Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable without a Digital-Twin?


 ,
, 
Abstract
:1. Introduction
- A database of physical properties is needed with in-house data on product related molecular process knowledge. Experimental methods have to be standardized in laboratories and thermodynamics experts need to maintain such a database and be expert advisors for modeling colleagues. The value of such in-house data is tremendous.
- Data need to provide accuracy and precision, which can be directly predefined by modeling.
- To build up reliable databases takes time. Each new molecular system has to be experimentally assessed with regard to physical properties for the unit operations feasible.
- Additional modeling and simulation methods require, of course, additional efforts and resources, i.e., costs. Those investments easily pay off if a reduction of experimental effort is achieved. As pilot run costs are quite high and time consuming, the easiest approach is to involve simulation experts into the piloting runs. A side benefit is that models can remain less complex as the colleagues are directly involved in process operation reality and the required measurement accuracy.
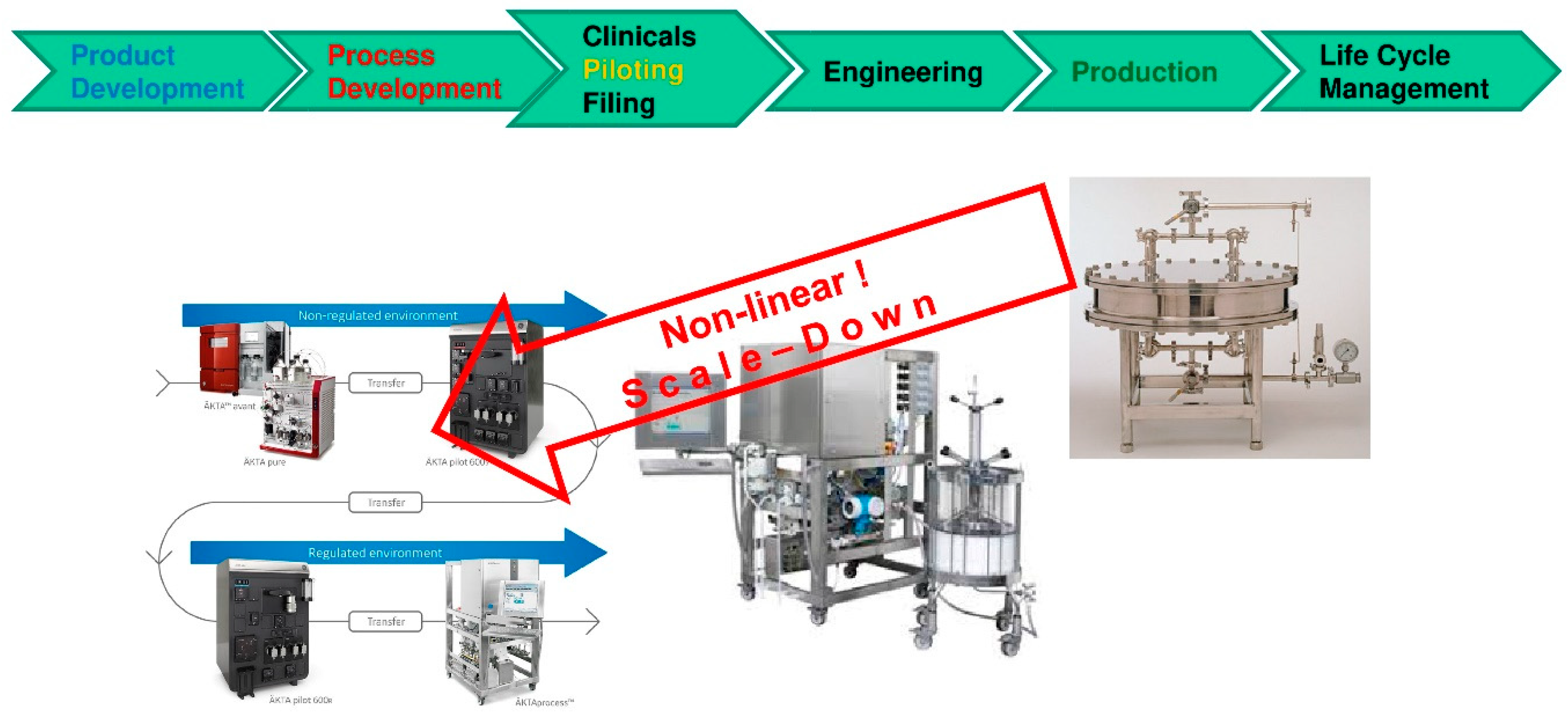
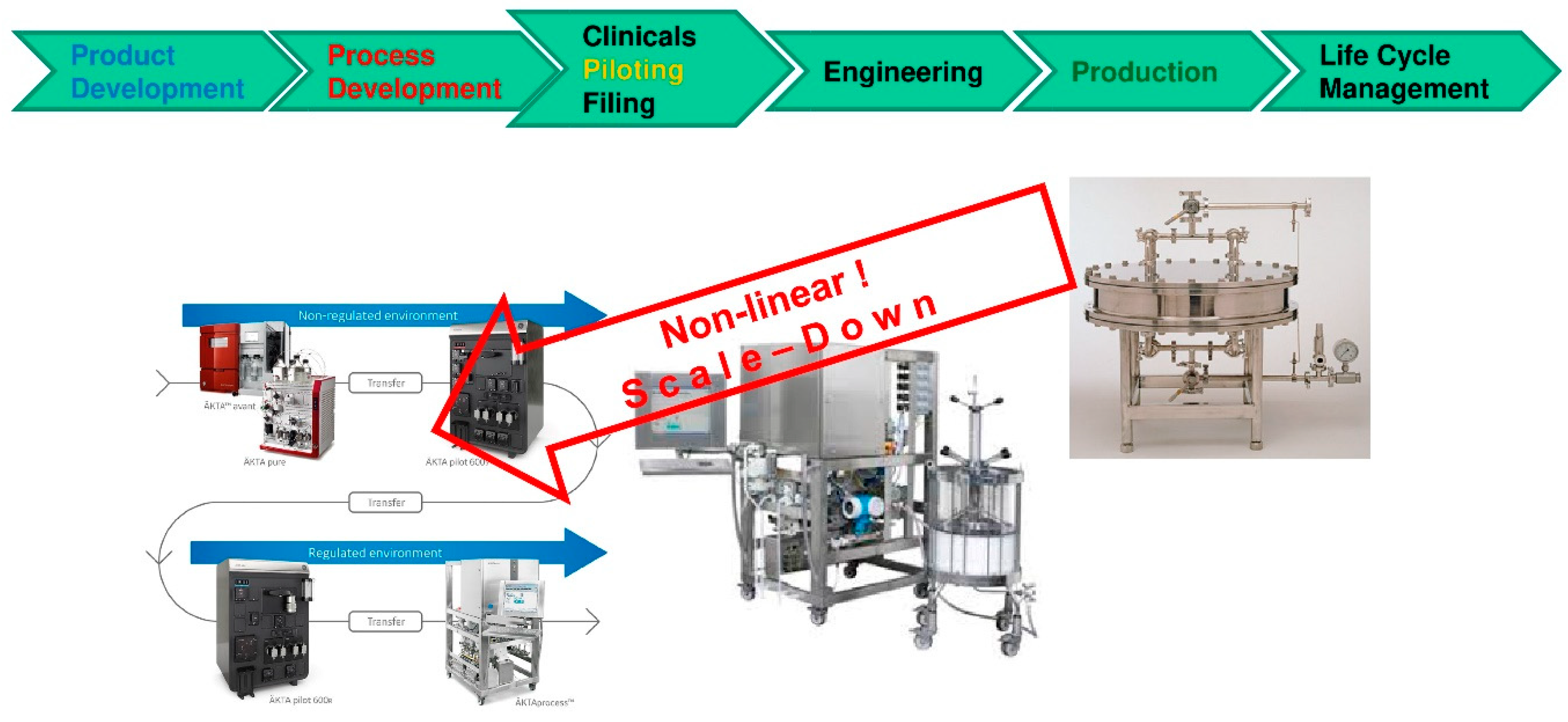
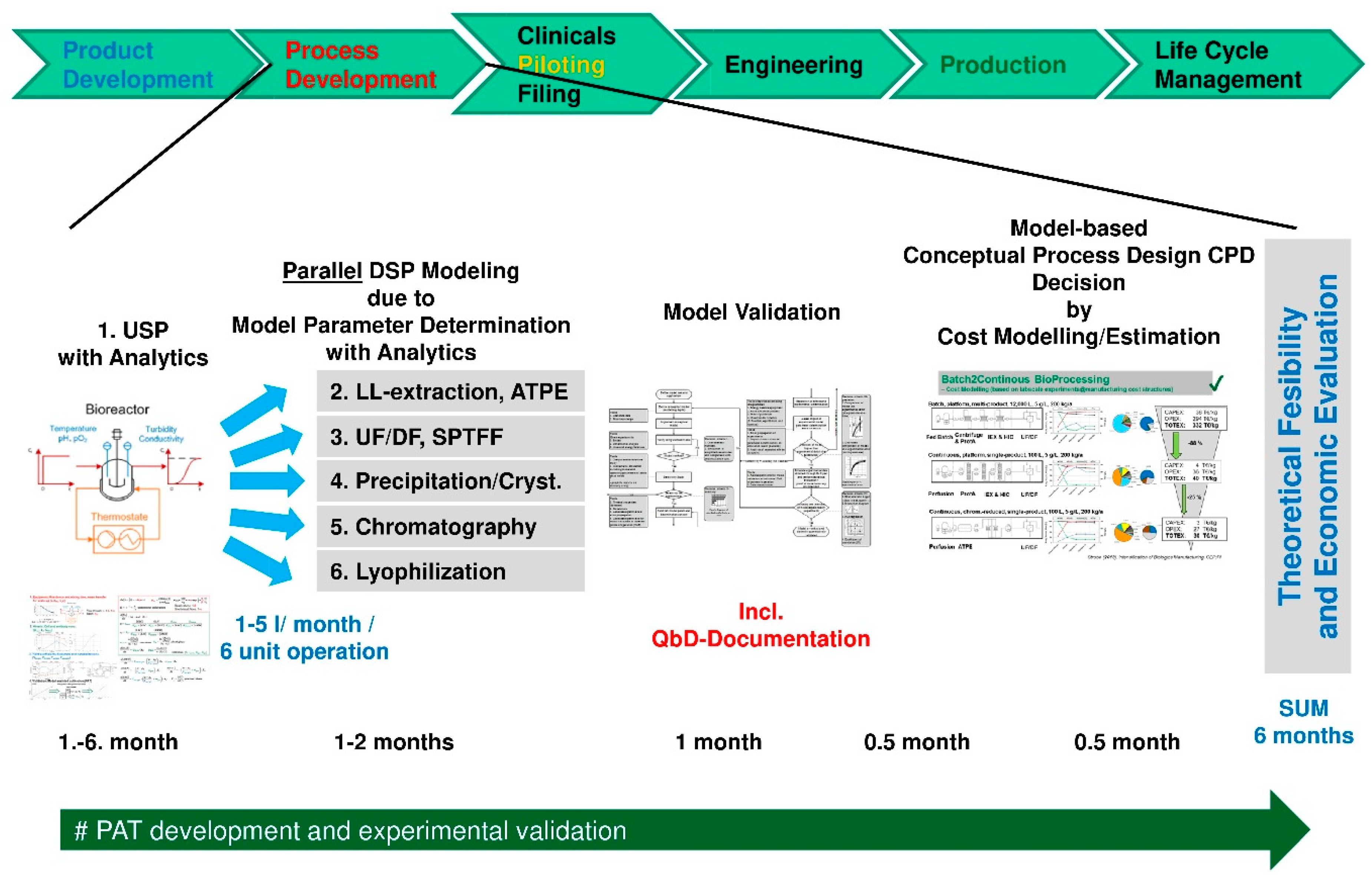
- Models derived in process development with miniaturized model parameter determination in laboratory scale follow clear workflows/recipes i.e., standard operation procedures (SOP). Direct benefits are gained if those models support piloting runs for experimental validation and afterwards move the product into engineering for equipment plant design and advanced process control concepts over the lifetime into manufacturing operation for process robustness analysis.
- Time reduction in process development is achieved by running the experimental plans for model parameter determination for each unit operation in parallel. Of course, the analytical methods have to be available and run in parallel for all methods considered. This may speed up conceptual process design towards experimental feasibility to less than a month for a well-trained interdisciplinary team.
- Necessary team know-how should include chemical engineering for model development. In addition, technicians running analytics and laboratory scale equipment for experimental model parameter determination should be trained on accuracy and precision needed for those experimental plans which differ from classical design of experiments (DoE) setups. Such laboratory scale equipment should ideally already be equipped with process analytical technology (PAT) methodologies and in addition with fractionation devices for sampling to quality assurance (QA).
2. Challenges with Regard to Models and Modeling
- At least three general model types have been defined before
- Physical-chemical based rigorous models
- Cost modeling i.e., cost estimation and
- (Advanced) process control e.g., statistical observers, neuronal networks and data mining as parts of artificial intelligence
Regression models of analytical data are left out. Each model type has its benefits and place within the workflow. They are totally different and should not be mismatched. - Cost modeling [86,87,88,89,90] is based on simple mass and component as well as macroscopic energy balances if needed. Data is taken from experimental operation of the units. Cost modeling is based more on classical flowsheet balancing than real modeling, therefore, in chemical engineering graduation it is called cost estimation (class 1–5) [91,92].
- Increasing modeling depth does not necessarily increase accuracy of prediction if the model parameters involved could not be determined sufficiently precise.Any model is a look at the specific view of its predefined aim from the point of view of the model developer with the needed, but of course limited accuracy towards a complex reality. Nevertheless, sufficient for the task. Therefore, objectives and accuracy have to be thoroughly defined first.
- Process control methods are well established in chemical engineering and industry. BASF reports to benefit from additional observer model training during plant start-up even with regard to the corresponding delay of manufacturing begin of about 3 months [93]. Those, observer or advanced process control methods based on statistics, such as artificial intelligence tools such as neuronal networks, are generally valid and available, which could be directly transferred to biologics operation. In line sensors are in most cases efficient, but there is the major challenge to cope with their natural drift.
- Here, again rigorous models—available already from process development with appropriate efficient organization—are the methodological solution of choice.
- The models discussed are available, all three of them. Any prejudices have to be rejected: Rigorous modeling of all unit operations needed for total process integration are on hand, as proven later on.
