Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates?


1
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK
2
School of Pharmacy, Institute of Clinical Sciences, University of Birmingham, Birmingham B15 2TT, UK
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(3), 31; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030031
Submission received: 29 November 2018
/
Revised: 6 February 2019
/
Accepted: 13 February 2019
/
Published: 1 March 2019
(This article belongs to the Special Issue Unconventional Anticancer Metallodrugs and Strategies to Improve their Pharmacological Profile)
{kind=link}
{kind=link}
Abstract
:After nearly 20 years of research on the use of ruthenium in the fight against cancer, only two Ru(III) coordination complexes have advanced to clinical trials. During this time, the field has produced excellent candidate drugs with outstanding in vivo and in vitro activity; however, we have yet to find a ruthenium complex that would be a viable alternative to platinum drugs currently used in the clinic. We aimed to explore what we have learned from the most prominent complexes in the area, and to challenge new concepts in chemical design. Particularly relevant are studies involving NKP1339, NAMI-A, RM175, and RAPTA-C, which have paved the way for current research. We explored the development of the ruthenium anticancer field considering that the mechanism of action of complexes no longer focuses solely on DNA interactions, but explores a diverse range of cellular targets involving multiple chemical strategies.
1. Introduction
Cancer is a major health burden in the developed world. Although many breakthroughs in biological targeted approaches have occurred (specifically, immunological therapy), an unmet clinical need remains for a large section of the patient population, so wide-spectrum chemotherapy agents are still required. This space is currently addressed with the use of platinum(II) coordination complexes, namely Cisplatin (CDDP), Oxaliplatin and Carboplatin (worldwide approval), alongside Nedaplatin and Lobaplatin, which are restricted to selected markets. Such platinum agents lack cellular selectivity, so rapidly proliferating cells (hair follicles, bone marrow, and gastrointestinal tract) are particularly vulnerable to off-target effects. This lack of cellular selectivity can lead to the development of severe side effects not limited to nausea, vomiting, hair loss, nephrotoxicity, neurotoxicity, and hearing loss [1,2].
The prevalence of platinum resistance in the clinic, both intrinsic and acquired [3,4,5], is an escalating clinical concern, especially considering that platinum drugs are currently used in over half of all chemotherapy regimens [3]. This has sparked the development of a new generation of cytotoxics based on different metals, and corresponding coordination complexes, which excel in cellular selectivity and retain their use against a wide range of malignancies, while exhibiting unique mechanism(s) of action. Given the chemical similarity of the platinum group of metals (Pt, Pd, Rh, Ir, Ru, and Os), given the successes of platinum therapies, interest in ruthenium(II/III) complexes has increased considerably.
2. Ruthenium Complexes as Anticancer Agents
Ruthenium compounds have been proven to be a starting point in the search for alternatives to platinum drugs in the clinic, as these compounds have both comparable ligand exchange kinetics and a greater number of accessible coordination geometries. Such comparison considers that Pt(II) compounds are limited to square planar geometries, akin to cisplatin, and that octahedral geometries comparable to ruthenium complexes can be only be achieved by a higher oxidation state, e.g., Pt(IV). The investigation around such complexes has been intense, with highly-renowned and international-leading research groups pouring resources into this fast growing field. The chemistry of Ru(II) and Ru(III) complexes is well established in both the materials and medicinal chemistry fields, and research into their use as prospective anticancer agents is widely spread. The latter oxidation state complexes are typically considered to be more inert, which is, to some extent, attributable to the higher effective nuclear charge. In combination with the highly reducing environment of a cancer cell, an “activation by reduction” mechanism has been speculated for many Ru(III) complexes [6], which are reduced to their more active Ru(II) form by cellular reductants such as ascorbate [7].
The in-cell mechanism of action (MoA) for many ruthenium complexes differs from the DNA-binding mechanism typically associated with platinum drugs [8]. With a wider range of intracellular targets, ruthenium anticancer complexes are well established, and many examples have shown promise in chemical model systems, in vitro, and in vivo [9,10,11]. However, despite widespread efforts, only three ruthenium complexes (NKP1339, NAMI-A, and TLD1433) have reached clinical trials. NKP1339, and NAMI-A are both being developed as chemotherapeutic agents, while octahedral Ru(II) complex TLD1433 has potential as a photosensitizer for photo-dynamic therapy [12]. Research into future anticancer agents should learn from both successes and difficulties encountered during the development of previous complexes in order to identify desirable physical, chemical, and biological properties associated with a successful future Ru drug candidate.
In this review, we critically evaluate four of the most significant Ru coordination complexes, NAMI-A, NKP1339, RM175, and RAPTA-C, and assess their contribution to the advancement of future ruthenium-based anticancer agents.
3. Case Study: RM175
RM175 [Ru(biphenyl)Cl(en)]+, where en = 1,2-ethylenediamine, was amongst the first ruthenium(II) complexes to be explored for anticancer activity (Figure 1). This pseudo-octahedral organometallic complex was developed by the Sadler group in 2001 [13]. RM175 is comprised of monodentate (chloride), bidentate (diamine), and arene (biphenyl) ligands, typical of a 3-legged “piano-stool” geometry. Originally synthesized with the aim of targeting DNA, it is intended to take advantage of the lower 2+ oxidation state that would not require activation by cellular reduction [14].
