

Sortase A Fusion Expression and mIFc2 Co-Expression of Bovine Lactoferricin and Analysis of Its Antibacterial Activity
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. E. coli Host Cells and Gram-Negative and Gram-Positive Bacteria Species
2.2. Amplification of LfcinB, SrtA Region of Staphylococcus aureus Transferase, and SrtA-LfcinB Coding Fragments
2.3. Construction of the pET32a Vector Carrying Multiple Expression Cassettes to Express the SrtA-LfcinB Fusion Protein and Development of LfcinB and mIFc2 Co-Overexpression Vectors
2.4. Expression of the SrtA-LfcinB Fusion Protein and Co-Expression of LfcinB and mIFc2
2.5. Protein Purification and Self-Cleavage of the SrtA-LfcinB Fusion Protein to Release LfcinB
2.6. SDS-Polyacrylamide Gel Analysis and Western Blot Assays
2.7. Treatments of Inclusion Bodies with Different Temperatures and pHs
2.8. Antibacterial Test Using the Agar Diffusion Method
2.9. Statistical Analysis
3. Results
3.1. Development of Two Different Expression Systems for Expression of LfcinB
3.2. Amplification of SrtA, LfcinB, SrtA-LfcinB, and the Full-Length mIFc2 DNA Fragment As Well As Construction of pET21b-SrtA-LfcinB and pET32a-SrtA-LfcinB and the LfcinB and mIFc2 Co-Expression Vectors
3.3. Expression of the TrxA-His-SrtA-LfcinB Fusion Protein
3.4. Co-Expression of mIFc2 and His-LfcinB
3.5. Enhancement of the Dissolution Rate of Inclusion Bodies by Treatments with Different Temperatures, pHs, and Resuspended Volumes
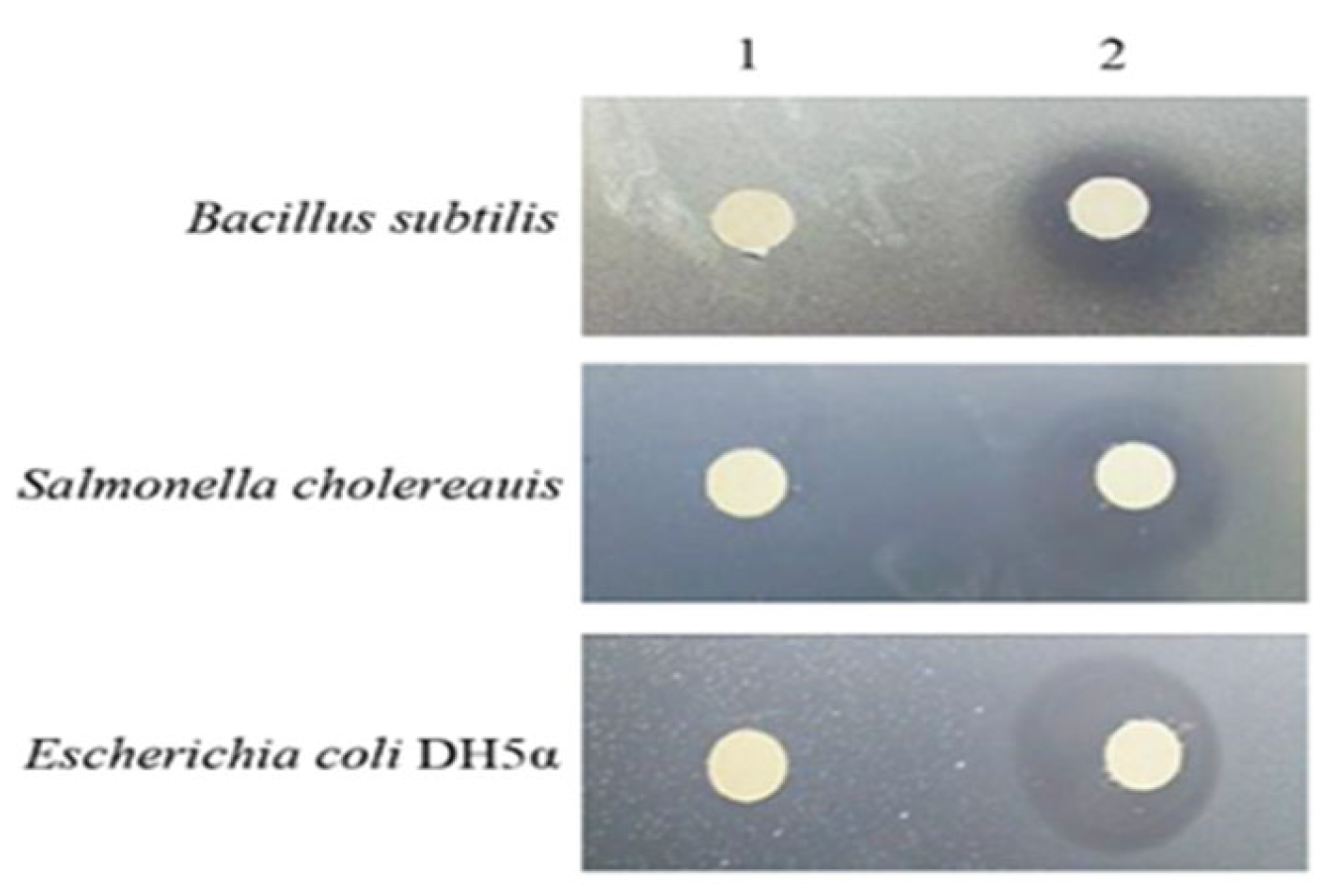
3.6. Antibacterial Activity Tests
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aarestrup, F.M.; Jensen, V.F.; Emborg, H.D.; Jacobsen, E.; Wegener, H.C. Changes in the use of antimicrobials and the effects on productivity of swine farms in Denmark. Am. J. Vet. Res. 2010, 71, 726–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, H.K.; Levine, U.Y.; Looft, T.; Bandrick, M.; Casey, T.A. Treatment, promotion, commotion: Antibiotic alternatives in food-producing animals. Trends Microbiol. 2013, 21, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, M.K. Use of antibiotics as feed additives: A burning question. Front. Microbiol. 2014, 5, 334. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.; Heritage, J. Antibiotic growth-promoters. Feed. Tech. 2002, 8, 20–22. [Google Scholar]
- Oliver, S.P.; Murinda, S.E.; Jayarao, B.M. Impact of antibiotic use in adult dairy cows on antimicrobial resistance of veterinary and human pathogens: A comprehensive review. Foodborne Pathog. Dis. 2011, 8, 337–355. [Google Scholar] [CrossRef]
- Lai, H.Z.; Chen, W.Y.; Wu, C.Y.; Chen, Y.C. Potent Antibacterial Nanoparticles for Pathogenic Bacteria. ACS Appl. Mater. Interfaces 2015, 7, 2046–2054. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic Tolerance Facilitates the Evolution of Resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef]
- Wang, F.; Fang, R.H.; Luk, B.T.; Hu, C.M.J.; Thamphiwatana, S.; Dehaini, D.; Angsantikul, P.; Kroll, A.V.; Pang, Z.; Gao, W.; et al. Nanoparticle-Based Antivirulence Vaccine for the Management of Methicillin-Resistant Staphylococcus aureus Skin Infection. Adv. Funct. Mater. 2016, 26, 1628–1635. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Dong, K.; Ren, J.; Qu, X. A b-Lactamase-Imprinted Responsive Hydrogel for the Treatment of Antibiotic-Resistant Bacteria. Angew. Chem. Int. Ed. 2016, 55, 8049–8053. [Google Scholar] [CrossRef]