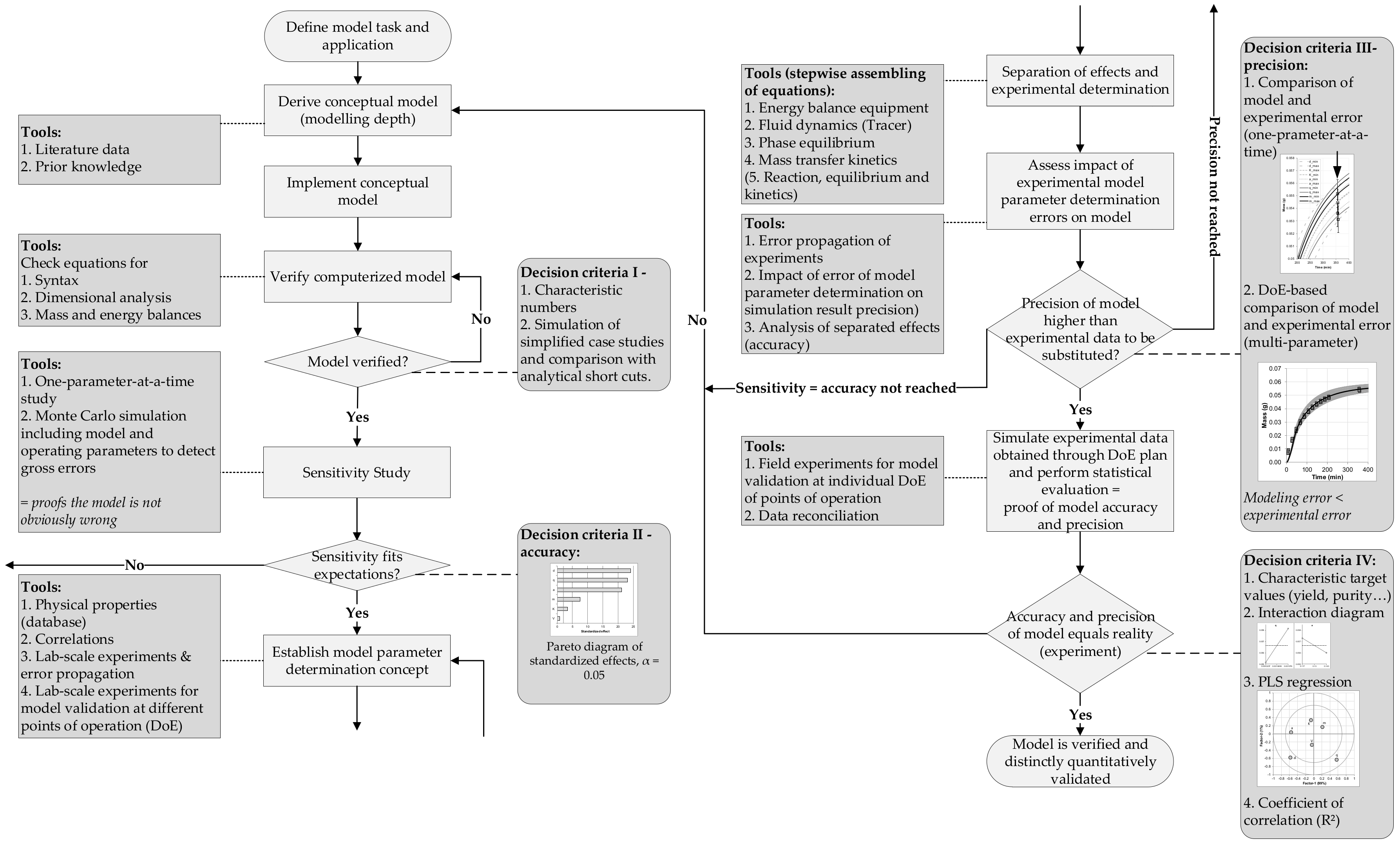
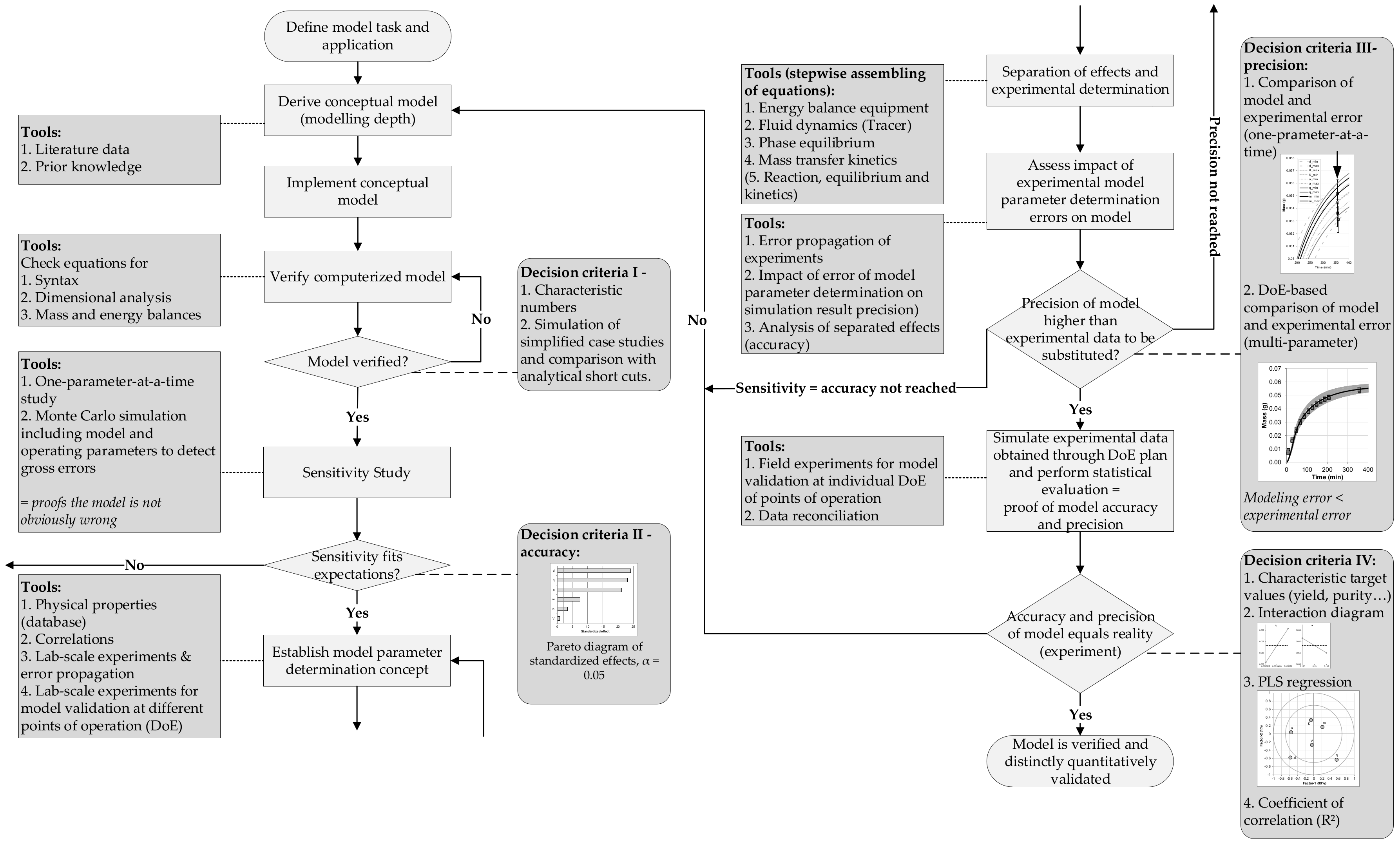
- Determination of model parameter data follows a distinct concept to guarantee accuracy and precision needed as well as the independence of each mechanism contributing within the model.
- Models are based on theory—and experiments! (as described above)—and industrial acceptance for theory is only given if it is validated by reality. Therefore, a distinct workflow with quantitative decision criteria is needed for model validation. This has been proposed and applied successfully [94,95,96]—and is available.
- An additional motivation for companies to industrialize modeling in biologics manufacturing should be the value of ownership of product related manufacturing data summarizing knowledge and experience of many hundreds, even thousands of man years, being a core asset for any manufacturing company. Moreover, taking into account that e.g., Samsung has already invested in 3 custom manufacturing organization (CMO) plants means that a more IT-based big-consortium company has direct access to manufacturing data [97]. Other IT-based companies even more dedicated to artificial intelligence may join that strategy of getting data access and generating value by product diversification.
3. Solutions
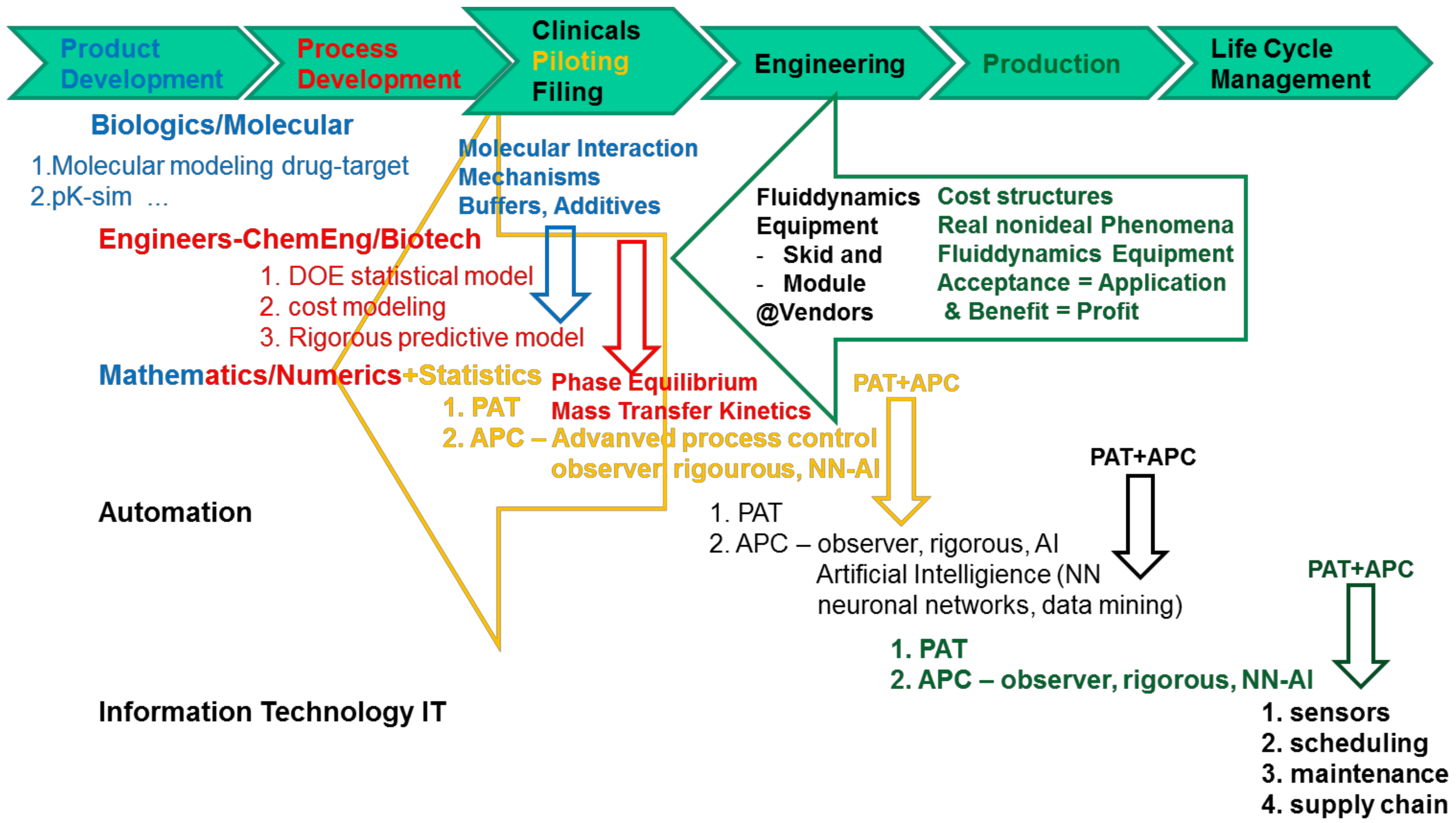
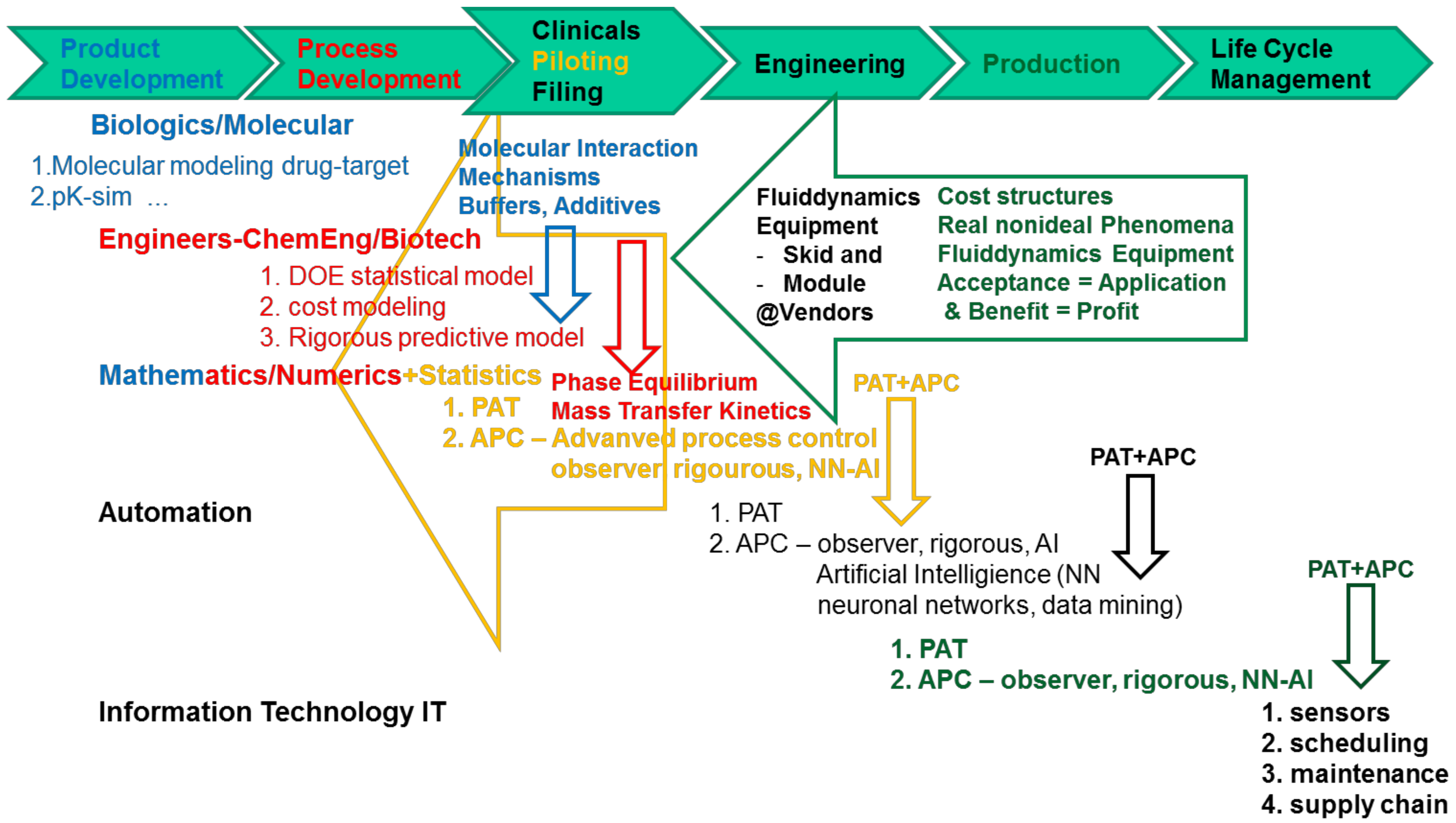
- Any interdisciplinary team must be set up to cover the following knowledge, background, and skills, see Figure 2:
- Biochemistry/Biotechnology for chemometrics on the molecular level for PAT sensors and PCA/PLS regression models, metabolomics for USP (up-stream processing) modeling
- Mathematics to support numerical solutions of process modeling, transferring models from process development towards engineering i.e., advanced (model-based) process control
- Statistical expertise for DoE design in process development, as well as regression modeling in analytical PAT via PCA/PLS
- Engineering i.e., Chemical/Biotechnology Engineering for process modeling combined with experimental model parameter determination in miniaturized laboratory scale, model validation experiments and process development and design
- as well as for operation of process development laboratory scale-down equipment automated and PAT equipped, piloting and manufacturing
- PAT sensor implementation
- Automation/process control system design including advanced process control and process analytical technology
- IT-experts for protocols, data bases, data access, interfaces, safety and security
- The largest challenge seems to be the necessary change in mind-set. Well established procedures and habits have to be changed by process modeling combined with experimental model parameter determination—instead of full experimental process development. The challenge lies not in manufacturing or piloting but in process development. This is a lesson learned from chemical industry in recent years/decades [45].