The arene substituent provides a hydrophobic surface, allowing for cellular diffusion through the lipophilic plasma membrane [14]. After entry into the cell, but prior to binding with DNA, complex activation is thought to occur by ligand exchange at the monodentate site [15,16]. The complex’s design envisaged the halogen atom acting like a leaving group, drawing similarities with the activation of cisplatin. The aquation reaction would then generate a coordinative vacancy, which, in turn, would allow for covalent binding to the N7 of guanine in the DNA double helix [14]. Although Ru(II) complexes are known to bind to the guanine residues in DNA [17], it is thought that the extended arene in this complex enables hydrophobic interactions to occur between RM175 and DNA via arene-intercalation between base pairs [18]. As a consequence of the free rotation of biphenyl about the Ru(II) centre, the structure is relatively flexible, which is anticipated to limit steric hindrance and increase the complex’s DNA-binding affinity [19]. Conformational flexibility may allow the complex to intercalate into the base pairs and bind to guanine simultaneously, as has been shown possible with other ruthenium complexes [19]. This may explain why the resulting RM175-DNA adduct is more resistant to DNA repair than platinated DNA; therefore, such observation begins to explain the lack of cross-resistance with platinum [20].
Further biological studies into the mechanism of action of RM175 revealed new cellular targets in addition to DNA binding, most notably the inhibition of matrix metalloproteinase-2 (MMP-2) [14]. MMPs are the most important members of the metalloproteinase family that contribute to tumor progression. They govern the conditions of the tumor microenvironment and use signalling pathways to assist in the modulation of cell growth and angiogenesis [21]. MMPs facilitate the migration and invasion of cancerous cells, through degradation of the extracellular matrix (ECM), suppression of the adaptive immune system, and other processes that promote cell survival and protect malignant cells from apoptosis [21,22]. Apoptosis, as a form of programmed cell death, does not procure an inflammatory response in principle; thus, MMPs are a noteworthy target for anticancer agents [23].
RM175 also exerts activity by disrupting the processes of cell invasion and migration. In order to assess the effect of metallodrugs on cellular detachment and re-adhesion, several proteins have been investigated including collagen IV, fibronectin, and poly-l-lysine. All these proteins may come into contact with malignant cells in vivo, since poly-l-lysine attracts cells through electrostatic interactions, and both fibronectin and collagen IV are constituents of the extracellular matrix. Upon exposure to RM175, invasive MDA-MB-231 breast adenocarcinoma cells became more resistant to detachment from either fibronectin or poly-l-lysine, depending on the substance on which the cells were originally grown. This effect was absent in non-tumorigenic and non-invasive cell lines (HBL-100 and MCF-7, respectively), suggesting that the reduction in cellular detachment after exposure to RM175 was selective for invasive cells that would otherwise metastasize. Adherence to cellular components is a vital stage in the formation of metastases, so the Ru(II) complex has the potential to expand this selective action across other cell lines. RM175 significantly inhibits cellular contact-induced movement (haptotaxis) in MDA-MB231 and HLB-100 cells, preventing metastatic formation induced by this stimulus [14].
The described effects on cell invasion and migration explain why the use of RM175 was directed toward preventing and treating metastases as opposed to the reduction in primary tumor volume. Though in vivo studies demonstrated a 50% reduction in primary mammary carcinoma (MCa) tumor mass, upon withdrawal of RM175, tumor growth resumed, demonstrating the limited effect of RM175 on primary tumors. MCa cells are also liable to spontaneously spread to lung tissue; therefore, the anti-metastatic activity was also assessed. The ruthenium complex was found to have greater potency against metastases over primary tumors, and the size of the administered dose was found to significantly impact the extent of metastatic growth. Although a high dose (10 mg/kg/day) resulted in an 85–95% reduction in metastatic mass, only 70% reduction was achieved at a lower dosage (7.5 mg/kg/day). Supplementation of the dose with human serum albumin (HSA) in ratios from 1:1 to 1:10 were found to elevate the drug’s cytotoxic profile and reduce cell viability to a greater extent, indicating that efficacy of RM175 may be greater in vivo than in vitro [14].
RM175 has been reported to up-regulate the tumor suppressor p53 and the pro-apoptotic protein Bax in HCT-116-wt cells, contributing to apoptosis. Significant apoptosis only occurred in cells that were not p53/Bax-null, demonstrating that both proteins are vital for early RM175-induced apoptosis (<48 h). Treatment with RM175 also induced long-term loss of cellular replication, independent of p53, p21 waf1/Cip1, and Bax [17].
