
A Validated RP-HPLC Method for the Determination of Butamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness



Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solvents
2.2. Instrumentation
2.3. Chromatographic Conditions
2.4. Preparation of Stock and Working Solutions
2.5. Sample Preparation
2.6. Validation Procedures
2.7. Robustness Testing Set-Up
3. Results and Discussion
3.1. Method Development
3.2. Validation Data
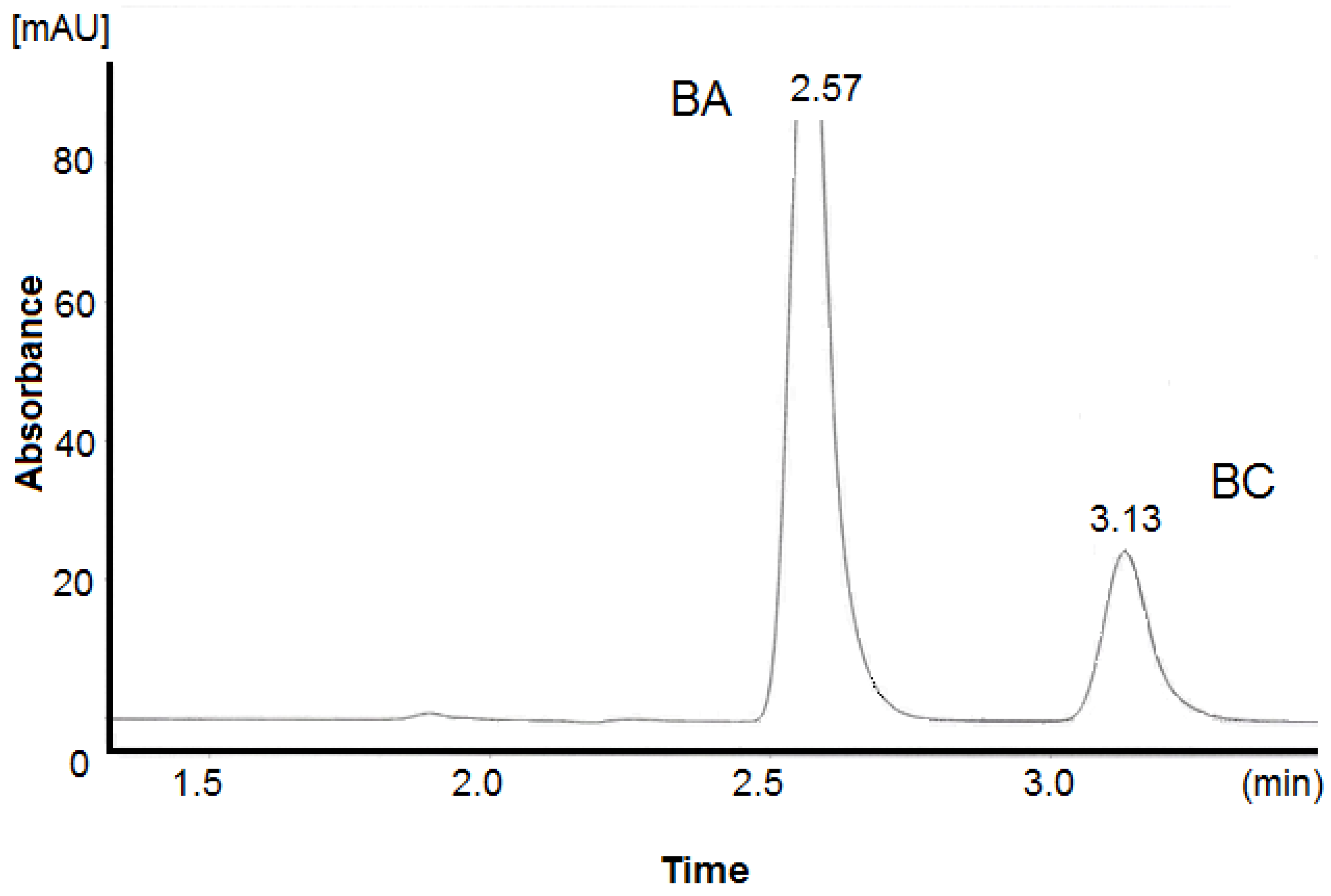
3.2.1. Specificity
3.2.2. Stability
3.2.3. Linearity
3.2.4. Accuracy
3.2.5. Precision
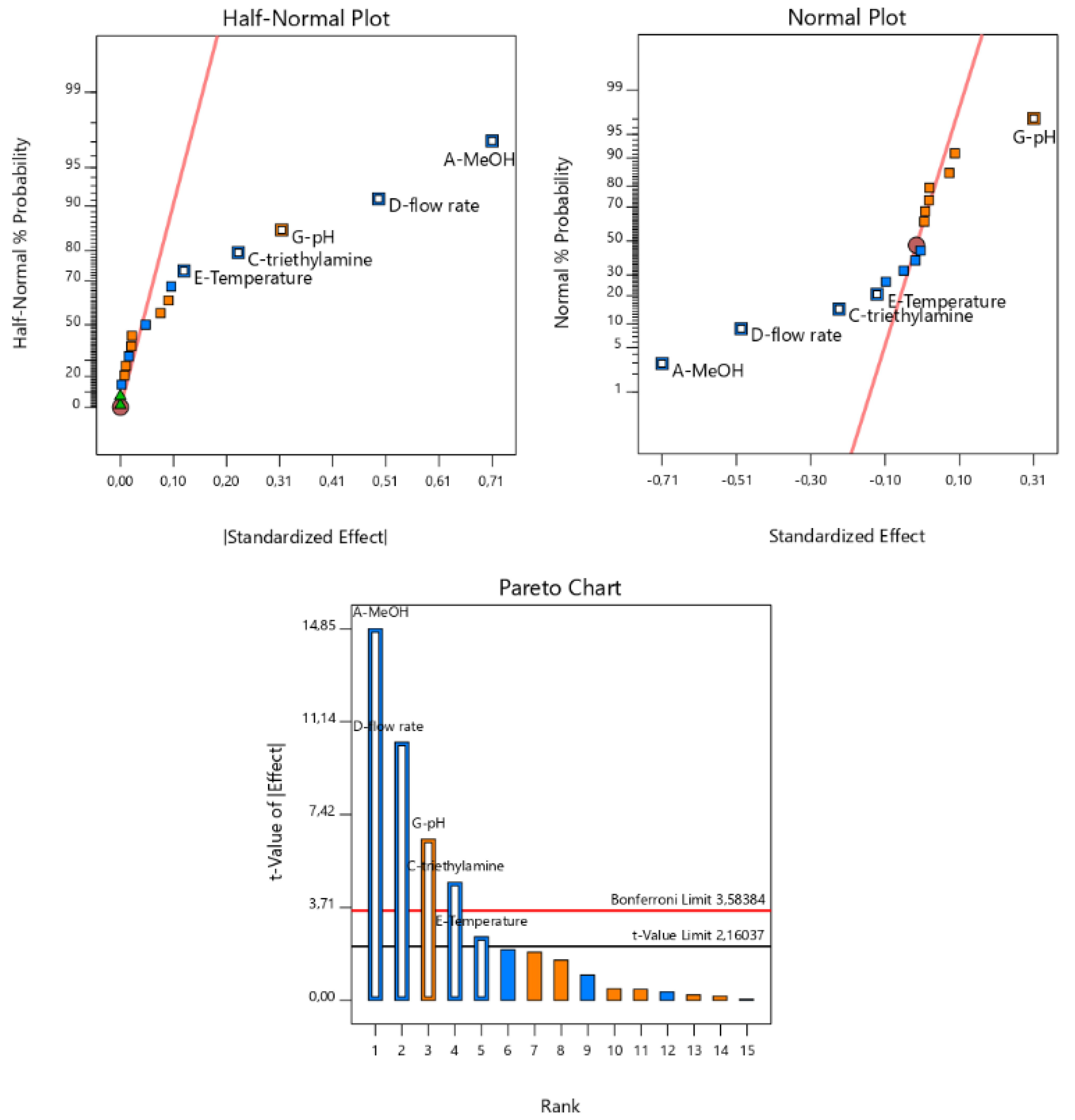
3.3. Robustness Testing Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Union of Pure and Applied Chemistry (IUPAC). Available online: https://iupac.org/ (accessed on 20 June 2021).
- Bayram, E.; Karakaya, P.; Topcu, Y.; Yis, U.; Hiz, S. Acute Cervical Dystonia After the First Dose of Butamirate Citrate. Pediatr. Emerg. Care 2013, 29, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Brayfield, A. Martindale: The Complete Drug Reference, 39th ed.; Pharmaceutical Press: London, UK, 2017. [Google Scholar]
- Haider, M. Development and Validation of a Stability Indicating HPLC Method for the Estimation of Butamirate Citrate and Benzoic Acid in Pharmaceutical Products. J. Chromatograph. Separat. Techniq. 2011, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Malliou, E.T.; Antoniou, C.G.; Koundourellis, J.E. Determination of Butamyrate Citrate in Cough Preparations by Derivative UV Spectrophotometry and High Performance Liquid Chromatography. Anal. Sci. 2003, 19, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greek National Organization for Medicines (eof). Definition of Summary of Product Characteristics (SPC) for Pharmaceutical Products Containing the API “BUTAMIRATE CITRATE”, Protocol No 45964, Athens, 30 June 2009. Available online: https://www.eof.gr/ (accessed on 15 July 2021).