- Access to data must be organized. To provide long-term development, an internal database is recommended, which must be maintained by experts who support the simulation team with recommendations and measurements for data quality control. Setting up any in-house cloud seems to be the key value-asset of any company future.
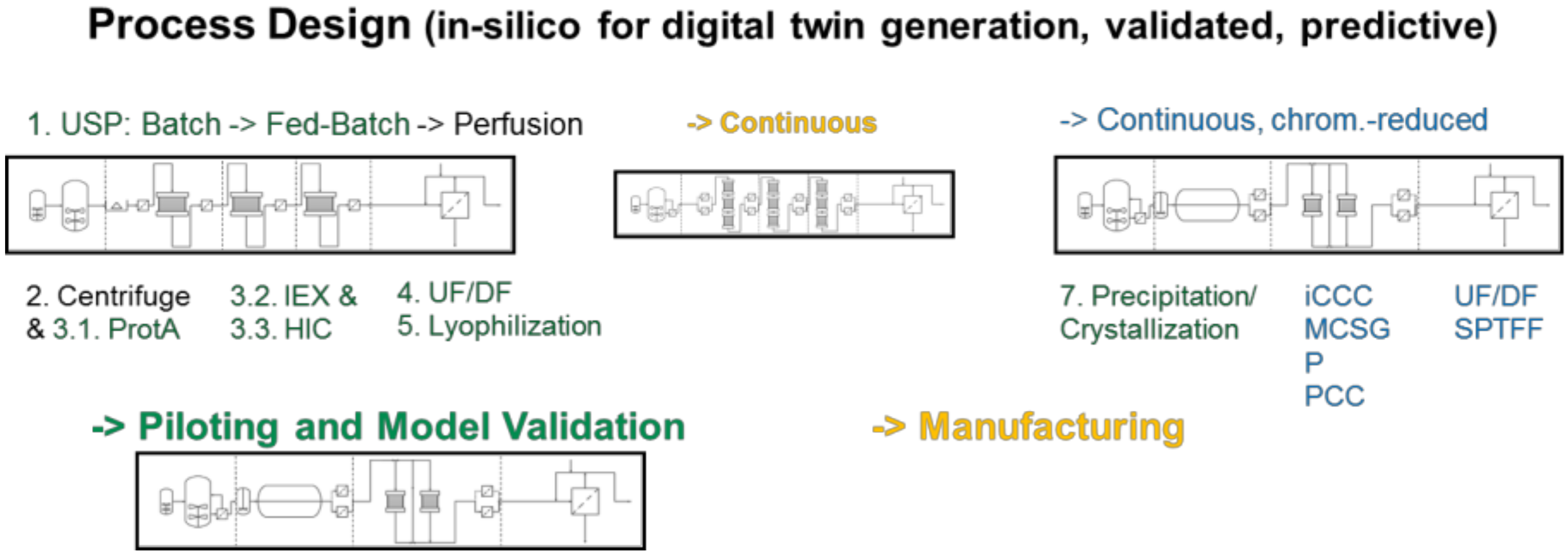
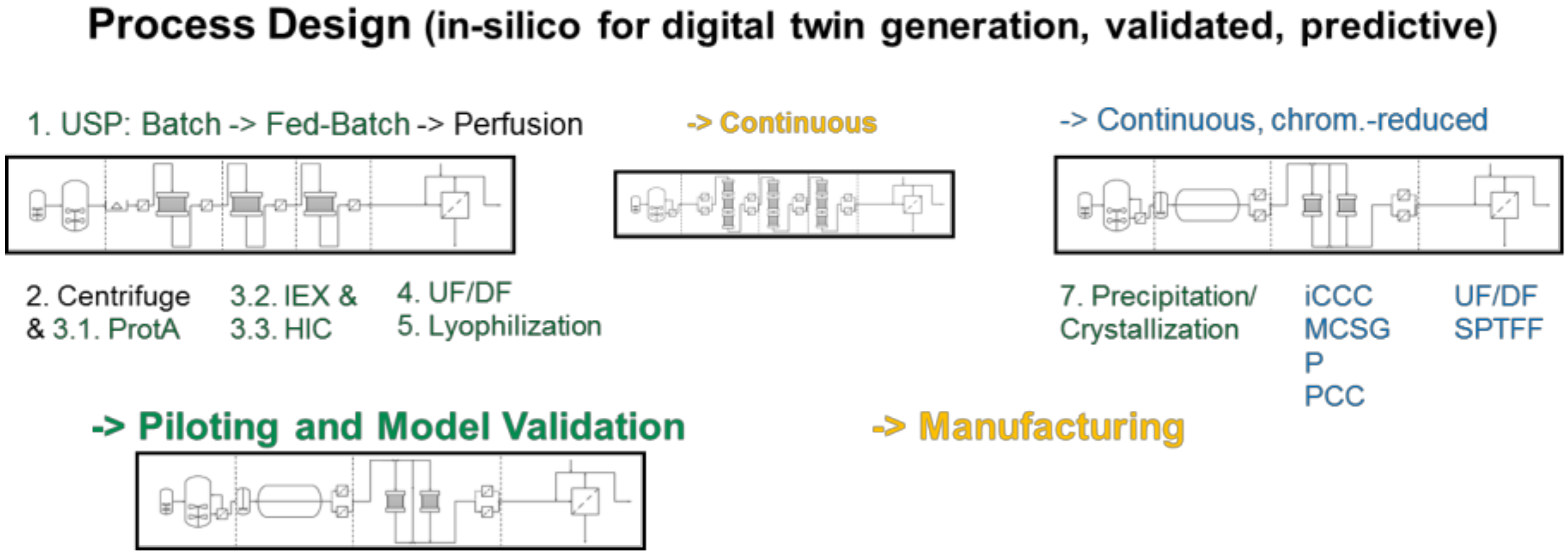
- At first, standardized laboratory equipment is used for each unit operation such as upstream fermentation (USP), Ultra-/Diafiltration (UF/DF), one or many chromatography steps, aqueous two-phase extraction (ATPE), precipitation or crystallization, as well as final lyophilization.
- This includes PAT-tools for model parameter determination and application of a DoE setup in the scale of few liters’ fermentation, few 100 cm2 membrane area and chromatography column volumes of few 10–100 ml.
- An existing DoE with risk analysis is applied for QbD-documentation and to validate the modeling and model parameter determination concept
- based on the validated process models process design studies with cost evaluations are performed.
- This leads to a decision for a best-in-class process in silico a priori, which
- is finally experimentally validated at pilot-scale to prove technical feasibility.
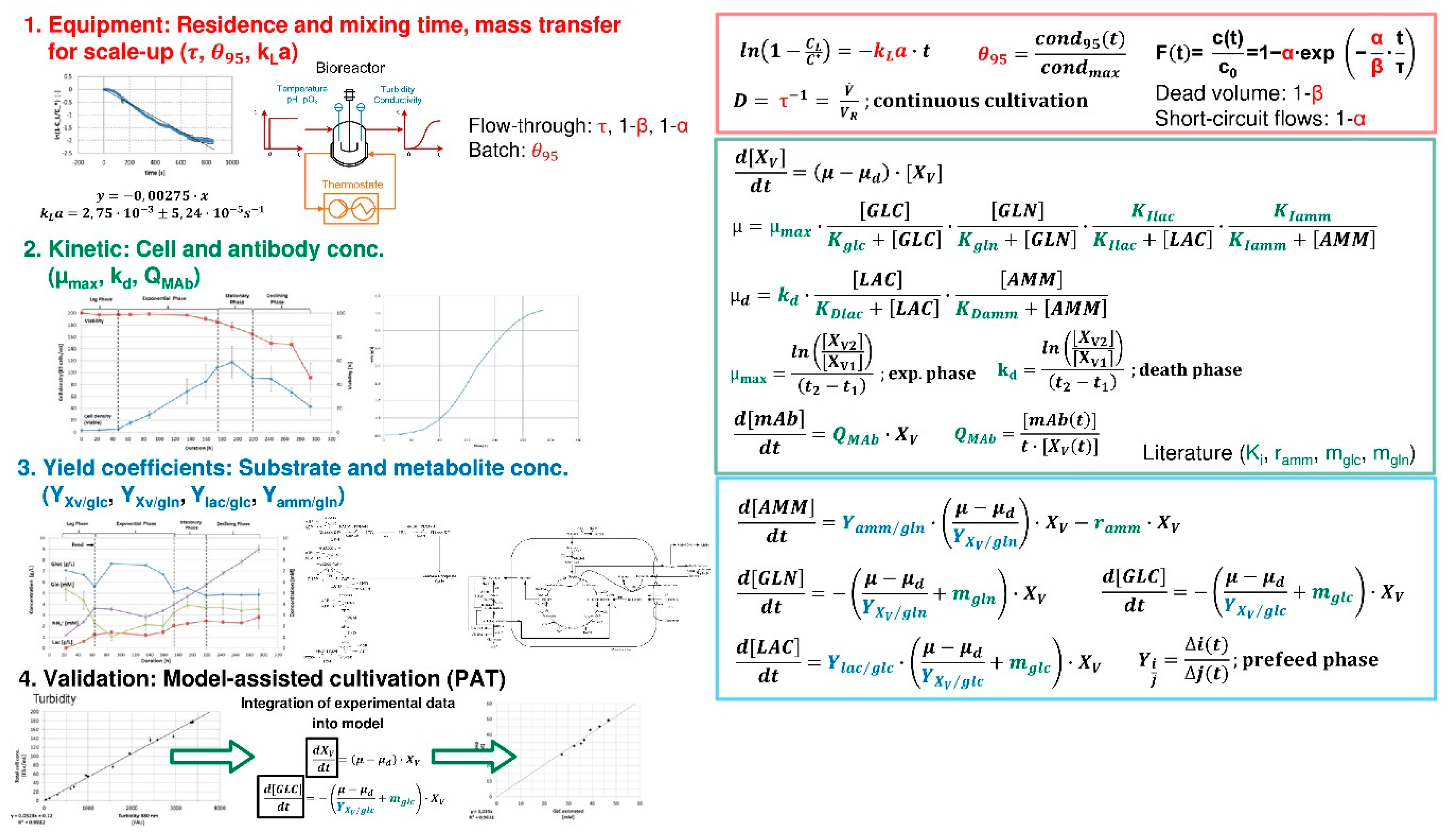
3.1. USP Fermentation Fed-Batch and Perfusion
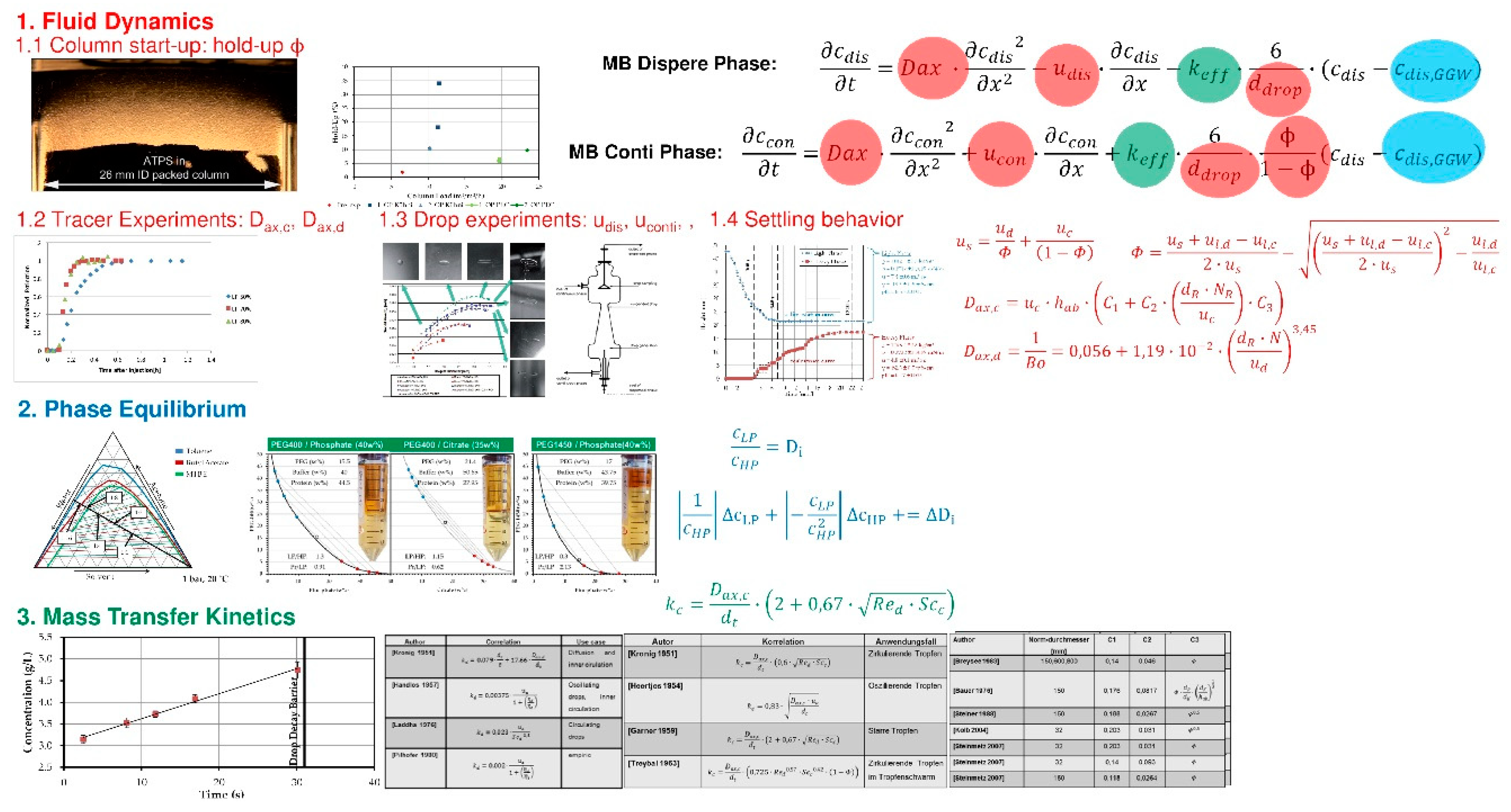
3.2. Capture, LLE, Cell Separation and Clarification
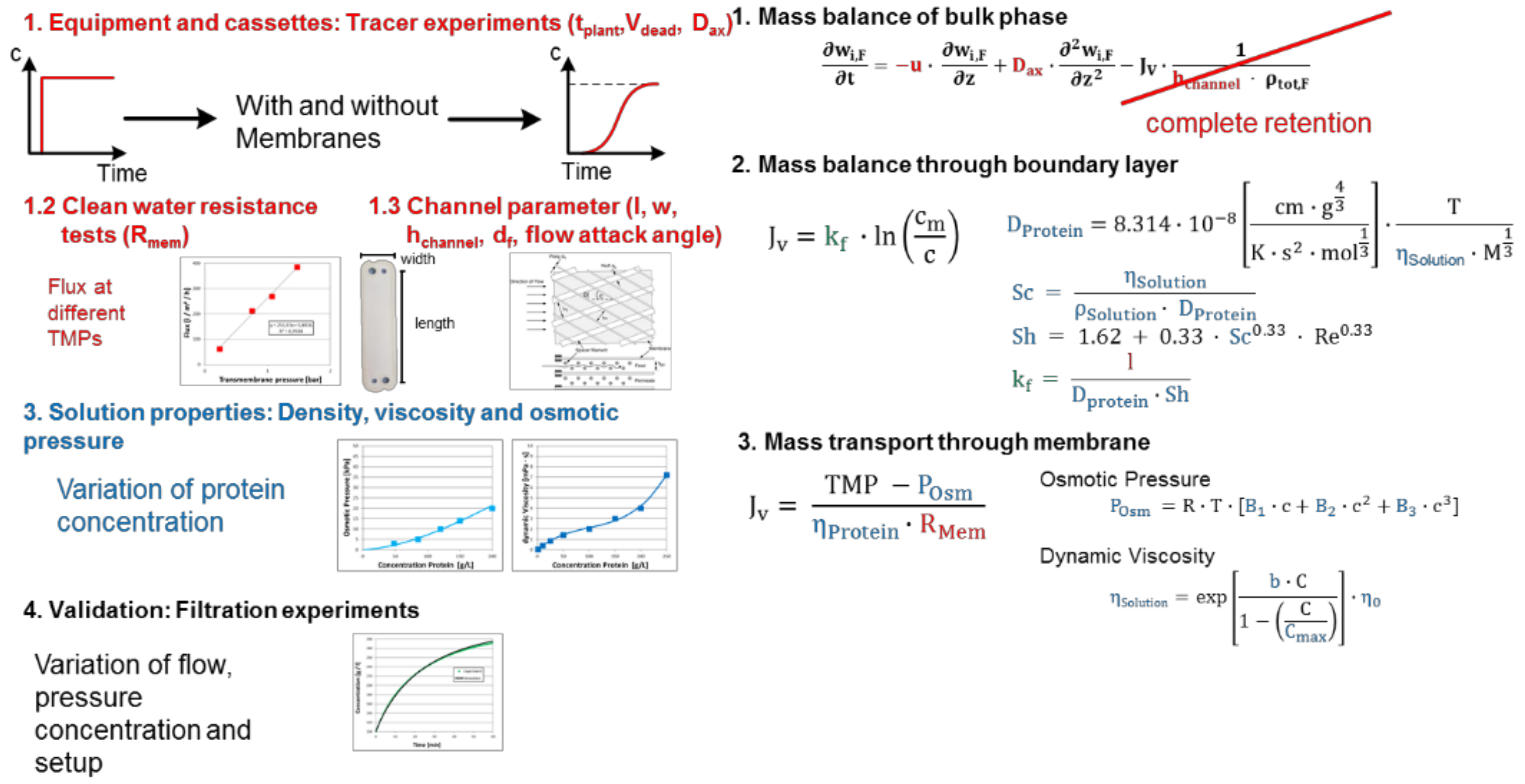
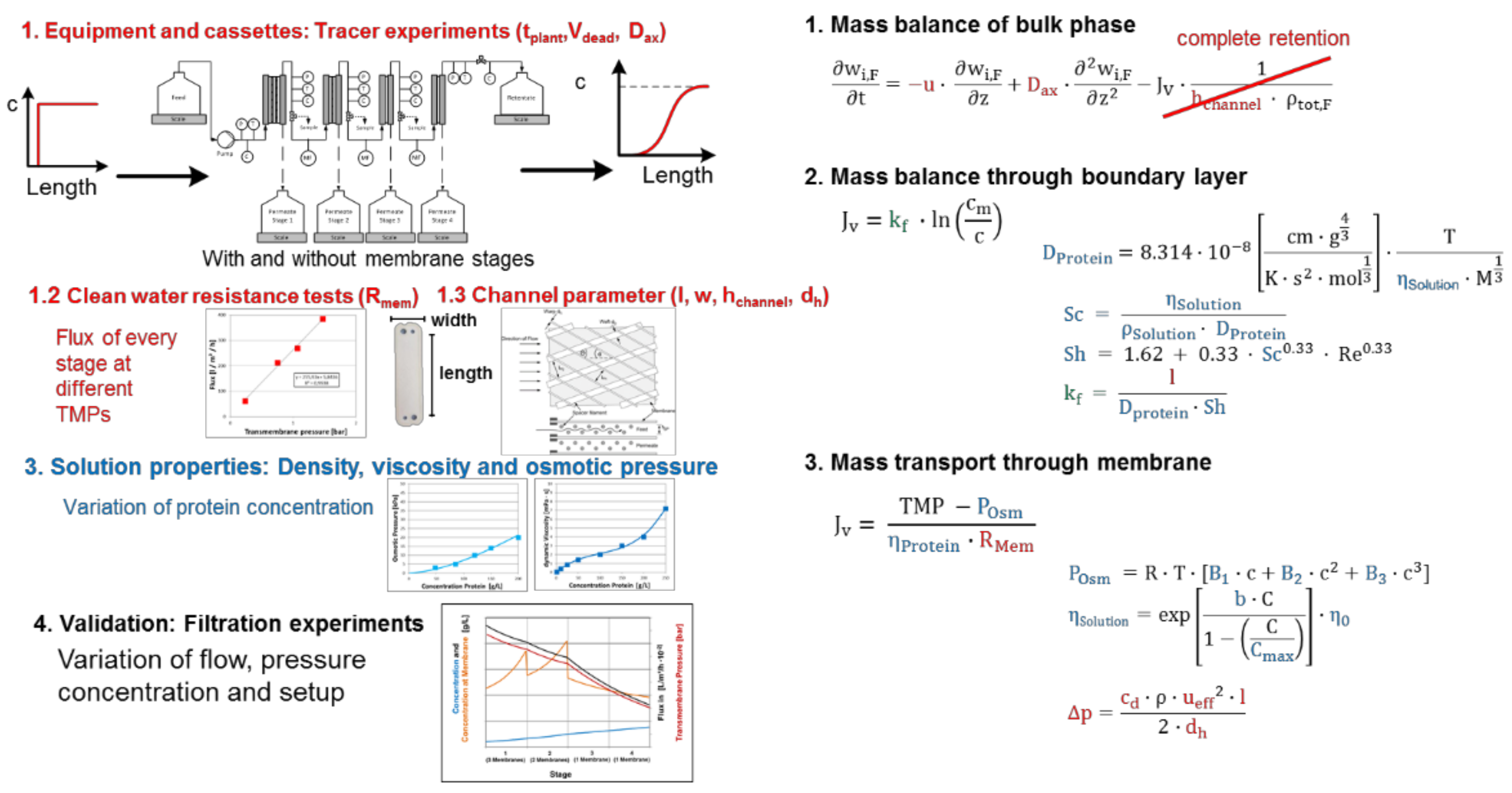
3.3. UF/DF, SPTFF for Concentration and Buffer Exchange
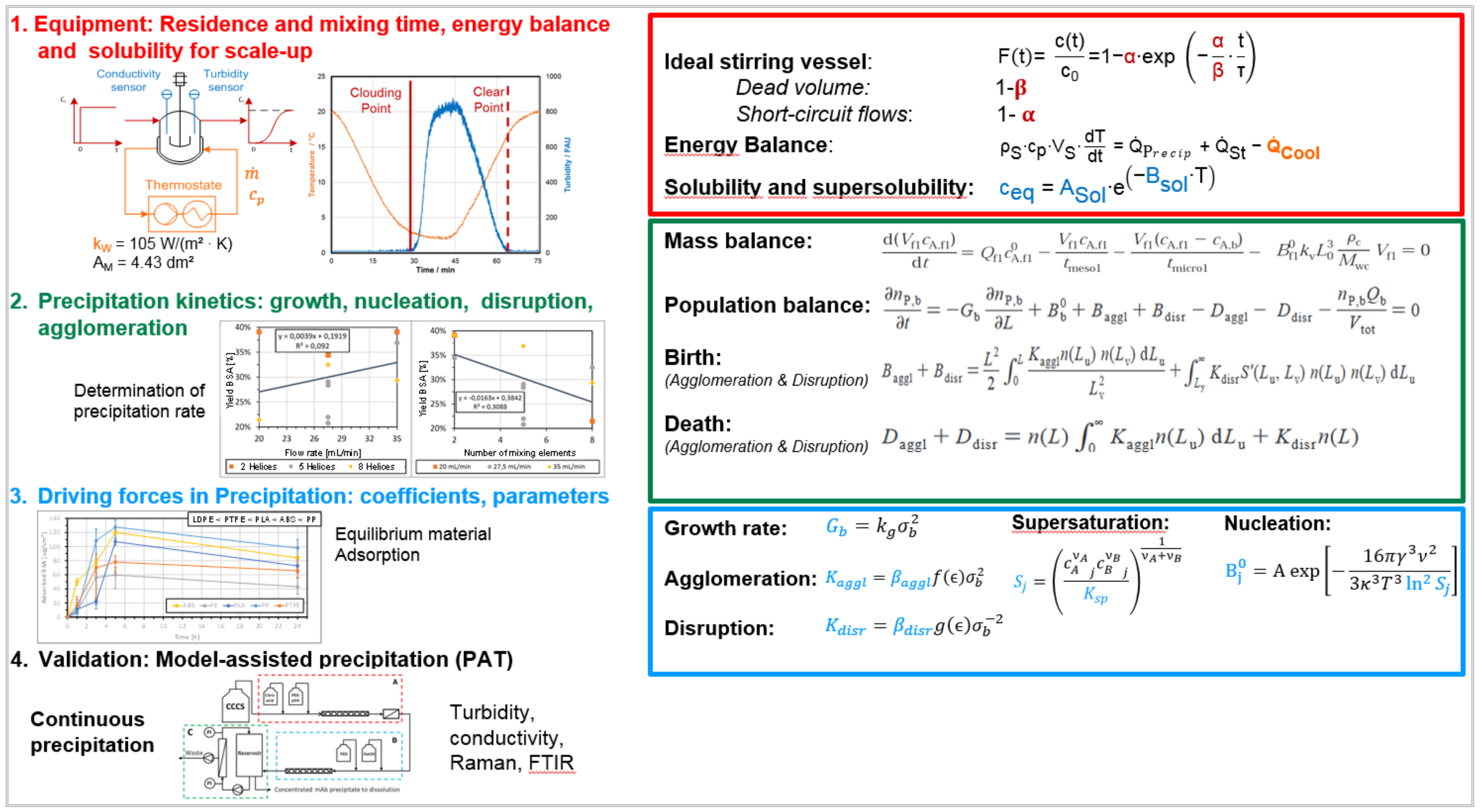
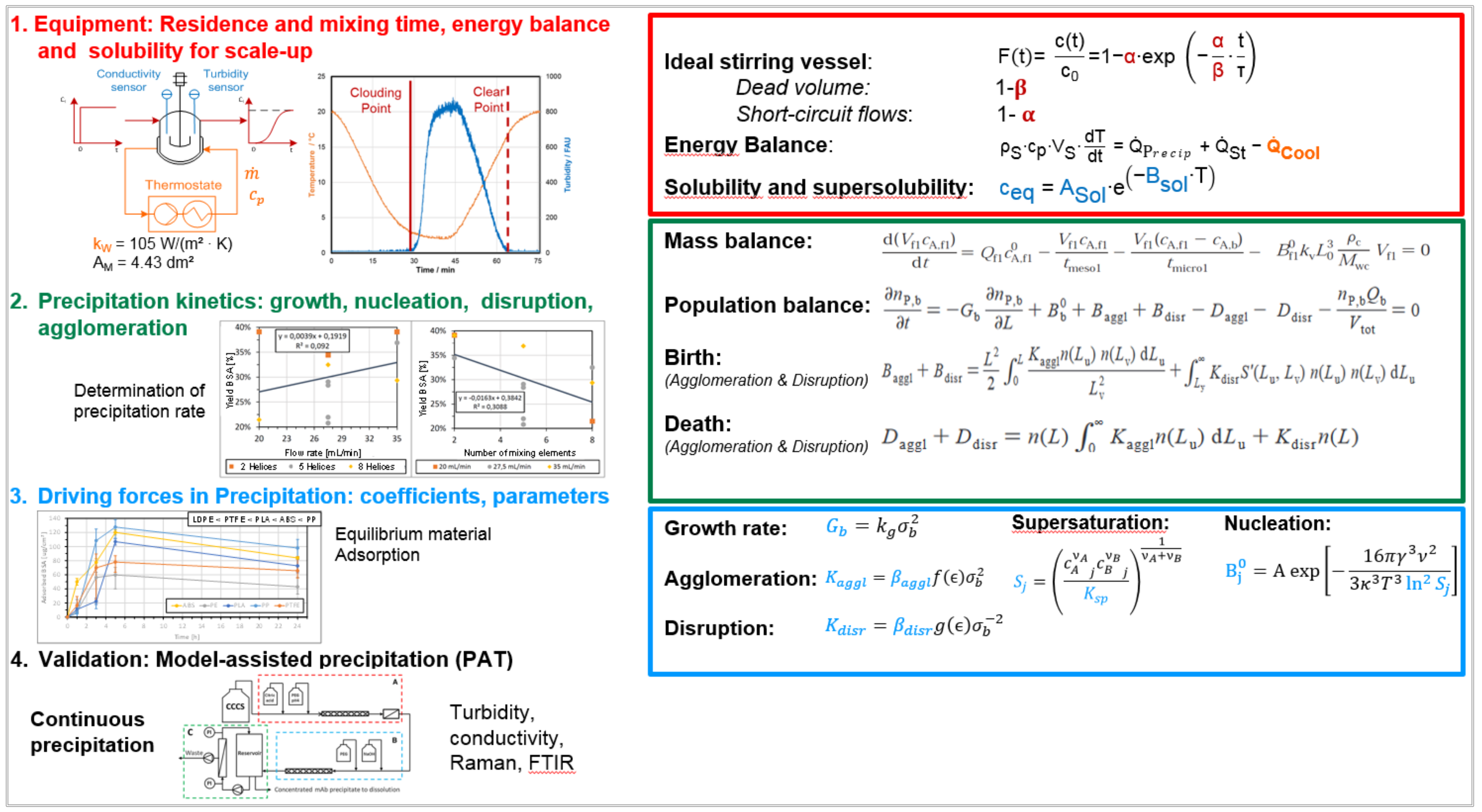
3.4. Precipitation/Crystallization
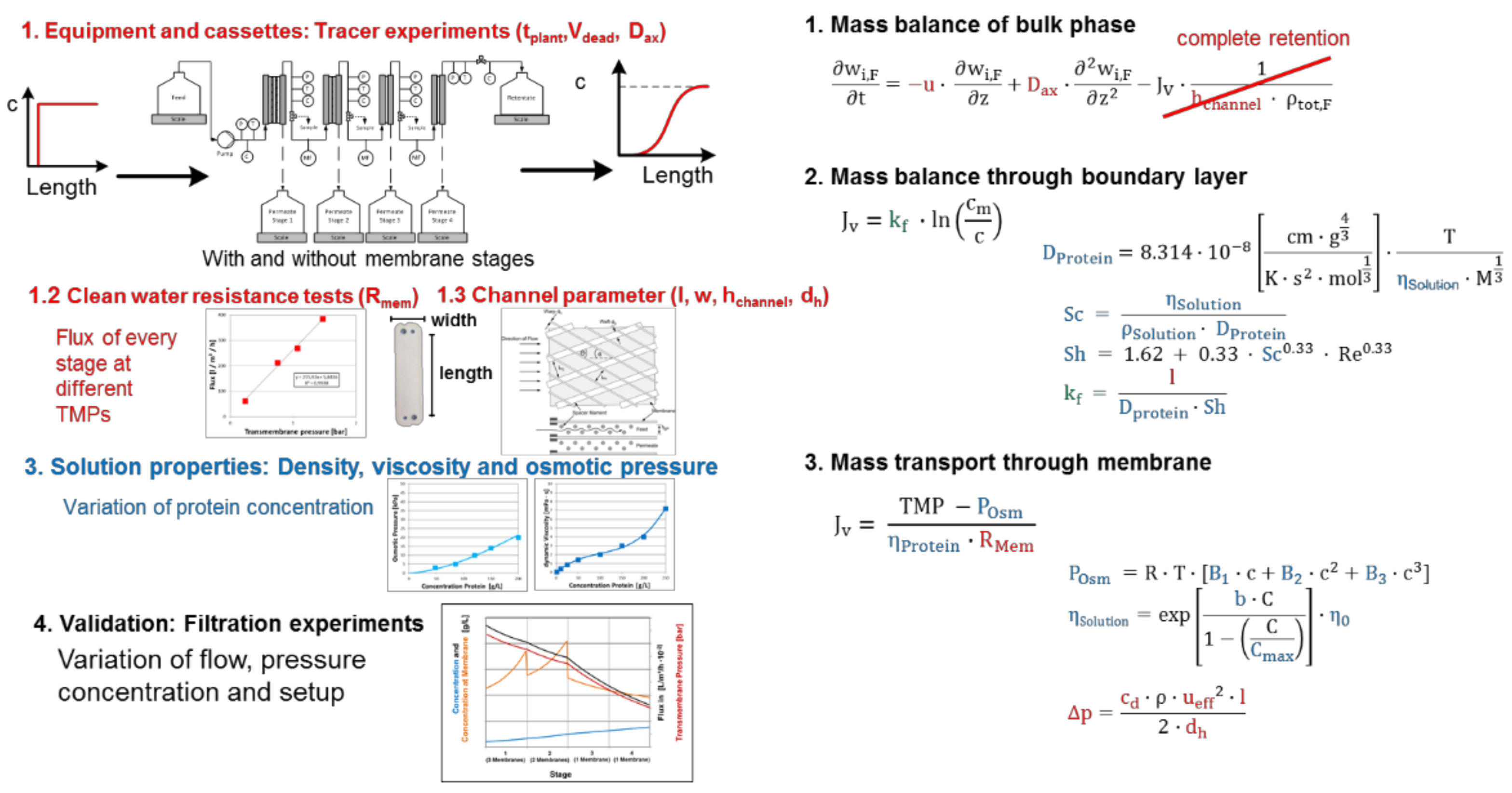
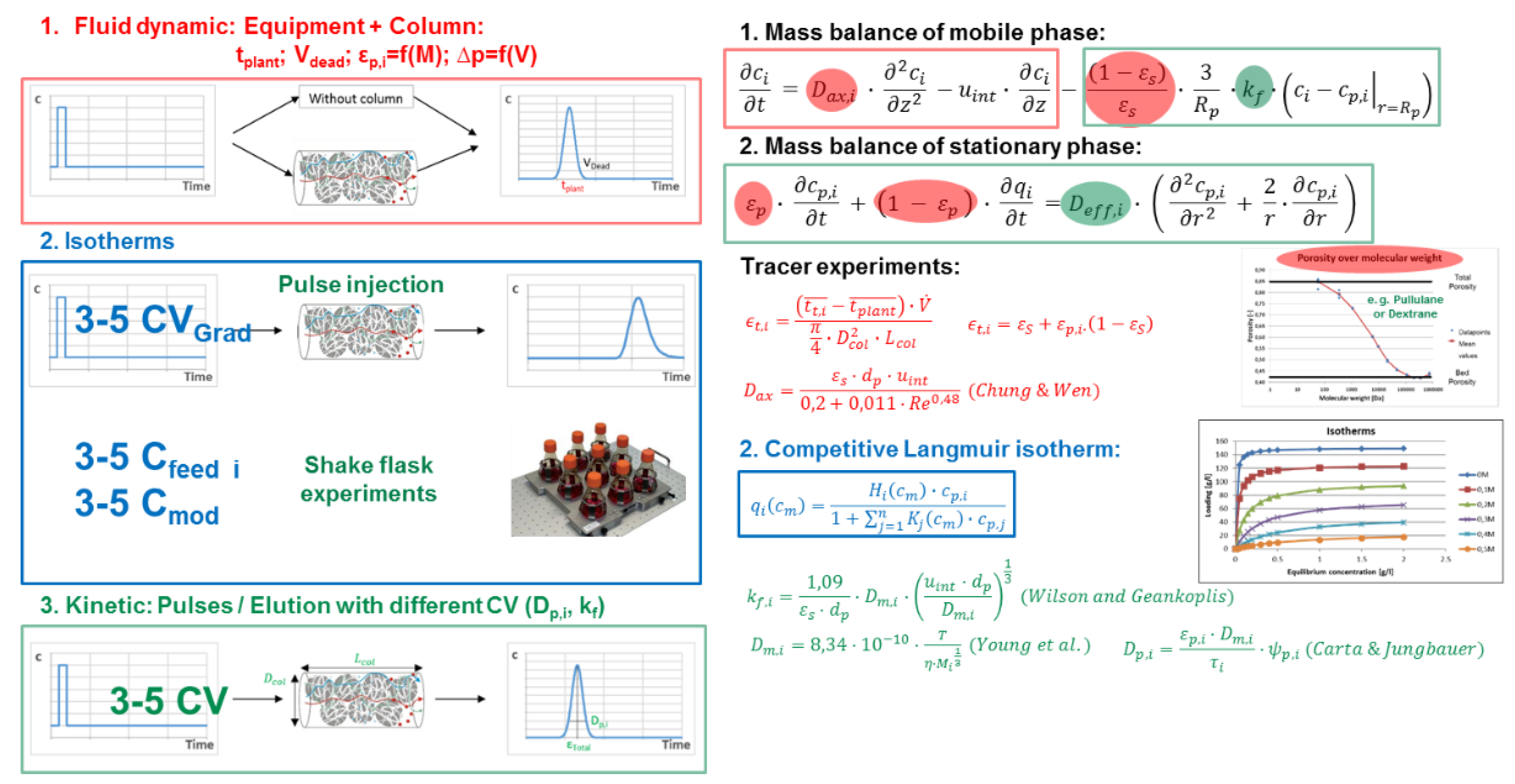
3.5. Chromatography, Membrane Adsorption
- Fluid dynamics (red)
- Isotherms (blue)
- Mass transfer and Kinetics (green)
