies Previously Associated Genes Including SHANK3. Eur. J. Hum. Genet. 2017, 25, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Amarasekera, M.; Martino, D.; Ashley, S.; Harb, H.; Kesper, D.; Strickland, D.; Saffery, R.; Prescott, S.L. Genome-wide DNA Methylation Profiling Identifies a Folate-sensitive Region of Differential Methylation Upstream of ZFP57-imprinting Regulator in Humans. FASEB J. 2014, 28, 4068–4076. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Cubas, C.; Sánchez-Hernández, B.E.; García-Ortiz, H.; Martínez-Hernández, A.; Barajas-Olmos, F.; Cid, M.; Mendoza-Caamal, E.C.; Centeno-Cruz, F.; Ortiz-Cruz, G.; Jiménez-López, J.C.; et al. Heterogenous Distribution of MTHFR Gene Variants among Mestizos and Diverse Amerindian Groups from Mexico. PLoS ONE 2016, 11, e0163248. [Google Scholar] [CrossRef]
- Ibarra-Lopez, J.J.; Duarte, P.; Antonio-Vejar, V.; Calderon-Aranda, E.S.; Huerta-Beristain, G.; Flores-Alfaro, E.; Moreno-Godinez, M.E. Maternal C677T MTHFR Polymorphism and Environmental Factors Are Associated With Cleft Lip and Palate in a Mexican Population. J. Investig. Med. 2013, 61, 1030–1035. [Google Scholar] [CrossRef]
- Pauwels, S.; Ghosh, M.; Duca, R.C.; Bekaert, B.; Freson, K.; Huybrechts, I.; Langie, S.A.S.; Koppen, G.; Devlieger, R.; Godderis, L. Maternal Intake of Methyl-Group Donors Affects DNA Methylation of Metabolic Genes in Infants. Clin. Epigenet. 2017, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Coker, E.S.; Gunier, R.; Huen, K.; Holland, N.; Eskenazi, B. DNA Methylation and Socioeconomic Status in a Mexican-American Birth Cohort. Clin. Epigenet. 2018, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, M.H.; Samms-Vaughan, M.; Ma, J.; Bressler, J.; Loveland, K.A.; Hessabi, M.; Dickerson, A.S.; Grove, M.L.; Shakespeare-Pellington, S.; Beecher, C.; et al. Interaction between GSTT1 and GSTP1 Allele Variants as a Risk Modulating-Factor for Autism Spectrum Disorders. Res. Autism Spectr. Disord. 2015, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, Y.; Seo, T.; Hashimoto, N.; Higa, Y.; Ishitsu, T.; Nakagawa, K. Glutathione-S-Transferase (GST) M1 Null Genotype and Combined GSTM1 and GSTT1 Null Genotypes Are Risk Factors for Increased Serum Gamma-Glutamyltransferase in Valproic Acid-Treated Patients. Clin. Chim. Acta 2008, 389, 98–102. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ASD | TD | p-Value | |
|---|---|---|---|
| Subject Age (mean, SD) | 5.2, 1.9 | 5.6, 1.09 | 0.7645 1 |
| (range) | 3–12 | 4–7 | |
| Gender | |||
| Male:Female | 22:5 | 6:9 | 0.01479 1 |
| Gene a | Chromosome | Width (Base Pairs) | False Discovery Rate Corrected p-Value | Methylation Differences (ASD-TD) b |
|---|---|---|---|---|
| FAIM, Fas Apoptotic Inhibitory Molecule 2 | chr12 | 469 | 3.18 × 10−3 | −0.087 |
| CPXM2, Carboxypeptidase X, M14 Family Member 2 | chr10 | 674 | 4.20 × 10−3 | 0.056 |
| NRIP2, Nuclear Receptor Interacting Protein 2 | chr12 | 592 | 8.99 × 10−3 | 0.052 |
| SOX7, CTD-2135J3.3 SRY-Box7 | chr8 | 205 | 3.03 × 10−2 | −0.061 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspra, Q.; Cabrera-Mendoza, B.; Morales-Marín, M.E.; Márquez, C.; Chicalote, C.; Ballesteros, A.; Aguilar, M.; Castro, X.; Gómez-Cotero, A.; Balboa-Verduzco, A.M.; et al. Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism. Children 2022, 9, 462. https://0-doi-org.brum.beds.ac.uk/10.3390/children9040462
Aspra Q, Cabrera-Mendoza B, Morales-Marín ME, Márquez C, Chicalote C, Ballesteros A, Aguilar M, Castro X, Gómez-Cotero A, Balboa-Verduzco AM, et al. Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism. Children. 2022; 9(4):462. https://0-doi-org.brum.beds.ac.uk/10.3390/children9040462
Chicago/Turabian StyleAspra, Queletzu, Brenda Cabrera-Mendoza, Mirna Edith Morales-Marín, Carla Márquez, Carlos Chicalote, Ana Ballesteros, Miriam Aguilar, Xochitl Castro, Amalia Gómez-Cotero, Ana María Balboa-Verduzco, and et al. 2022. "Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism" Children 9, no. 4: 462. https://0-doi-org.brum.beds.ac.uk/10.3390/children9040462