- Sudha, T.; Manthena, K.; Ravikumar, V.R.; Ganesan, V. HPLC method for the determination of ambroxol HCl in the presence of antimicrobial preservatives in oral liquid formulation. J. Pharm. Sci. Innov. 2015, 4, 134–139. [Google Scholar] [CrossRef]
- Mahmoud, A.; Sayed, N.; Mosaad, F.; Souaya, E.R. Chromatographic methods for the determination of butamirate citrate in presence of its degradation product. Chem. Pap. 2019, 73, 1309–1320. [Google Scholar] [CrossRef]
- Dol, I.; Altesor, C.; Knochen, M. Application of an optical compensation method to the simultaneous determination of butamirate citrate and sodium benzoate by derivative spectrophotometry in the ultraviolet. Quim. Anal. 1996, 15, 148–153. [Google Scholar]
- Gouda, A.A.; El-Sheikh, R.; Amin, A.S. Spectrophotometric determination of some anti-tussive drugs and its applications to pharmaceutical formulations. Anal. Chem. Ind. J. 2008, 7, 757–767. [Google Scholar]
- Taşkin, D.; Erensoy, G.; Sungur, S. Optimized and validated spectrophotometric determination of butamirate citrate in bulk and dosage forms using ion-pair formation with methyl orange and bromothymol blue. Farmacia 2017, 65, 761–765. [Google Scholar]
- Koppenhoefer, B.; Jakob, A.; Zhu, X.; Lin, B. Separation of Enantiomers of Drugs by Capillary Electrophoresis with Permethyl-gamma-Cyclodextrin as Chiral Solvating Agent. J. High Resolut. Chromatogr. 2000, 23, 413–429. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.K.; Rahman, M.; Seraj, S.; Paul, A.; Arif, H.A.; Islam, T.; Arifuzzaman, S. Development and validation of a new stable HPLC method for the assay of butamirate citrate in pharmaceutical formulations. Adv. Nat. Appl. Sci. 2011, 5, 75–84. [Google Scholar]
- Mostafa, A.; El-Gindy, A.; Emara, S. Development, application and validation of RP-HPLC method for the simultaneous determination of butamirate citrate and its main degradation product in pharmaceutical dosage forms. Anal. Methods 2011, 3, 1643–1651. [Google Scholar] [CrossRef]
- Hassib, S.T.; El-Zaher, A.A.; Fouad, M.A. Development and validation of RP-HPLC stability-indicating methods for the determination of butamirate citrate and sodium cromoglycate. J. Chem. Pharm. Res. 2011, 3, 243–258. [Google Scholar]
- Heinänen, M.; Barbas, C. Validation of an HPLC method for the quantification of ambroxol hydrochloride and benzoic acid in a syrup as pharmaceutical form stress test for stability evaluation. J. Pharm. Biomed. Anal. 2001, 24, 1005–1010. [Google Scholar] [CrossRef]
- Madhu, M.; Sreeram, V.; Nagendrakumar, A.V.D.; Reddy, T.V. Validated Rp-Hplc method for the estimation of benzoic acid in bulk and pharmaceutical Formulation. Chem. Sin. 2014, 5, 52–60. [Google Scholar]
- Dönmez, Ö.A.; Dinç-Zor, Ş.; Aşçı, B.; Şen, E. Simultaneous HPLC-DAD determination of pseudoephedrine HCl, sodium benzoate, sunset yellow, and methyl paraben in syrup preparation by use of partial least squares and principal component regression. J. Liq. Chrom. Relat. Technol. 2019, 42, 648–653. [Google Scholar] [CrossRef]
- Watson, D.G. Pharmaceutical Analysis: A Textbook for Pharmacy Students and Pharmaceutical Chemists, 3rd ed.; Elsevier/Churchill: Livingstone, UK, 2012. [Google Scholar]
- Snyder, L.R.; Kirkland, J.J.; Dolan, J.W. Introduction to Modern Liquid Chromatography, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Vander Heyden, Y.; Nijhuis, A.; Smeyers-Verbeke, J.; Vandeginste, B.G.M.; Massart, D.L. Guidance for robustness/ruggedness tests in method validation. J. Pharm. Biomed. Anal. 2001, 24, 723–753. [Google Scholar] [CrossRef]
- Restek Chromatography Products and Solutions. Available online: https://www.restek.com/ (accessed on 15 April 2021).