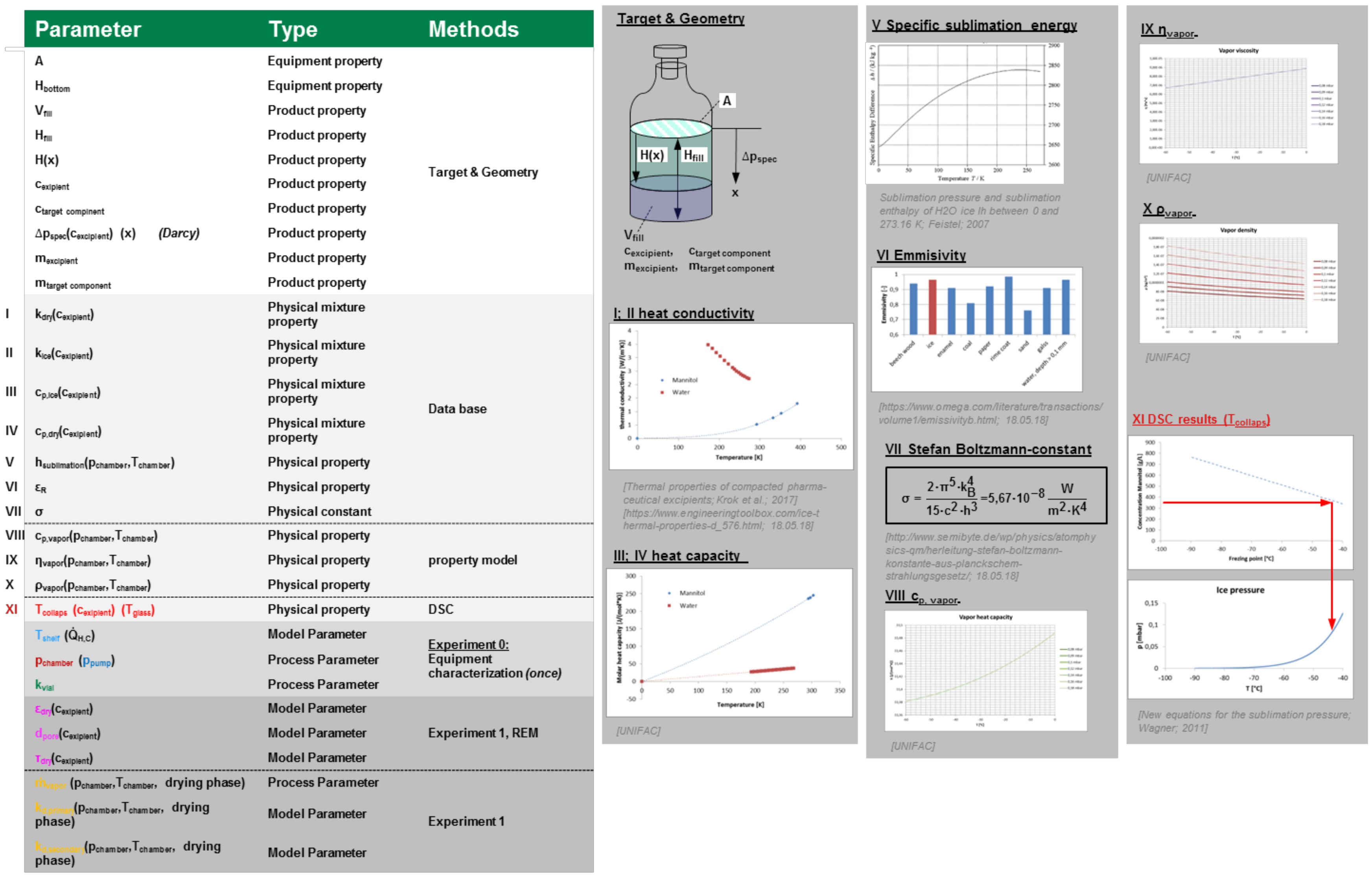
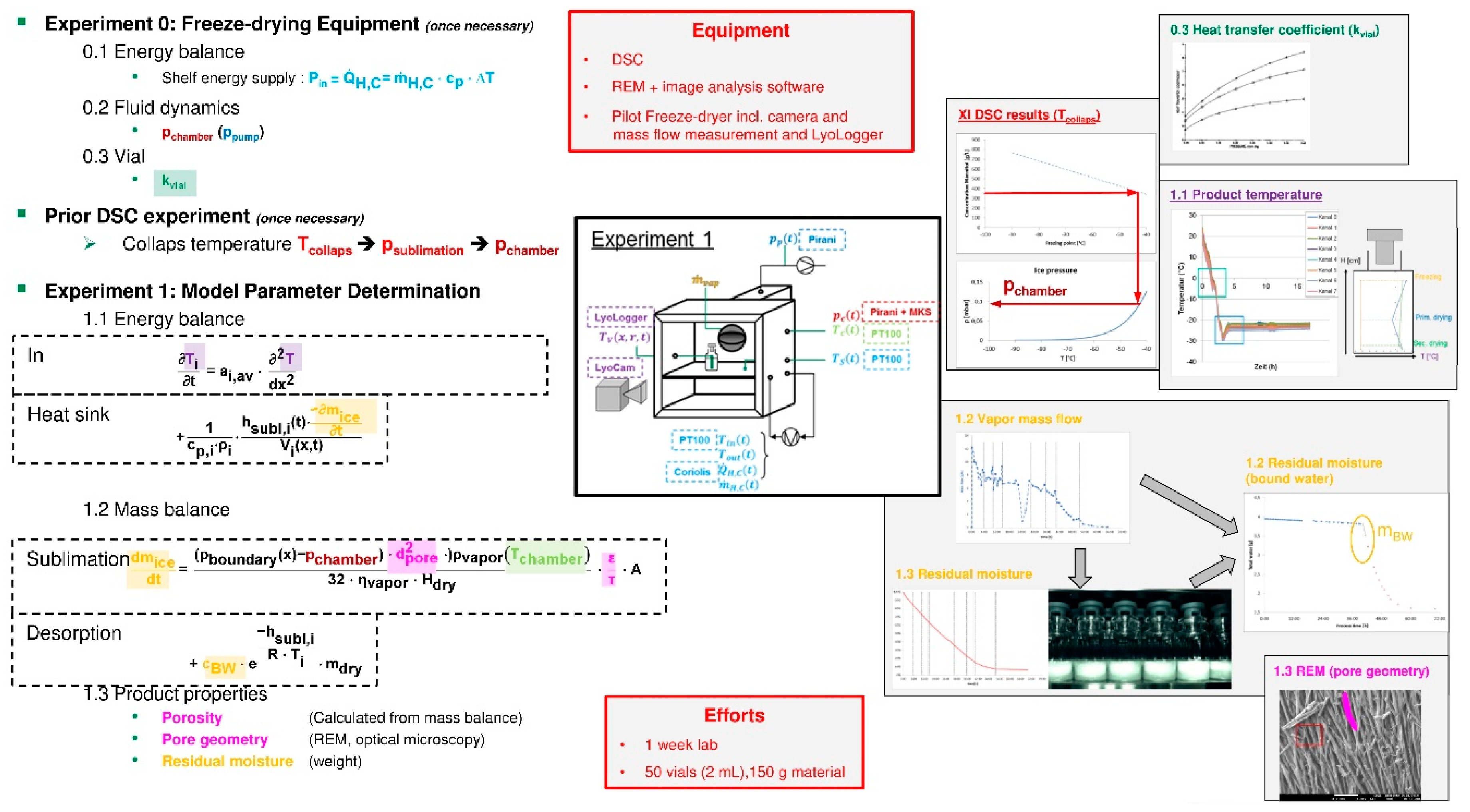
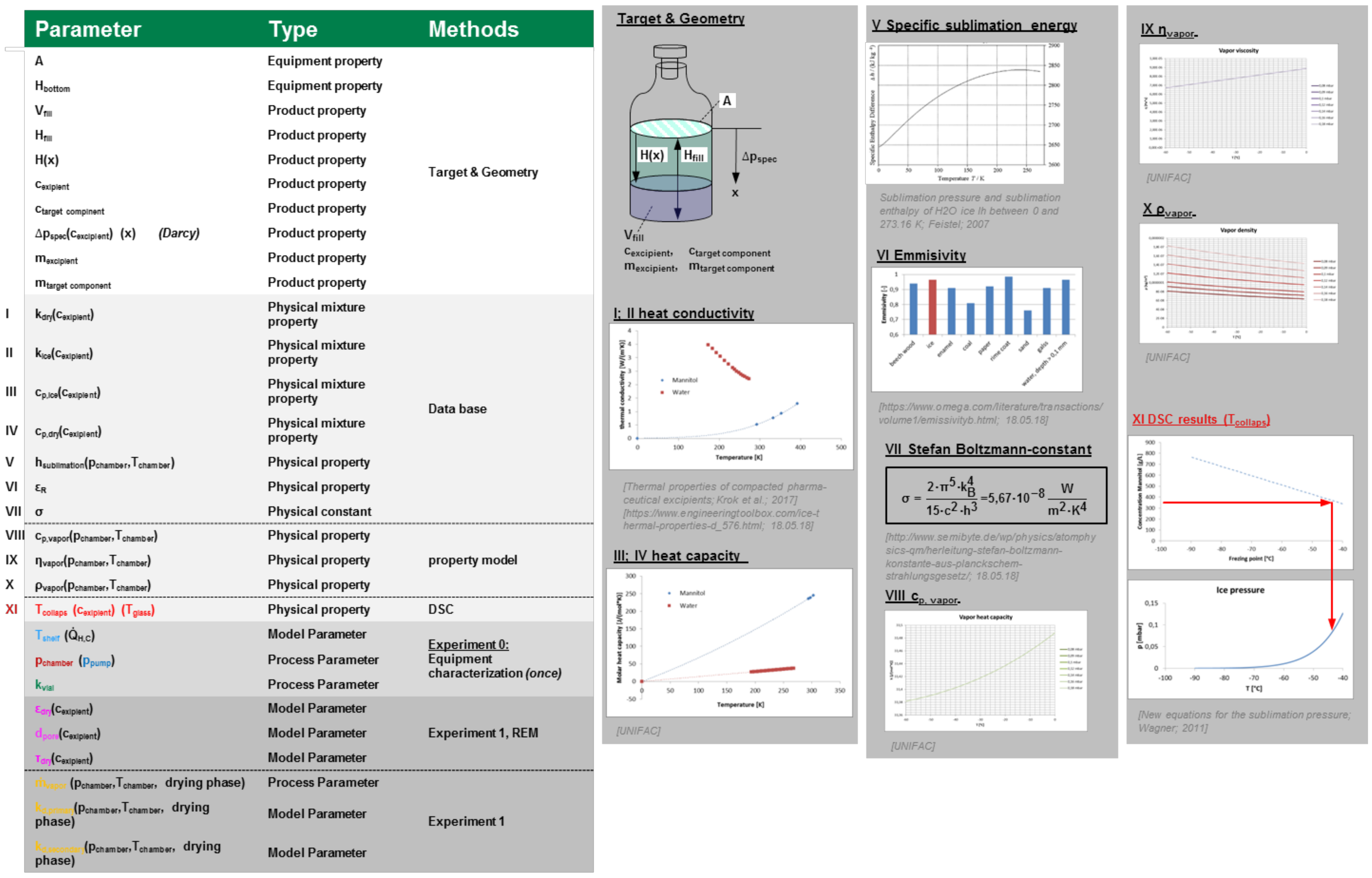
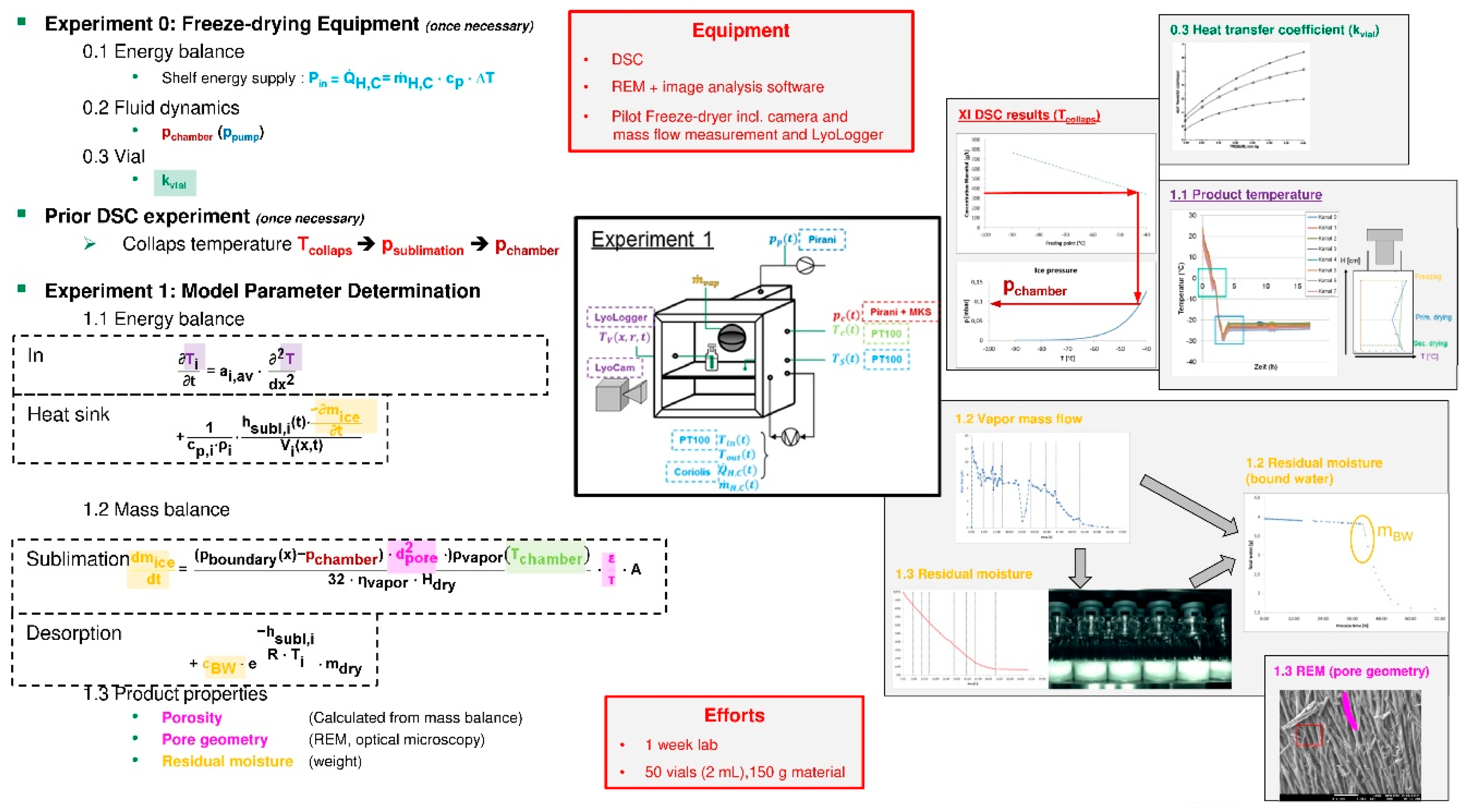
3.6. Lyophilization
- A distinct workflow is recommended for quantitative model validation in [94]
4. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | artificial intelligence |
| APC | advanced process control |
| ATF | alternating tangential flow filtration |
| ATPE | aqueous two-phase extraction |
| ATR | Attenuated total reflectance |
| Dax | axial dispersion coefficient |
| DF | diafiltration |
| DoE | design of experiments |
| DSC | differential scanning calorimetry |
| DSP | downstream processing |
| EMA | European Medicines Agency |
| FDA | Food and Drug administration |
| FTIR | Fourier-transformed infrared spectroscopy |
| HIC | hydrophobic interaction chromatography |
| iCCC | integrated counter-current chromatography |
| IEX | ion exchange chromatography |
| IOT | internet of things |
| IPC | inline process control analytics |
| keff | effective mass transfer coefficient |
| LLE | liquid–liquid extraction |
| mAb | monoclonal antibody |
| MCSGP | multicolumn countercurrent solvent gradient purification |
| MPC | model-based process control |
| MRSA | methicilline-resistent Staphylococcus aureus |
| MS | mass spectrometry |
| NN | neuronal networks |
| OQ | Operation qualification |
| PAT | process analytical technology |
| PCA | principle component analysis |
| PCS | process control system |
| Prot A | protein A chromatography |
| QA | quality assurance |
| QbD | quality-by-design |
| SOP | standard operation procedure |
| SPTFF | single-pass tangential flow filtration |
| UF | ultrafiltration |
| USP | upstream processing |
| VLP | virus-like particles |
References