RM175 also induces G1/G2-phase cell cycle arrest in vitro [17]. G1 inhibition of cell cycle progression was found to be dependent on tumor suppressors and cell cycle regulators, p53 and p21, which binds to CDK1, as well as to, CDK2 [24], yet independent from the Bcl-2 family member, Bax [17]. This highlights the vital role of both tumor suppressors in initiating cell cycle arrest in contrast to RM175-mediated apoptosis, which only requires p53 and Bax. Exposure resulted in the accumulation of the aforementioned genetic components [17], particularly p21, as expression of this cell cycle regulator typically leads to cell cycle arrest [25]. Activation of p53 initiates a cascade of anti-mitogenic signals, promoting the p21-induced inhibition of CDK2 activity. This consequently prevents expression of various genes (some of which are involved in DNA replication) [25] and inhibits progression to S phase. Alternatively, the cell cycle arrest in the G2 phase could be attributed to p53-mediated activation of either the GADD45 protein or p21 [24], both of which inhibit CDK1, thereby inducing G2/M cell cycle arrest [26]. RM175 exerts its anticancer activity by both preventing progression through the cell cycle leading to apoptosis, and “traditional” disruption of DNA replication by strand intercalation and nucleobase binding.
4. Case Study: RAPTA-C
A second notable piano-stool complex is RAPTA-C, a ruthenium(II) arene complex developed by the Dyson group that comprises an amphiphilic 1,3,5-triaza-7-phosphaadamantane (PTA) ligand and two labile chloride ligands additional to the η6-coordinated arene, which helps to stabilize the +2 oxidation state (Figure 1) [27]. In contrast to other phosphine ligands [28], PTA is relatively sterically undemanding and is anticipated to contribute to the increased water solubility of RAPTA-C relative to other Ru(II) arene complexes [29]. Similarly to cisplatin, RAPTA-C undergoes rapid hydrolysis of Ru–Cl at low (intracellular) chloride concentrations (4–5 mM), predominantly yielding the mono-aquated complex, [Ru(p-cymene)Cl(H2O)(PTA)]+. The Ru–Cl bond remains intact at higher chloride concentrations (100 mM, i.e., in blood) and, like cisplatin, RAPTA-C may be considered a pro-drug in its di-chlorido form [28]. Initial studies of RAPTA-C indicated that damage to supercoiled pBR322 DNA was pH-dependent (occurring only below physiological pH; <7.0), so it was hypothesized that this may invoke selective targeting of the typically more acidic environment of cancer cells [27].
Unlike cisplatin, preliminary in vitro studies found RAPTA-C to be remarkably inactive toward TS/A adenocarcinoma and non-cancerous epithelial (HBL-100) cell lines, as well as initially having limited activity against primary tumors in vivo. However, later in vivo studies using pre-clinical models (using chicken chorioallantoic membrane and, more recently, mice) found that RAPTA-C was able to inhibit tumor growth by 50–75% [30,31]. In both instances, analysis of the treated tumor identified the significant anti-angiogenic properties of the complex. Both RAPTA-C and structurally similar RAPTA-B (in which the arene unit has been changed to a benzene ring) showed promise for size reduction of solid lung metastases in vivo [30]. These pre-clinical studies clearly demonstrate the requirement for careful optimization and control of experimental conditions, and for scientists to be mindful of the fact that in vitro experiments often do not accurately predict in vivo activities. Complexes, such as RAPTA-B or HC11, that contain cyclic, non-substituted hydrocarbons in their structure have the potential to cause liver toxicity after P450 poly-hydroxylation; hence, in vivo experiments need to closely monitor phase I metabolism of the ruthenium drugs [32]. In the case of RAPTA complexes, even at the highest dose administered to mice, negligible side effects were observed. Ruthenium was found to be excreted rapidly via the renal system and did not significantly accumulate in vital organs [28,33].
Later structural iterations of RAPTA-C included tethering of organic substituents on the coordinated arene. Planar anthracene groups were investigated to enhance DNA interaction by intercalation with concurrent fluorescence to map intracellular localization, with limited success [34,35,36]. In contrast, similar naphthalimide (a DNA intercalator) complexes were found to possess modest potencies against A2780 ovarian cancer cells (and a platinum-resistant-derived cell line, A2780Cis) (IC50 2.3–9.1 μM) and displayed some selectivity over non-cancerous (HEK-293) cells [37]. Conjugation of ethacrynic acid, a glutathione transferase inhibitor, to RAPTA-C yielded a new ruthenium complex that was found to bind to the enzyme’s H-site. Importantly, the ruthenium center is involved in the inhibition of glutathione transferase through cysteine residue binding [38]. After an extended incubation time, the ethacrynic acid moiety remained in the H-site of the enzyme but the ruthenium center had been released, which was also found to occur in vitro [39,40].
Most recently, a comparative metallomic study of the metabolism of RAPTA-C and cisplatin was reported. Despite RAPTA-C and cisplatin having similar labile chloride ligands with comparable exchange kinetics, RAPTA-C was found to be more inert to extracellular reactions despite being administered at a dose 40-fold higher than cisplatin. RAPTA-C was found to predominantly bind albumin, though extracellular metal speciation was found to be time-dependent [41]. This highlights the need to better understand speciation, and consequently drug pharmacokinetics and dynamics, to form a well-rounded pre-clinical research portfolio.