- Seo, M.D.; Won, H.S.; Kim, J.H.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial Peptides for Therapeutic Applications: A Review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef] [Green Version]
- Jiri Patocka, J.; Nepovimova, E.; Klimova, B.; Wu, Q.; Kuca, K. Antimicrobial peptides: Amphibian host defense peptides. Curr. Med. Chem. 2019, 26, 5924–5946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Peng, Y.; Gao, X.; Song, Q.; Zhang, H.; Elhag, O.; Cai, M.; Zheng, L.; Yu, Z.; et al. Structural and functional characterizations and heterogenous expression of the antimicrobial peptides, Hidefensins, from black soldier fly, Hermetia illucens (L.). Protein Expr. Purif. 2022, 192, 106032. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Mohamed, G.; Polash, S.A.; Hasan, M.A.; Sultana, R.; Saiara, N.; Dong, W. Antimicrobial Peptides from Plants: A cDNA-library based isolation, purification, characterization approach and elucidating their modes of action. Int. J. Mol. Sci. 2021, 22, 8712. [Google Scholar] [CrossRef] [PubMed]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sonksen, C.P.; Ludvigsen, S.; Raventos, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- Van’t Hof, W.; Veerman, E.C.; Helmerhorst, E.J.; Amerongen, A.V. Antimicrobial peptides: Properties and applicability. Biol. Chem. 2001, 382, 597–619. [Google Scholar]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef]
- Baker, H.M.; Anderson, B.F.; Kidd, R.D.; Shewry, S.C.; Baker, E.N. Lactoferrin three-dimensional structure: A framework for interpreting function. In Lactoferrin: Structure, Function and Applications; Shimazaki, K., Tsuda, H., Tomita, M., Kunata, T., Perraudin, J.P., Eds.; Elsevier Science: New York, NY, USA, 2000; pp. 3–15. [Google Scholar]
- Bellamy, W.; Takase, M.; Yamauchi, K.; Wakabayashi, H.; Kawase, K.; Tomita, M. Identification of the bactericidal domain of lactoferrin. Biochim. Biophys. Acta BBA-Protein Struct. Mol. Enzym. 1992, 1121, 130–136. [Google Scholar] [CrossRef]
- Schibli, D.J.; Vogel, H.J. Structural studies of lactoferricin B and its antimicrobial active peptide fragments. In Lactoferrin, Structure, Function and Application; Shimazaki, K., Ed.; Elsevier Science: New York, NY, USA, 2000; pp. 27–35. [Google Scholar]
- Hwang, P.M.; Zhou, N.; Shan, X.; Arrowsmith, C.H.; Vogel, H.J. Three-Dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry 1998, 37, 4288–4298. [Google Scholar] [CrossRef]
- Vogel, H.J.; Schibli, D.J.; Jing, W.; Lohmeier-Vogel, E.M.; Epand, R.F.; Epand, R.M. Towards a structure-function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem. Cell Biol. 2002, 80, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.; Takase, M.; Wakabayashi, H.; Kawase, K.; Tomita, M. Antibactericidal spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J. Appl. Bacteriol. 1992, 73, 472–479. [Google Scholar] [CrossRef]