- Shukla, A.A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Kumar, A. Upstream processes in antibody production: evaluation of critical parameters. Biotechnol. Adv. 2008, 26, 46–72. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Elvin, J.G.; Couston, R.G.; van der Walle, C.F. Therapeutic antibodies: market considerations, disease targets and bioprocessing. Int. J. Pharm. 2013, 440, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Vijayasankaran, N.; Shen, A.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. mAbs 2010, 2, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Gronemeyer, P.; Ditz, R.; Strube, J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering 2014, 1, 188–212. [Google Scholar] [CrossRef]
- Subramanian, G. (Ed.) Continuous Processing in Pharmaceutical Manufacturing; WILEY-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. mAbs 2010, 2, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.R.; Racher, A.J. Antibody production. Adv. Drug Delivery Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef]
- Thiess, H.; Zobel-Roos, S.; Gronemeyer, P.; Ditz, R.; Strube, J. Engineering Challenges of Continuous Biomanufacturing Processes (CBP). In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2017; pp. 69–107. [Google Scholar]
- Zobel-Roos, S.; Thiess, H.; Gronemeyer, P.; Ditz, R.; Strube, J. Continuous Chromatography as a Fully Integrated Process in Continuous Biomanufacturing. In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2017; pp. 369–393. [Google Scholar]
- Gronemeyer, P.; Thiess, H.; Zobel-Roos, S.; Ditz, R.; Strube, J. Integration of Upstream and Downstream in Continuous Biomanufacturing. In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2017; pp. 481–510. [Google Scholar]
- Strube, J.; Ditz, R.; Kornecki, M.; Huter, M.; Schmidt, A.; Thiess, H.; Zobel-Roos, S. Process Intensification in Biologics Manufacturing. Chem. Eng. Process. 2018, 133, 278–293. [Google Scholar] [CrossRef]
- Hanna, E.; Rémuzat, C.; Auquier, P.; Toumi, M. Advanced therapy medicinal products: current and future perspectives. J. Mark. Access Health Policy 2016, 4, 31036. [Google Scholar] [CrossRef]