Various combination therapies involving RAPTA-C have also been explored in vivo. When administered alongside erlotinib (epidermal growth factor receptor (EGFR) inhibitor) and BEZ-235 (Phosphoinositide 3-kinase PI3K/mTOR inhibitor), the combination was found to synergistically inhibit tumor growth (up to 11-fold relative to single-drug experiments) [42]. An alternative combination therapy involving RAPTA-C and axitinib (vascular endothelial growth factor receptor (VEGFR) targeting tyrosine kinase inhibitor) led to a ca. 90% inhibition of tumor growth with a dose of only 0.4 mg/kg, despite negligible toxicity observed at doses of 100 mg/kg [43].
5. Case Study: NAMI-A
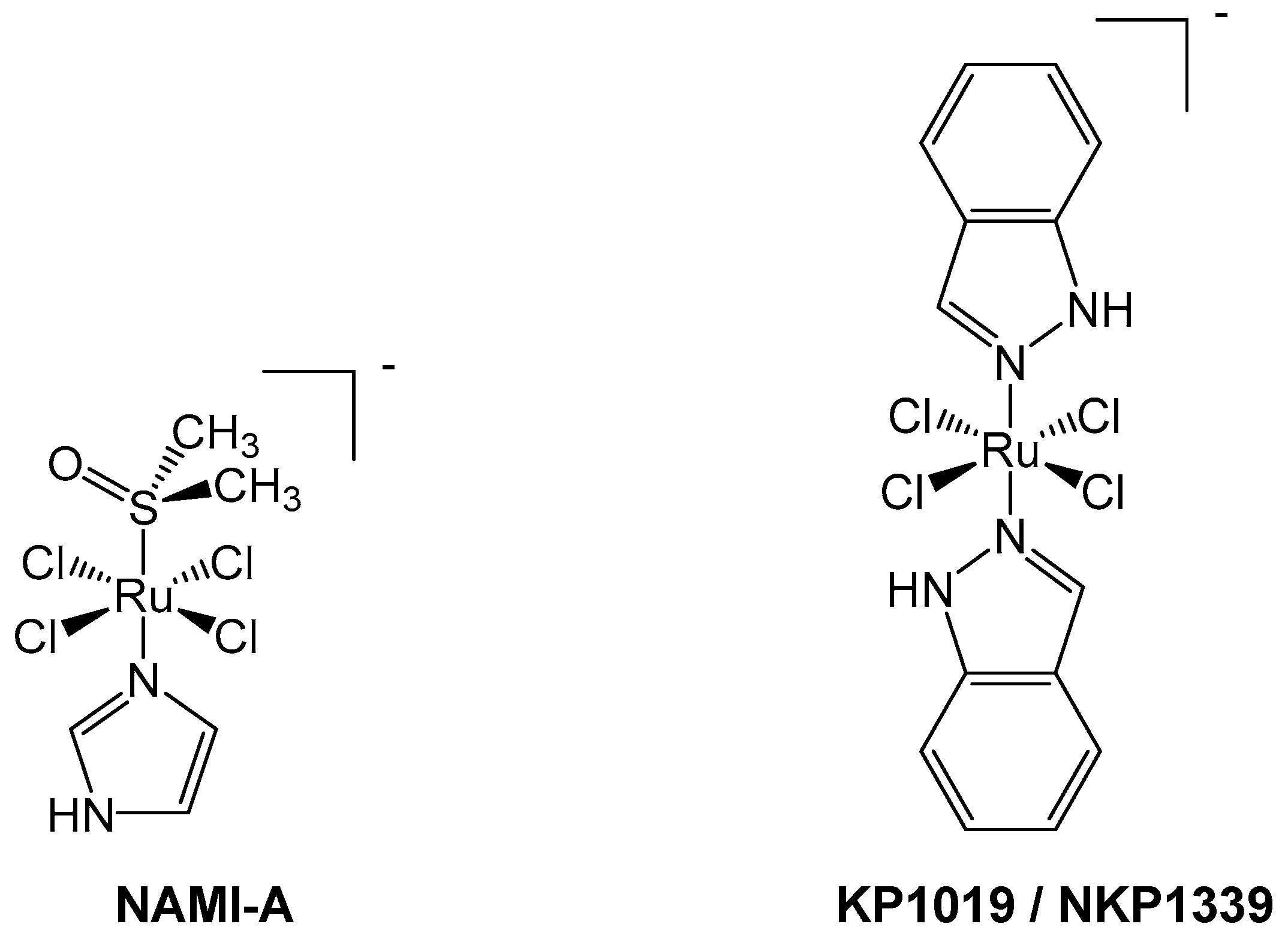
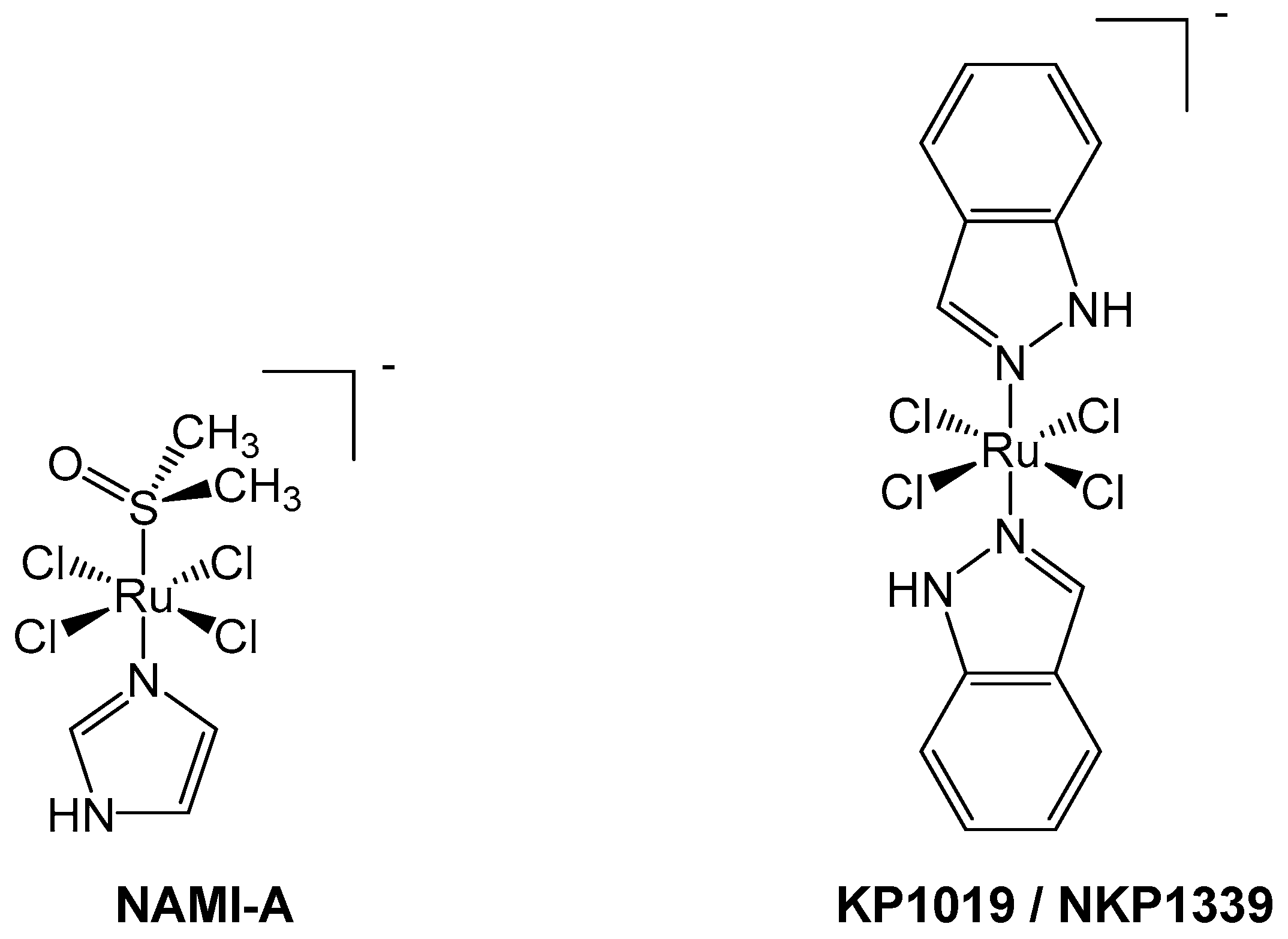
NAMI-A, originally synthesized by the groups of Alessio and Sava (Figure 2), differs from the two previously described complexes, being composed of chloride, imidazole, and dimethylsulfoxide (DMSO) in an octahedral arrangement around a Ru(III) center. Ruthenium(III) complexes are often considered to be pro-drugs, since they are typically less reactive than Ru(II) congeners and require activation via reduction to the 2+ oxidation state [44,45]. Such activation improves tumor targeting as the hypoxic cellular environment favors reduction of the metal center, thereby generating antiproliferative selectivity for cancerous cells compared to healthy cells [45,46]. Reduction of ruthenium from +3 to +2 also leads to kinetic lability; hence, the hydrolysis of chlorido ligands occurs at a faster rate, which may facilitate the reaction of ruthenium complexes with DNA [47]. In highly reactive cases, such substitution process, like hydrolysis, can effectively occur before the activation via reduction [48].
Having entered clinical trials in 2008, NAMI-A constitutes one success story in the development of ruthenium anticancer complexes. NAMI-A was first used in a phase I/II study in 32 patients suffering from an advanced form of non-small cell lung cancer (NSCLC) [49]. The metallodrug was administered intravenously in combination with gemcitabine, which is regularly used alongside cisplatin in this type of cancer [44]. Trial results indicated that such combination of NAMI-A and gemcitabine affected the quality of life of the patients, with side effects including gastrointestinal (GI) disturbances, neutropenia, and elevated liver enzymes [44]. Although the regimen was deemed to be “insufficiently effective for further use” [49], this problem may yet have a solution.