- Tomita, S.; Shirasaki, N.; Hayashizaki, H.; Matsuyama, J.; Benno, Y.; Kiyosawa, I. Binding characteristics of bovine lactoferrin to the cell surface of Clostridium species and identification of the lactoferrin-binding protein. Biosci. Biotechnol. Biochem. 1998, 62, 1476–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strøm, M.B.; Rekdal, O.; Svendsen, J.S. Antibacterial activity of 15-residue lactoferricin derivatives. J. Pept. Res. 2000, 56, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.M.; Smart, A.; Bloomberg, G.; Burgess, L.; Millar, M.R. Lactoferricin, a new antimicrobial peptide. J. Appl. Bacteriol. 1994, 77, 208–214. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, M.S.; Cho, J.H.; Kim, S.C. Enhanced expression of tandem multimers of the antimicrobial peptide buforin II in Escherichia coli by the DEAD-box protein and trxB mutant. Appl. Microbiol Biotechnol. 2002, 58, 790–796. [Google Scholar] [PubMed]
- Kim, J.M.; Jang, S.A.; Yu, B.J.; Sung, B.H.; Cho, J.H.; Kim, S.C. High-level expression of an antimicrobial peptide histonin as a natural form by multimerization and furin-mediated cleavage. Appl. Microbiol. Biotechnol. 2008, 78, 123–130. [Google Scholar] [CrossRef]
- Mao, H. A self-cleavable sortase fusion for one-step purification of free recombinant proteins. Protein Expr. Purif. 2004, 37, 253–263. [Google Scholar] [CrossRef]
- Yin, Z.X.; He, W.; Chen, W.J.; Yan, J.H.; Yang, J.N.; Chan, S.M.; He, J.G. Cloning, expression and antimicrobial activity of an antimicrobial peptide, epinecidin-1, from the orange-spotted grouper, Epinephelus coioides. Aquaculture 2005, 253, 204–211. [Google Scholar] [CrossRef]
- Jang, S.A.; Sung, B.H.; Cho, J.H.; Kim, S.C. Direct expression of antimicrobial peptides in an intact form by a translationally coupled two-cistron expression system. Appl. Environ. Microbiol. 2009, 75, 3980–3986. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 149–158. [Google Scholar] [CrossRef]
- Gong, C.; Sun, J.; Xiao, Y.; Qu, X.; Lang, M. Synthetic mimics of antimicrobial peptides for the targeted therapy of multidrug-resistant bacterial infection. Adv. Healthc. Mater. 2021, 10, e2101244. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Yamauchi, K.; Teraguchi, S.; Hayasawa, H.; Tomita, M.; Otsuka, Y.; Yamazaki, S. Antibacterial activity of bovine lactoferrin and its peptides against enterohaemorrhagic O157:H7. Lett. Appl. Microbiol. 1998, 26, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Shestakov, A.; Jenssen, H.; Nordström, I.; Eriksson, K. Lactoferricin but not lactoferrin inhibit herpes simplex virus type 2 infection in mice. Antivir. Res. 2012, 93, 340–345. [Google Scholar] [CrossRef]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- McManus, M.C. Mechanisms of bacterial resistance to antimicrobial agents. Am. J. Health Syst. Pharm. 1997, 54, 1420–1433. [Google Scholar] [CrossRef]
- Liu, L.; Xu, K.; Wang, H.; Tan, P.K.; Fan, W.; Venkatraman, S.S.; Li, L.; Yang, Y.Y. Self-Assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 2009, 4, 457–463. [Google Scholar] [CrossRef]
- Chen, C.; Pan, F.; Zhang, S.; Hu, J.; Cao, M.; Wang, J.; Xu, H.; Zhao, X.; Lu, J.R. Antibacterial activities of short designer peptides: A link between propensity for nanostructuring and capacity for membrane destabilization. Biomacromolecules 2010, 11, 402–411. [Google Scholar] [CrossRef]