- Martins, J.P.; Santos, J.M.; de Almeida, J.M.; Filipe, M.A.; de Almeida, M.V.T.; Almeida, S.C.P.; Água-Doce, A.; Varela, A.; Gilljam, M.; Stellan, B.; et al. Towards an advanced therapy medicinal product based on mesenchymal stromal cells isolated from the umbilical cord tissue: Quality and safety data. Stem Cell Res. Ther. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Ramqvist, T.; Andreasson, K.; Dalianis, T. Vaccination, immune and gene therapy based on virus-like particles against viral infections and cancer. Expert Opin. Biol. Ther. 2007, 7, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Warnock, J.N.; Merten, O.-W.; Al-Rubeai, M. Cell culture processes for the production of viral vectors for gene therapy purposes. Cytotechnology 2006, 50, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, N.; Streatfield, S.J.; Yusibov, V. Virus-like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine 2012, 31, 58–83. [Google Scholar] [CrossRef]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.T.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Vicente, T.; Roldão, A.; Peixoto, C.; Carrondo, M.J.T.; Alves, P.M. Large-scale production and purification of VLP-based vaccines. J. Invertebr. Pathol. 2011, 107, S42–S48. [Google Scholar] [CrossRef]
- Cho, B.S.; Kim, J.O.; Ha, D.H.; Yi, Y.W. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res. Ther. 2018, 9, 187. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef]
- Watson, D.C.; Bayik, D.; Srivatsan, A.; Bergamaschi, C.; Valentin, A.; Niu, G.; Bear, J.; Monninger, M.; Sun, M.; Morales-Kastresana, A.; et al. Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials 2016, 105, 195–205. [Google Scholar] [CrossRef] [PubMed]
- ICH. Quality Risk Management Q9, 2005 (Step 4 Version). Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf (accessed on 17 January 2018).