NAMI-A has a synergistic ability to prevent cell invasion and hinder neo-angiogenesis [50], making it selective for metastasis rather than fully formed tumors [46]. Compared to cisplatin, NAMI-A has a wide variety of biological targets, most of which are extracellular rather than DNA-based [44]. One of the main mechanisms through which NAMI-A exerts its anti-angiogenic effects is thought to be the scavenging of nitric oxide [51]. The nitric oxide synthetase (NOS) pathway stimulates angiogenesis and endothelial cell migration and also includes VEGF-activated enzymes that catalyze the generation of NO—a signaling molecule involved in these processes [51,52]. The ruthenium center of NAMI-A, as well as its albumin adducts, have been proven to bind strongly to NO through displacement of the DMSO ligand [53]. This reaction is thought to be irreversible in vivo as NO release is unfavorable, even in the presence of glutathione or other reducing agents. Nitric oxide is a downstream mediator of VEGF and is implicated in endothelial cell migration [52]. NO scavengers, including NAMI-A, are reported to cause potent blockade of VEGF-mediated endothelial cell processes related to angiogenesis, including cell migration [51]. This may be due to NAMI-A’s observed effects on NO, since NAMI-A has no effect on the phosphorylation status of the downstream proteins, PKB (more commonly known as Akt) and ERK½ [54]. NAMI-A does not affect VEGF itself but instead is thought to enable cells to overcome its inductive properties, supporting the theory that NAMI-A prevents VEGF-mediated activity through NO scavenging [54].
Another angiogenic process effected by NAMI-A is inhibition of endothelial cellular proliferation [51], since NO is also a signaling molecule in the mitogen-activated protein kinase (MAPK) pathway [55]. This pathway includes a cascade of kinases that partake in cellular signaling, which coordinate various biological processes including cellular proliferation, migration, and survival [56]. After treatment with NAMI-A, cellular proliferation was inhibited for at least 48 h [51], an effect that could be attributed to NO’s role in the MAPK pathway. Angiogenesis is essential for tumor progression, since oxygen and nutrients are needed to sustain malignant growth. Angiogenic-inhibition provides another means of suppressing metastatic development. NAMI-A also reduces the VEGF-dependent migration of cancerous cells [57]. Inhibition of VEGF activity hence provides a selective mechanism through which NAMI-A can prevent neo-angiogenesis and the formation of metastases.
NAMI-A has also been reported to act on cells via selective inhibition of the Ca2+-activated potassium ion channel, KCa3.1 [58]. In most cells, the concentration of potassium ions regulates the membrane potential, which, in turn, governs cell cycle progression [59]. However, the gene that encodes KCa3.1 (KCNN4) is overexpressed in many cancer cells [58]. The downstream biological processes governed by KCa3.1 differ depending on the cell type: (1) proliferation in leukemia and lymphoma cells or (2) migration in epithelial and glial cancer cells [58,60]. Such cell-type-dependent function of the KCa ion channel may explain why the activity of NAMI-A is typically limited in primary tumors, yet the complex remains potent against leukaemia and metastases. In leukemia cells, NAMI-A-induced inhibition of KCa3.1 was found to induce G2/M cell cycle arrest, leading to apoptosis [58]. Ion channels such as KCa3.1 are considered to be indispensable in relation to cellular migration [61]; hence, NAMI-A is also able to inhibit cell motility. Proliferation and migration of NSCLC cells is also thought to be dictated by KCa3.1, as more aggressive cancers typically display up-regulation of this ion channel [62]. NAMI-A-mediated inhibition of KCa3.1 effects malignant cell migration, either by diminishing or completely preventing the migration of epithelial cancer cells [61].
Another pathway thought to be involved in the mechanism of action of NAMI-A is the ATM/ATR (Ataxia telangiectasia mutated / RAD3-related) kinase pathway, which is activated after DNA damage and works to regulate the cellular response [63]. NAMI-A activates both kinases, though appears more selective for phosphorylation of ATR [54]. The downstream effects include phosphorylation of the tumor suppressor p53 and kinases CHK1/CHK2, which are normally activated in response to genetic irregularities to coordinate cell cycle arrest [54]. NAMI-A also increases the expression of tumors suppressors such as p15INK4b, p21, and p27Kip1—CDK inhibitors that contribute to cell cycle arrest [54,64].