- Wang, Y.D.; Kung, C.W.; Chen, J.Y. Antiviral activity by fish antimicrobial peptides of epinecidin-1 and hepcidin 1-5 against nervous necrosis virus in medaka. Peptides 2010, 31, 1026–1033. [Google Scholar] [CrossRef]
- Peng, C.C. Application of antimicrobial peptides in biotechnology. Plant Pathol. Bull. 2016, 15, 69–75. [Google Scholar]
- Liu, Y.; Han, F.; Xie, Y.; Wang, Y. Comparative antimicrobial activity and mechanism of action of bovine lactoferricin-derived synthetic peptides. Biometals 2011, 24, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Vorland, L.H.; Ulvatne, H.; Andersen, J.; Haukland, H.H.; Rekdal, O.; Svendsen, J.S.; Gutteberg, T.J. Antibacterial effects of lactoferricin B. Scand. J. Infect. Dis. 1999, 31, 179–184. [Google Scholar]
- Wakabayashi, H.; Teraguchi, S.; Tamura, Y. Increased Staphylococcus-killing activity of an antimicrobial peptide, Lactoferricin B, with minocycline and monoacylglycerol. Biosci. Biotechnol. Biochem. 2002, 66, 2161–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliot, J.I.; Senft, B.; Erhardt, G.; Fraser, D. Isolation of lactoferrin and its concentration in sow’s colostrum and milk during a 21-day lactation. J. Anim. Sci. 1984, 59, 1080–1084. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganisms | LfcinB ug/mL |
|---|---|
| Gram negative bacteria | |
| Escherichia coli 10675 | 100 |
| Salmonella choleraesuis | 37 |
| Salmonella typhimurium | 84 |
| Klebsiella oxytoca 13985 | 62 |
| Enterobacter aerogenes 10370 | 62 |
| Aeromonas hydrophila Ah | 37 |
| Vibrio alginolyticus 12829 | 75 |
| Listonella anguillarum 13810 | 37 |
| Gardnerella vaginalis 17040 | 75 |
| Yersinia enterocolitica subsp. 13999 | 75 |
| Pseudomonas aerruginosa | 150 |
| Vibrio parahaemolyticus | 75 |
| Vibrio vulnificus | 75 |
| Gram positive bacteria | |
| Bacillus subtilis | 75 |
| Micrococcus luteus 11034 | 75 |
| Streptococcus agalactiae 10787 | 37 |
| Listeria monocytogenes 14845 | 37 |
| Streptococcus pyogenes Rosenbach 10797 | 37 |
| Staphylococcus aureus 10451 | 75 |
| Staphylococcus sp. 10783 | 75 |
| Streptococcus pneumoniae 10794 | 75 |
| Staphylococcus haemolyticus 15237 | 75 |
| Enterococcus faecalis 10066 | 62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-Y.; Hsieh, C.-Y.; Yang, C.-Y.; Chang, Y.-K.; Shih, W.-L.; Yeh, C.-M.; Hu, N.-J.; Chen, M.-S.; Nielsen, B.L.; Liu, H.-J. Sortase A Fusion Expression and mIFc2 Co-Expression of Bovine Lactoferricin and Analysis of Its Antibacterial Activity. Processes 2022, 10, 2470. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10122470
Hsu C-Y, Hsieh C-Y, Yang C-Y, Chang Y-K, Shih W-L, Yeh C-M, Hu N-J, Chen M-S, Nielsen BL, Liu H-J. Sortase A Fusion Expression and mIFc2 Co-Expression of Bovine Lactoferricin and Analysis of Its Antibacterial Activity. Processes. 2022; 10(12):2470. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10122470
Chicago/Turabian StyleHsu, Chao-Yu, Chung-Yiu Hsieh, Cheng-Yao Yang, Yu-Kang Chang, Wen-Ling Shih, Chuan-Ming Yeh, Nien-Jen Hu, Ming-Shan Chen, Brent L. Nielsen, and Hung-Jen Liu. 2022. "Sortase A Fusion Expression and mIFc2 Co-Expression of Bovine Lactoferricin and Analysis of Its Antibacterial Activity" Processes 10, no. 12: 2470. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10122470