- ICH. Pharmaceutical Quality System Q10, 2008 (Step 4 Version). Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf (accessed on 17 January 2018).
- ICH. Pharmaceutical Development Q8 (R2), 2009 (Step 4 Version). Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf (accessed on 2 January 2015).
- ICH. Development and Manufacturing of Drug Substances Q11, 2013 (Step 4 Version). Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf (accessed on 17 January 2018).
- Express-scripts.com. The Need for U.S. Biosimilars. Available online: http://lab.express-scripts.com/lab/insights/drug-options/~/link.aspx?_id=905e58d6e6494fb4ae1b2581566b3538&_z=z (accessed on 17 November 2018).
- PharmTech. Biosimilars and Follow-on-Biologics Market to Hit $35 Billion Globally by 2020. Available online: http://www.pharmtech.com/biosimilars-and-follow-biologics-market-hit-35-billion-globally-2020 (accessed on 3 November 2018).
- Epstein, M.S.; Ehrenpreis, E.D.; Kulkarni, P.M. Biosimilars: The need, the challenge, the future: The FDA perspective. Am. J. Gastroenterol. 2014, 109, 1856–1859. [Google Scholar] [CrossRef]
- JSR Life Sciences. Chromassette. Available online: https://www.jsrlifesciences.com/bioprocess/chromassette (accessed on 6 November 2018).
- Subramanian, G. (Ed.) Continuous Biomanufacturing. In Innovative Technologies and Methods; WILEY-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Schofield, M. Current state of the art in continuous bioprocessing. Biotechnol. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bisschops, M.; Schofield, M.; Grace, J. Two Mutually Enabling Trends: Continuous Bioprocessing and Single-Use Technologies. In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2017; pp. 149–170. [Google Scholar]
- Clutterbuck, A.; Beckett, P.; Lorenzi, R.; Sengler, F.; Bisschop, T.; Haas, J. Single-Pass Tangential Flow Filtration (SPTFF) in Continuous Biomanufacturing. In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2017; pp. 423–456. [Google Scholar]
- Pollock, J.; Coffman, J.; Ho, S.V.; Farid, S.S. Integrated continuous bioprocessing: Economic, operational, and environmental feasibility for clinical and commercial antibody manufacture. Biotechnol. Prog. 2017, 33, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Karst, D.J.; Steinebach, F.; Morbidelli, M. Continuous integrated manufacturing of therapeutic proteins. Curr. Opin. Biotechnol. 2018, 53, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Bioprozesstechnik; Chmiel, H. (Eds.) 3., neu bearb. Aufl.; Spektrum Akademischer Verlag: Heidelberg, Germany, 2011. [Google Scholar]
- Ladisch, M.R. Bioprocess Engineering (Biotechnology). In Van Nostrand’s Scientific Encyclopedia; John Wiley & Sons, Inc.: New York, NY, USA, 2005. [Google Scholar]
- Ben Yahia, B.; Malphettes, L.; Heinzle, E. Macroscopic modeling of mammalian cell growth and metabolism. Appl. Microbiol. Biotechnol. 2015, 99, 7009–7024. [Google Scholar] [CrossRef] [PubMed]
- Schuler, H. Prozessimulation; WILEY-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Carta, G.; Jungbauer, A. Protein Chromatography. Process Development and Scale-Up; WILEY-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Strube, J. Prädiktive Modellierung von Trennverfahren. Chem. Ing. Tech. 2012, 84, 867. [Google Scholar] [CrossRef]
- Kroll, P.; Hofer, A.; Ulonska, S.; Kager, J.; Herwig, C. Model-Based Methods in the Biopharmaceutical Process Lifecycle. Pharm. Res. 2017, 34, 2596–2613. [Google Scholar] [CrossRef]
- Nfor, B.K.; Verhaert, P.D.E.M.; van der Wielen, L.A.M.; Hubbuch, J.; Ottens, M. Rational and systematic protein purification process development: The next generation. Trends Biotechnol. 2009, 27, 673–679. [Google Scholar] [CrossRef]
- Zobel-Roos, S. Entwicklung, Modellierung und Validierung von Integrierten Kontinuierlichen Gegenstrom-Chromatographie-Prozessen, 1. Auflage; Shaker: Herzogenrath, Germany, 2018. [Google Scholar]
- Strube, J. Technische Chromatographie: Auslegung, Optimierung, Betrieb und Wirtschaftlichkeit; Shaker: Aachen, Germany, 2000. [Google Scholar]
- Wiesel, A.; Schmidt-Traub, H.; Lenz, J.; Strube, J. Modelling gradient elution of bioactive multicomponent systems in non-linear ion-exchange chromatography. J. Chromatogr. A 2003, 1006, 101–120. [Google Scholar] [CrossRef]
- Thiess, H.; Leuthold, M.; Grummert, U.; Strube, J. Module design for ultrafiltration in biotechnology: Hydraulic analysis and statistical modeling. J. Membr. Sci. 2017, 540, 440–453. [Google Scholar] [CrossRef]
- Kornecki, M.; Strube, J. Process Analytical Technology for Advanced Process Control in Biologics Manufacturing with the Aid of Macroscopic Kinetic Modeling. Bioengineering 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Richter, M.; Rudolph, F.; Strube, J. Integration of Aqueous Two-Phase Extraction as Cell Harvest and Capture Operation in the Manufacturing Process of Monoclonal Antibodies. Antibodies 2017, 6, 21. [Google Scholar] [CrossRef]
- Thiess, H.; Schmidt, A.; Strube, J. Development of a Scale-up Tool for Pervaporation Processes. Membranes 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Bayer. Open Systems Pharmacology Suite with PK-Sim and MoBi: Unmatched Flexibility—Unlimited Transparency. Available online: http://www.systems-biology.com/products/pk-sim.html (accessed on 2 November 2018).
- Meyer, E.F.; Swanson, S.M.; Williams, J.A. Molecular modelling and drug design. Pharmacol. Ther. 2000, 85, 113–121. [Google Scholar] [CrossRef]
- Duch, W.; Swaminathan, K.; Meller, J. Artificial Intelligence Approaches for Rational Drug Design and Discovery. CPD 2007, 13, 1497–1508. [Google Scholar] [CrossRef]
- Minsky, M. Matter, Mind and Models. In Proceedings of the IFIP Congress, New York, NY, USA, 24–29 May 1965; pp. 45–49. [Google Scholar]
- Guidance for Industry—Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice; FDA: Silver Spring, MD, USA, 2004.
- Guidance for Industry CMC Postapproval Manufacturing Changes to Be Documented in Annual Reports; FDA: Silver Spring, MD, USA, 2014.
- Guidance for Industry Changes to an Approved NDA or ANDA; FDA: Silver Spring, MD, USA, 2004.
- Boschert, S.; Rosen, R. 5. Digital Twin—The Simulation Aspect. In Mechatronic Futures; Hehenberger, P., Bradley, D., Eds.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Rosen, R.; von Wichert, G.; Lo, G.; Bettenhausen, K.D. About the Importance of Autonomy and Digital Twins for the Future of Manufacturing. Ifac-Pap. 2015, 48, 567–572. [Google Scholar] [CrossRef]
- Eigner, M.; Koch, W.; Muggeo, C. Modellbasierter Entwicklungsprozess Cybertronischer Systeme; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Brown, J. Siemens Digital Twin Strategy. Available online: https://tech-clarity.com/siemens-plm-twin/7017 (accessed on 3 November 2018).
- Galvanauskas, V.; Simutis, R.; Lübbert, A. Hybrid process models for process optimisation, monitoring and control. Bioprocess Biosyst. Eng. 2004, 26, 393–400. [Google Scholar] [CrossRef]
- Ji, Y. Model Based Process Design for Bioprocess Optimisation: Case Studies on Precipitation with Its Applications in Antibody Purification; University College London: London, UK, 2012. [Google Scholar]
- McCullagh, P. What is a statistical model? Ann. Stat. 2002, 30, 1225–1310. [Google Scholar] [CrossRef]
- Dreyfus, H.L. What Computers Can’t Do. The Limits of Artificial Intelligence; Harper & Row: New York, NY, USA, 1979. [Google Scholar]
- Domingos, P. The Master Algorithm. How the Quest for the Ultimate Learning Machine Will Remake Our World, First Paperback ed.; Basic Books: New York, NY, USA, 2018. [Google Scholar]
- Nielsen, H.; Brunak, S.; von Heijne, G. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. Des. Sel. 1999, 12, 3–9. [Google Scholar] [CrossRef]
- CNN. AI Set to Exceed Human Brain Power. Available online: http://edition.cnn.com/2006/TECH/science/07/24/ai.bostrom/ (accessed on 3 November 2018).
- Dreyfus, H.L. What Computers Still Can’t Do. A Critique of Artificial Reason; 6th print; MIT Press: Cambridge, MA, USA, 1999. [Google Scholar]
- Bostrom, N. Superintelligence. Paths, Dangers, Strategies, 1st ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Charaniya, S.; Hu, W.-S.; Karypis, G. Mining bioprocess data: Opportunities and challenges. Trends Biotechnol. 2008, 26, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Burghaus, R.; Leineweber, D.; Lippert, J. Einsatz von Data-Mining zur Analyse eines Polymerprozesses. Chem. Ing. Tech. 2003, 75, 897–900. [Google Scholar] [CrossRef]
- Balakin, K.V. Pharmaceutical Data Mining. Approaches and Applications for Drug Discovery; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Müller, F.-J.; Schuppert, A. Few inputs can reprogram biological networks. Nature 2011, 478, E4. [Google Scholar] [CrossRef]
- Wolkenhauer, O.; Auffray, C.; Brass, O.; Clairambault, J.; Deutsch, A.; Drasdo, D.; Gervasio, F.; Preziosi, L.; Maini, P.; Marciniak-Czochra, A.; et al. Enabling multiscale modeling in systems medicine. Genome Med. 2014, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Kuepfer, L.; Schuppert, A. Systems Medicine in Pharmaceutical Research and Development. In Systems Medicine, 1st ed.; Schmitz, U., Wolkenhauer, O., Eds.; Humana Press: New York, NY, USA, 2016; pp. 87–104. [Google Scholar]
- Schuppert, A. Data Mining. Bayer Res. Mag. 2000, 16. [Google Scholar]
- Helling, C.; Strube, J. Modeling and Experimental Model Parameter Determination with Quality by Design for Bioprocesses. In Biopharmaceutical Production Technology, 1. Aufl.; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany, 2012; pp. 409–443. [Google Scholar]
- Helling, C.; Dams, T.; Gerwat, B.; Belousov, A.; Strube, J. Physical characterization of column chromatography: Stringent control over equipment performance in biopharmaceutical production. Trends Chromatogr. 2013, 2013, 55–71. [Google Scholar]
- Strube, J.; Zobel-Roos, S. ITVP Training Course; DSP. Purification of Biomolecules: Clausthal-Zellerfeld, Germany, 2016. [Google Scholar]
- Strube, J.; Ditz, R. ITVP Training Course; Process Chromatography: Clausthal-Zellerfeld, Germany.