The anti-metastatic properties of NAMI-A arise from its ability to inhibit certain processes that are vital for metastatic formation and survival, including the adhesion and migration of cells. In addition to the mechanisms explained above, NAMI-A can inhibit these processes through protein interference, such as via α5β1 integrin [65]. Integrins are transmembrane-bound proteins that bind to other cellular components to facilitate the adhesion and migration of cells. Studies indicate that NAMI-A not only blocks α5β1 but significantly reduces the number of integrin receptors by modulating the expression of ITGA5 and ITGB1—the coding genes for the integrin subunits α5 and β1, respectively. This reduces downstream phosphorylation of focal adhesion kinase (FAK) and both of these processes are thought to contribute to decreased cellular adherence. Pre-treatment of HCT-116 colon cancer cells with NAMI-A significantly reduced adherence to Fibronectin and collagen I; an effect that was less apparent in α5β1 integrin-null cells. Notably, the observed effects on cellular adhesion and pFAK occurred in a manner that was inversely correlated with NAMI-A concentration, further supporting the hypothesis that lower doses of NAMI-A have greater efficacy [65].
MMP targeting is another anti-metastatic mechanism associated with NAMI-A. It inhibits the production of MMP-2 and the activity of MMP-2 and MMP-9 [66]. Within the functions of these MMPs, those of particular interest include chemotaxis and the activation of transforming growth factor (TGF β1) [67]. Since NAMI-A inhibits endothelial chemotaxis, this may be mediated via MMP inhibition [66]. The MMPs inhibited by NAMI-A and TGF-β1 partake in a feedback mechanism as TGF-β1 is also responsible for regulating MMP expression [68], which may explain the effect of NAMI-A on MMP-2 generation. TGF-β1 is a growth factor that selectively targets cancerous cells, inducing the epithelial–mesenchymal transition (EMT), invasion and migration; all integral processes needed for metastatic growth. NAMI-A counteracts this stimulus as it supposedly shares some of the pathways used by TGF-β1 but leads to opposing outcomes. Therefore, a metastatic response to TGF-β1 is prevented in tumor cells by hindering the processes mentioned above, leaving non-cancerous cell lines unaffected [69].
6. Case Study: KP1019/NKP1339
Another success story in the development of Ru(III) compounds that have reached clinical trials is NKP1339, reported by the Keppler group (Figure 2). The original form, KP1019, was modified to increase its aqueous solubility, generating the sodium salt equivalent, NKP1339 [46]. This Ru(III) complex has some structural similarity to NAMI-A due to its octahedral geometry and chloride ligands [46]. NKP1339 is a pro-drug and relatively inert compared to NAMI-A, as ligand loss/exchange does not occur as readily [44,47].
Both NAMI-A and NKP1339 bind non-covalently with proteins in the blood, likely via hydrophobic interactions [44,46]. NKP1339 undergoes rapid binding with albumin, so this is an important factor to consider when the metal complex is administered intravenously. The metal-protein adduct does account for the low side effect profile documented during the phase I trial of NKP1339, as the complex remains in its pro-drug form until it undergoes activation by reduction, after internalization by cells and release from albumin [70]. Albumin binding may also improve the selective accumulation of NKP1339 through two possible mechanisms: the EPR effect or by binding to the glycoprotein, gp60 [44]. The former occurs due to leaky vasculature within malignant cells, resulting in a greater uptake of albumin and other substances, which are then prevented from leaving by poor lymphatic drainage. In contrast, gp60 is present along the tumor endothelium and allows receptor-mediated transcytosis of the drug-albumin adduct to occur through to the extracellular tumor matrix [44]. Adduct formation with blood proteins is more significant for NKP1339 than NAMI-A since cellular uptake for the latter is limited [44], whereas uptake of NKP1339 is considered significantly more efficient [71].
Other ruthenium complexes, including RM175 [17], have been previously shown to interact with DNA through binding to guanine bases after reduction to their active hydrolyzed form [72], and NKP1339 is no exception. Due to the ability of NKP1339 to accumulate within the nucleus after activation [47], DNA may be one of the drug’s intracellular targets [46]. During activation, the metal ion is reduced, and the Ru(II) species is responsible for the DNA-adduct formation. Only weak adduct signals were emitted when NKP1339 was incubated with oligonucleotides, indicating low levels of interaction. This may be due, at least in part, to the slow ligand exchange rate of NKP1339, and a significant proportion of the drug remains intact [47]. In contrast with cisplatin, neither drug uptake nor genetic mutations, such as those involving p53 and k-ras, are correlated with the efficacy of NKP1339. This p53-independent mechanism implies that DNA binding is not likely to be the primary mechanism of action of NKP1339 [70], which may account for the lack of platinum-cross resistance observed with this Ru(III) complex in pre-clinical studies [73].
NKP1339 induces cell cycle arrest through mechanisms attributed to its redox activity in cancer cells [46]. Intracellular reactive oxygen species (ROS) concentrations (such as hydroxide radicals and superoxide) are already elevated in neoplastic cells compared with healthy cells [74] and this is thought to increase the vulnerability of cancer cells to further ROS fluctuations [75]. As such, redox-targeting metal complexes have attracted significant attention in recent years, and often exhibit significant selectivity for cancer cells over healthy cells [74,76,77,78]. NKP1339 has been shown to increase the intracellular ROS concentration [79] and upregulate the pro-apoptotic p38 MAPK pathway [80]. This biological cascade is normally activated in response to cellular stress, such as cytokines, DNA damage and ROS, and is implicated in cell cycle progression [81]. Activation of this pathway leads to downstream regulation of gene expression, including those that encode for cytokines, transcription factors, and cellular receptors [82]. More importantly, this pathway also regulates the G1/S and G2/M check points within the cell cycle [81]. By generating ROS and altering the cellular redox balance, NKP1339 induces G2/M cell cycle arrest [46].