{kind=link}
{kind=link}
| Experimental Factor | Interval | Experimental Response | ||
| −1 | 0 (nominal value) | +1 | Area of BA Area of BC As of BA As of BC Theoretical plates (N) of BA Theoretical plates (N) of BC tR of BA tR of BC critical peak pair resolution (Rs) | |
| Flow | 1.30 | 1.50 | 1.70 | |
| % MeOH | 48 | 50 | 52 | |
| T | 34 | 36 | 38 | |
| λ | 208 | 210 | 212 | |
| pH | 2.8 | 3.0 | 3.2 | |
| Csalt | 48 | 50 | 52 | |
| % Et3N | 0.95 | 1.00 | 1.05 | |
| Experiment Nr | Experimental Factor | ||||||
|---|---|---|---|---|---|---|---|
| Flow | pH | % MeOH | Csalt | T | λ | % Et3N | |
| 1 | 1.30 | 2.8 | 48 | 52 | 38 | 212 | 0.95 |
| 2 | 1.70 | 2.8 | 52 | 52 | 34 | 208 | 0.95 |
| 3 | 1.30 | 3.2 | 52 | 52 | 34 | 212 | 0.95 |
| 4 | 1.70 | 3.2 | 48 | 52 | 38 | 208 | 0.95 |
| 5 | 1.30 | 2.8 | 48 | 48 | 34 | 208 | 0.95 |
| 6 | 1.70 | 2.8 | 48 | 48 | 38 | 208 | 1.05 |
| 7 | 1.30 | 2.8 | 52 | 52 | 38 | 208 | 1.05 |
| 8 | 1.50 | 3.0 | 50 | 50 | 36 | 210 | 1.00 |
| 9 | 1.70 | 2.8 | 52 | 48 | 38 | 212 | 0.95 |
| 10 | 1.70 | 2.8 | 48 | 52 | 34 | 212 | 1.05 |
| 11 | 1.70 | 3.2 | 52 | 52 | 38 | 212 | 1.05 |
| 12 | 1.70 | 3.2 | 52 | 48 | 34 | 208 | 1.05 |
| 13 | 1.50 | 3.0 | 50 | 50 | 36 | 210 | 1.00 |
| 14 | 1.50 | 3.0 | 50 | 50 | 36 | 210 | 1.00 |
| 15 | 1.30 | 3.2 | 48 | 48 | 38 | 212 | 1.05 |
| 16 | 1.30 | 3.2 | 48 | 52 | 34 | 208 | 1.05 |
| 17 | 1.30 | 3.2 | 52 | 48 | 38 | 208 | 0.95 |
| 18 | 1.70 | 3.2 | 48 | 48 | 34 | 212 | 0.95 |
| 19 | 1.30 | 2.8 | 52 | 48 | 34 | 212 | 1.05 |
| Experiment Nr | Response | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BA Area (mAUxmin) | BC Area (mAUxmin) | Af of BA | Af of BC | Rs | NBA | NBC | tR of BA (min) | tR of BC (min) | |
| 1 | 674.70 | 149.95 | 1.60 | 1.67 | 4.19 | 6787 | 6738 | 3.03 | 3.72 |
| 2 | 502.19 | 143.82 | 1.75 | 1.60 | 3.25 | 5108 | 5245 | 2.23 | 2.67 |
| 3 | 716.44 | 151.04 | 1.70 | 1.54 | 3.92 | 6232 | 6890 | 2.91 | 3.53 |
| 4 | 536.26 | 142.42 | 1.44 | 1.46 | 3.90 | 4579 | 5251 | 2.30 | 2.87 |
| 5 | 655.01 | 186.07 | 1.55 | 1.50 | 4.37 | 6085 | 7155 | 3.09 | 3.83 |
| 6 | 510.32 | 147.32 | 1.44 | 1.33 | 3.50 | 4686 | 5129 | 2.33 | 2.84 |
| 7 | 660.53 | 190.45 | 1.60 | 1.50 | 3.41 | 7213 | 6429 | 2.89 | 3.41 |
| 8 | 568.17 | 148.30 | 1.67 | 1.42 | 3.72 | 4859 | 6244 | 2.57 | 3.13 |
| 9 | 516.77 | 116.37 | 1.44 | 1.50 | 2.97 | 4215 | 5089 | 2.21 | 2.63 |
| 10 | 513.16 | 114.54 | 1.30 | 1.55 | 3.72 | 4848 | 5398 | 2.37 | 2.91 |
| 11 | 543.37 | 113.49 | 1.44 | 1.50 | 3.15 | 4956 | 5063 | 2.19 | 2.62 |
| 12 | 544.56 | 142.98 | 1.45 | 1.46 | 3.16 | 4266 | 5245 | 2.22 | 2.67 |
| 13 | 567.32 | 148.39 | 1.67 | 1.50 | 3.72 | 5703 | 5439 | 2.57 | 3.13 |
| 14 | 567.18 | 148.72 | 1.67 | 1.42 | 3.72 | 5703 | 5439 | 2.57 | 3.13 |