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—Generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process Intensif. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Farid, S.S. Cost-effective bioprocess design for the manufacture of allogeneic CAR-T cell therapies using a decisional tool with multi-attribute decision-making analysis. Biochem. Eng. J. 2018, 137, 192–204. [Google Scholar] [CrossRef]
- Pereira Chilima, T.D.; Moncaubeig, F.; Farid, S.S. Impact of allogeneic stem cell manufacturing decisions on cost of goods, process robustness and reimbursement. Biochem. Eng. J. 2018, 137, 132–151. [Google Scholar] [CrossRef]
- Toumi, A.; Jürgens, C.; Jungo, C.; Maier, B.A.; Papavasileioum, V.; Petrides, D.P. Design and Optimization of a Large Scale Biopharmaceutical Facility Using Process Simulation and Scheduling Tools. Pharm. Eng. 2010, 30, 1–9. [Google Scholar]
- Strube, J.; Sommerfeld, S.; Lohrmann, M. Processes Development and Optimization for Biotechnology Production—Monoclonal Antibodies. In Bioseparation and Bioprocessing: A Handbook, 2.; Subramanian, G., Ed.; WILEY-VCH: Weinheim, Germany; New-York, NY, USA, 2007; pp. 65–99. [Google Scholar]
- Green, D.W.; Perry, R.H. (Eds.) Perry’s Chemical Engineers’ Handbook, 8th ed.; McGraw-Hill: New York, NY, USA, 2008. [Google Scholar]
- Peters, M.S.; Timmerhaus, K.D.; West, R.E. Plant Design and Economics for Chemical Engineers, 5th ed.; McGraw-Hill: Boston, MA, USA, 2003. [Google Scholar]
- Netzer, F. Digitale Transformation bei BASF—Fallstricke und Erfolgsbeispiele aus der Umsetzungspraxis. Chem. Ing. Tech. 2018, 90, 1293–1294. [Google Scholar] [CrossRef]
- Sixt, M.; Uhlenbrock, L.; Strube, J. Toward a Distinct and Quantitative Validation Method for Predictive Process Modelling—On the Example of Solid-Liquid Extraction Processes of Complex Plant Extracts. Processes 2018, 6, 66. [Google Scholar] [CrossRef]
- Sargent, R.G. Verification and Validation of Simulation Models. In Proceedings of the 2011 Winter Simulation Conference: (WSC), Phoenix, AZ, USA, 11–14 December 2011, Including the MASM (Modeling and Analysis for Semiconductor Manufacturing) Conference; Jain, S., Ed.; IEEE: Piscataway, NJ, USA, 2011; pp. 183–198. [Google Scholar]
- Schleisinger, S.; Crosbie, R.E.; Gagné, R.E. Terminology for model credibility. Simulation 1979, 32, 103–104. [Google Scholar] [CrossRef]
- Pharmaceutical Technology. Samsung BioLogics’ Third Manufacturing Facility, Songdo. Available online: https://www.pharmaceutical-technology.com/projects/samsung-biologics-third-manufacturing-facility-songdo/ (accessed on 17 November 2018).
- Strube, J.; Zobel-Roos, S.; Ditz, R. Chapter X: Process Chromatography. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chichester, UK, 2019. [Google Scholar]
- GE Healthcare. Available online: https://www.gehealthcare.com/ (accessed on 3 November 2018).
- Bio Rad. Available online: http://www.bio-rad.com/ (accessed on 3 November 2018).
- Pall Corporation. Available online: https://www.pall.com/ (accessed on 3 November 2018).
- Merck Millipore. Available online: http://www.merckmillipore.com (accessed on 3 November 2018).
- Kornecki, M.; Strube, J. Process analytical technology mechanisms in biologics manufacturing. Chem. Ing. Tech. 2018, 90, 1270. [Google Scholar] [CrossRef]
- Kornecki, M. Host Cell Proteins in Biologics Manufacturing. A Methodical and Systematic Integration of Upstream and Downstream Processing; Achema: Frankfurt am Main, Germany, 2018. [Google Scholar]
- Kornecki, M. Process Analytical Technology Mechanisms in Biologics Manufacturing; Achema: Frankfurt am Main, Germany, 2018. [Google Scholar]
- Kornecki, M.; Mestmäcker, F.; Zobel-Roos, S.; Heikaus de Figueiredo, L.; Schlüter, H.; Strube, J. Host Cell Proteins in Biologics Manufacturing: The Good, the Bad, and the Ugly. Antibodies 2017, 6, 13. [Google Scholar] [CrossRef]
- Huter, M.; Strube, J. Model-Based Optimization of SPTFF Ultrafiltration for Integration in Continuous Biopharmaceutical Processing. Chem. Ing. Tech. 2018, 90, 1251. [Google Scholar] [CrossRef]
- Huter, M. Modeling of Continuous Ultrafiltration for Biopharmaceutical Processes; Achema: Frankfurt am Main, Germany, 2018. [Google Scholar]
- Lucke, M.; Koudous, I.; Sixt, M.; Huter, M.J.; Strube, J. Integrating crystallization with experimental model parameter determination and modeling into conceptual process design for the purification of complex feed mixtures. Chem. Eng. Res. Des. 2018, 133, 264–280. [Google Scholar] [CrossRef]
- Altenhöner, U.; Meurer, M.; Strube, J.; Schmidt-Traub, H. Parameter estimation for the simulation of liquid chromatography. J. Chromatogr. A 1997, 769, 59–69. [Google Scholar] [CrossRef]
- Rouquerol, J.; Baron, G.V.; Denoyel, R.; Giesche, H.; Groen, J.; Klobes, P.; Levitz, P.; Neimark, A.V.; Rigby, S.; Skudas, R.; et al. The characterization of macroporous solids: An overview of the methodology. Microporous Mesoporous Mater. 2012, 154, 2–6. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Seidel-Morgenstern, A. Experimental determination of single solute and competitive adsorption isotherms. J. Chromatogr. A 2004, 1037, 255–272. [Google Scholar] [CrossRef]
- Mazzotti, M. Equilibrium theory based design of simulated moving bed processes for a generalized Langmuir isotherm. J. Chromatogr. A 2006, 1126, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Guiochon, G.; Felinger, A.; Shirazi, D.G.; Katti, A.M. Fundamentals of Preparative and Nonlinear Chromatography, 2th ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Klepzig, L.; Strube, J. Rigorous modeling of lyophilization for botanicals and biologics process integration. Chem. Ing. Tech. 2018, 90, 1299. [Google Scholar] [CrossRef]
- Klepzig, L. Rigorous Modelling of Lyophilisation for Botanicals and Biologics Process Integration; Achema: Frankfurt am Main, Germany, 2018. [Google Scholar]
- Klepzig, L. Process Modelling in Combination with Experimental Model Parameter Determination; Parenteral Drug Association: Sevilla, Spain, 2018. [Google Scholar]
- Gronemeyer, P.; Ditz, R.; Strube, J. DoE based integration approach of upstream and downstream processing regarding HCP and ATPE as harvest operation. Biochem. Eng. J. 2016, 113, 158–166. [Google Scholar] [CrossRef]
- Meyer, U.A.; Zanger, U.M.; Schwab, M. Omics and drug response. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 475–502. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.; Clemens, C.; Kaufmann, H.; Schulz, T.W. Advancing Biopharmaceutical Process Development by System-Level Data. Analysis and Integration of Omics Data. In Genomics and Systems Biology of Mammalian Cell Culture; Hu, W.S., Zeng, A.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 133–163. [Google Scholar]
- Schaub, J.; Clemens, C.; Schorn, P.; Hildebrandt, T.; Rust, W.; Mennerich, D.; Kaufmann, H.; Schulz, T.W. CHO gene expression profiling in biopharmaceutical process analysis and design. Biotechnol. Bioeng. 2010, 105, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Zeng, A.-P. (Eds.) Genomics and Systems Biology of Mammalian Cell Culture; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Wiesel, A.; Schmidt-Traub, H.; Lenz, J.; Strube, J. Modellierung der Trennung von Mehrkomponentensystemen mittels Gradientenelution in der präparativen Ionenaustausch-Chromatographie. Chem. Ing. Tech. 2002, 74, 677. [Google Scholar] [CrossRef]
- Sixt, M.; Strube, J. Pressurized hot water extraction of 10-deacetylbaccatin III from yew for industrial application. Resour-Effic. Technol. 2017. [Google Scholar] [CrossRef]
- Koudous, I.; Sixt, M.; Strube, J. Model-Based Systematic Interpretation of the Extraction and Purification of 10-Deacetylbaccatin III from Taxus baccata; Berichte aus dem Julius Kühn-Institut: Quedlinburg, Germany, 2016. [Google Scholar]
- Sixt, M.; Strube, J. Systematic and Model-Assisted Evaluation of Solvent Based- or Pressurized Hot Water Extraction for the Extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86. [Google Scholar] [CrossRef]
- Sixt, M.; Schmidt, A.; Mestmäcker, F.; Huter, M.; Uhlenbrock, L.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part I: Conceptual Process Design and Cost Estimation. Processes 2018, 6, 161. [Google Scholar] [CrossRef]
- Schmidt, A.; Sixt, M.; Huter, M.; Mestmäcker, F.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing. Processes 2018, 6, 179. [Google Scholar] [CrossRef]
- Mestmäcker, F.; Schmidt, A.; Huter, M.; Sixt, M.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes 2018, 6, 180. [Google Scholar] [CrossRef]
- Huter, M.; Schmidt, A.; Mestmäcker, F.; Sixt, M.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181. [Google Scholar] [CrossRef]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and Operation of Continuous Countercurrent Chromatography in Biotechnological Production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Schmidt, A.; Strube, J. Application and Fundamentals of Liquid-Liquid Extraction Processes: Purification of Biologicals, Botanicals, and Strategic Metals. In Encyclopedia of Chemical Technology; Kirk, R.E., Othmer, D.F., Eds.; Wiley: New York, NY, USA, 2003; pp. 1–52. [Google Scholar]
- Sixt, M.; Gudi, G.; Schulz, H.; Strube, J. In-line Raman spectroscopy and advanced process control for the extraction of anethole and fenchone from fennel (Foeniculum vulgare L. MILL.). Comptes Rendus Chim. 2018, 21, 97–103. [Google Scholar] [CrossRef]
- Gudi, G.; Krähmer, A.; Koudous, I.; Strube, J.; Schulz, H. Infrared and Raman spectroscopic methods for characterization of Taxus baccata L.—Improved taxane isolation by accelerated quality control and process surveillance. Talanta 2015, 143, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbrock, L.; Sixt, M.; Strube, J. Quality-by-Design (QbD) process evaluation for phytopharmaceuticals on the example of 10-deacetylbaccatin III from yew. Resour-Effic. Technol. 2017. [Google Scholar] [CrossRef]
- CMC Biotech Working Group. A-Mab: A Case Study in Bioprocess Development. Available online: http://www.casss.org/?page=286 (accessed on 17 November 2018).
- CMC-Vaccines Working Group. A-VAX: Applying Quality by Design to Vaccines. Available online: http://qbdworks.com/wp-content/uploads/2014/06/a-vax-applying-qbd-to-vaccines.pdf (accessed on 17 November 2018).
- Zobel-Roos, S.; Mouellef, M.; Siemers, C.; Strube, J. Process Analytical Approach towards Quality Controlled Process Automation for the Downstream of Protein Mixtures by Inline Concentration Measurements Based on Ultraviolet/Visible Light (UV/VIS) Spectral Analysis. Antibodies 2017, 6, 24. [Google Scholar] [CrossRef]
- Zobel-Roos, S.; Stein, D.; Strube, J. Evaluation of Continuous Membrane Chromatography Concepts with an Enhanced Process Simulation Approach. Antibodies 2018, 7, 13. [Google Scholar] [CrossRef]
- Schwellenbach, J.; Zobel, S.; Taft, F.; Villain, L.; Strube, J. Purification of Monoclonal Antibodies Using a Fiber Based Cation-Exchange Stationary Phase: Parameter Determination and Modeling. Bioengineering 2016, 3, 24. [Google Scholar] [CrossRef]
- Mahler, A. Die Reifeprüfung. Der Spiegel. 13 October 2016, pp. 74–78. Available online: https://magazin.spiegel.de/SP/2018/42/159904415/index.html (accessed on 7 November 2018).
- Bardt, H.; Bertenrath, R.; Demary, V.; Fritsch, M.; Grömling, M.; Klös, H.-P.; Kolev, G.V.; Kroker, R.; Lichtblau, K.; Matthes, J.; et al. Wohlstand in der Digitalen Welt. Erster IW-Strukturbericht; Institut der deutschen Wirtschaft Medien GmbH: Köln, Germany, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process Model Type | Definition | Required Input | Falsification |
|---|---|---|---|
| Rigorous | taking all relevant physical-chemical process effects of fluid dynamics, phase equilibrium and mass transfer into account by separating those effects from each other | 1. experimental model parameter determination with separated effects of fluid dynamics, phase equilibrium and mass transfer | short-cut models, statistical models, hybrid models |
| 2. validated model | |||
| 3. predictive in scale | |||
| Short-cut | macroscopic equipment mass (and energy) balances overall parameter such as separation factor, capacity etc. | 1. non predictive in scale and dimensions | physical chemical effects not all separated and overall factors |
| 2. overall parameter | |||
| Cost modelling | use short cut models add cost correlations for operation and investment costs | 1. short-cut models | better name: cost estimation |
| 2. overall parameter | |||
| 3. cost correlations operation and investment costs | |||
| 4. non predictive outside trainings range | |||
| Hybrid models | combining short-cut or rigorous models with statistical parts | 1. short-cut or partially rigorous models | nonsense |
| 2. statistical part | |||
| 3. non-predictive due to statistical part | |||
| Observer models | for process control statistically trained | 1. operation data sets | data sets strictly separated for training and operation |
| 2. not fully predictive | sensor drifts cause problems | ||
| 3. inline, at-line PAT needed | not process design | ||
| Model based process control | belong to advanced process control | 1. rigorous models | cope with sensor drift |
| not process design, but APC | |||
| PCA/PLS regression model | statistical model for analytical description | 1. analytical data sets | no model but analytics data regression equation |
| Artificial intelligence | mostly neuronal networks | 1. data set to train | |
| 2. non predictive outside trainings range |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable without a Digital-Twin? Processes 2019, 7, 94. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7020094
Zobel-Roos S, Schmidt A, Mestmäcker F, Mouellef M, Huter M, Uhlenbrock L, Kornecki M, Lohmann L, Ditz R, Strube J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable without a Digital-Twin? Processes. 2019; 7(2):94. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7020094
Chicago/Turabian StyleZobel-Roos, Steffen, Axel Schmidt, Fabian Mestmäcker, Mourad Mouellef, Maximilian Huter, Lukas Uhlenbrock, Martin Kornecki, Lara Lohmann, Reinhard Ditz, and Jochen Strube. 2019. "Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable without a Digital-Twin?" Processes 7, no. 2: 94. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7020094