| 15 | 719.17 | 151.36 | 1.60 | 1.46 | 4.35 | 6697 | 6762 | 3.01 | 3.73 |
| 16 | 690.48 | 184.10 | 1.55 | 1.62 | 4.45 | 5981 | 7105 | 3.07 | 3.82 |
| 17 | 697.12 | 186.25 | 1.78 | 1.58 | 3.93 | 7114 | 6658 | 2.87 | 3.47 |
| 18 | 540.17 | 114.79 | 1.56 | 1.42 | 4.27 | 5641 | 4876 | 2.34 | 2.97 |
| 19 | 686.77 | 150.42 | 1.60 | 1.58 | 3.26 | 6260 | 5784 | 2.91 | 3.45 |
| Experimental Factor | Ex on BA Area | Ex on BA Area | Ex on Af of BA | Ex on Af of BC | Ex on Rs | Ex on NBA | Ex on NBC | Ex on tR of BA | Ex on tR of BC |
|---|---|---|---|---|---|---|---|---|---|
| Flow | −161.67 | −39.24 | −0.14 | −0.080 | −0.49 | −1758.75 | −1528.13 | −0.70 | −0.85 |
| pH | 33.52 | −1.56 | 0.029 | −0.025 | 0.31 | 33.00 | 110.38 | −0.019 | 0.027 |
| % MeOH | 3.56 | 0.53 | 0.091 | 0.033 | −0.71 | 7.50 | −251.38 | −0.14 | −0.28 |
| Csalt | −4.10 | −0.72 | 0.00 | 0.073 | 0.022 | 92.50 | 117.63 | 0.00 | 0.00 |
| T | 1.18 | 1.23 | −0.011 | −0.032 | −0.12 | 228.25 | −72.38 | −0.039 | −0.070 |
| λ | 14.26 | −32.68 | −0.039 | 0.021 | −0.016 | 75.50 | −202.13 | 0.00 | 0.00 |
| % Et3N | 3.71 | 0.49 | −0.10 | −0.033 | −0.23 | −106.75 | −123.38 | 0.00 | −0.030 |
| Response | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| BA Area | BC Area | Af of BA | Af of BC | Rs | NBA | NBC | tR of BA | tR of BC | |
| Ecritical | 1.15 | 0.48 | 0.00 | 0.10 | 0.00 | 1048.39 | 999.95 | 0.00 | 0.00 |
| Factor | BA Area | BC Area | Af of BA | Af of BC | Rs | NBA | NBC | tR of BA | tR of BC |
|---|---|---|---|---|---|---|---|---|---|
| Flow | ● ○ | ● ○ | ● □ | □ | ● ○ | ● ○ | ● ○ | ● ○ | ● ○ |
| pH | ● ○ | ● | ● | ● ○ | ● ○ | ● | |||
| % MeOH | ● | ● | ● | ● ○ | ● ○ | ● ○ | |||
| Csalt | ● | ● | ● | ||||||
| T | ● | ● | ● | ● □ | ● ○ | ● ○ | |||
| λ | ● ○ | ● ○ | ● | ● | |||||
| % Et3N | ● | ● | ● □ | ● ○ | ● |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neofotistos, A.-D.G.; Gkountanas, K.; Boutsikaris, H.; Dotsikas, Y. A Validated RP-HPLC Method for the Determination of Butamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness. Separations 2021, 8, 163. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8100163
Neofotistos A-DG, Gkountanas K, Boutsikaris H, Dotsikas Y. A Validated RP-HPLC Method for the Determination of Butamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness. Separations. 2021; 8(10):163. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8100163
Chicago/Turabian StyleNeofotistos, Antonios-Dionysios G., Kostas Gkountanas, Haris Boutsikaris, and Yannis Dotsikas. 2021. "A Validated RP-HPLC Method for the Determination of Butamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness" Separations 8, no. 10: 163. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8100163