Treatment of sensitive cell lines with NKP1339 induced cell death, typically within 20–30 h [70]. Accumulation of ROS prompts the loss of the mitochondrial membrane potential by inducing membrane permeabilisation, leading to activation of the intrinsic apoptosis pathway [83,84], which is typically caspase-dependent, and involves the cleavage of the zinc finger protein, poly-(ADP-ribose)-polymerase (PARP). [70,85] However, in the case of NKP1339, the extent of mitochondrial membrane depolarization is limited, suggesting another mechanism of apoptosis may be at work. Caspase-8 was cleaved as part of NKP1339-induced apoptosis [70], a key feature of the extrinsic apoptosis pathway. Combination treatment with NKP1339 and a caspase-8 inhibitor increased the extent of apoptosis observed within less responsive cell lines. The researchers deduced that the majority of apoptosis occurs not by the mitochondrial-intrinsic pathway, but via the extrinsic pathway [70]. Though mitochondria are a target of NKP1339, the effect of this interaction is limited as the majority of apoptotic activity is mediated by either death receptors on the cell surface or via other extrinsic pathways [86]. Since NKP1339 does not enhance the expression of these death receptors or their ligands, the Ru(III) complex may act via an alternative extrinsic pathway involving endoplasmic reticulum (ER) homeostasis [70].
Since the anticancer activity of NKP1339 is predominantly mediated through changes in redox homeostasis [46], another notable target is the endoplasmic reticulum (ER), which is also influenced by the cellular redox environment. This organelle controls the maturation, folding, and release of proteins, and initiates the unfolded protein response (UPR) as part of a feedback system, suspending the cell cycle either temporarily until the proteins are restored or, permanently, prior to apoptosis [79]. The protein Nrf2 is able to trigger the expression of antioxidant genes to reduce the cell’s exposure to oxidative stress [87]. After treatment with NKP1339, Nrf2 translocates to the nucleus to commence its protective activity. This observation may be ROS-mediated and demonstrates that drug-induced hyper-oxidation is sufficiently substantial to initiate a cellular response. The redox properties of NKP1339 lead to dysregulation of various other proteins that affect the ER. NKP1339 increases ROS levels in vitro in colon carcinoma cell lines (HCT116 and SW480), causing upregulation of CHOP mRNA [79]. The targets of this transcription factor include (1) GADD34, implicated in cell cycle arrest; (ii) DR5, which promotes cell death through caspase activation and initiation of the extrinsic pathway; and (3) Ero1α, which leads to ER hyper-oxidation and stimulates cell death [88]. CHOP also reduces the expression of the anti-apoptotic protein Bcl2, and consequently, excess CHOP generation favors the induction of apoptosis [79].
Sustained ER stress may result in cellular dysfunction or death [88], so alterations in the UPR could be exploited for antiproliferative activity. In vitro treatment with NKP1339 was shown to downregulate various ER-based proteins, including signaling molecules PERK and Ire1α and the chaperone proteins calnexin and GRP78 [70]. NKP1339 is an effective inhibitor of the last one by preventing its activation [70,89]. These proteins all have an active role in UPR regulation: calnexin assists with protein folding, whereas the signaling molecules PERK and Ire1α are two of the main transmembrane receptors bound to the heat shock protein, GRP78 [79]. When GRP78 senses unfolded proteins, it simultaneously binds them and liberates the signaling molecules that drive the UPR [79]. GRP78 is often viewed as the “master regulator” of the UPR [89] and functions to protect the cell with the aim of restoring the ER to its original condition [90]. Downregulation or inhibition of ER proteins results in a lack of control over protein folding and accumulation of damaged proteins. Drug-induced ROS generation damages proteins, further enhancing the ER’s exposure to stress [79]. Another advantage of this mechanism is that GRP78 is associated with chemo-resistance, so inhibition of this protein may lead to a better prognosis and prevent the occurrence of GRP78-mediated resistance [89]. In vitro use of NKP1339 is also able to increase drug-sensitivity in cells that are already resistant; a promising indication that drug-resistance might be reversed.
Another reactive oxygen species, nitric oxide, can be scavenged by NKP1339, though its effect on NO-mediated processes is weak in comparison to NAMI-A [51]. Therefore, there is little evidence to support its interference with VEGF/NO-stimulated angiogenesis or cell migration [51], explaining why NKP1339 is more effective against solid tumors than NAMI-A. By unsettling redox homeostasis, NKP1339 is able to inhibit DNA synthesis, induce G2/M cell cycle arrest and initiate both intrinsic [46] and extrinsic apoptosis [70]. Owing to its multi-targeting mechanism of action, overexpression of proteins associated with multi-drug resistance (MRP1, BCRP, LRP, and the transferrin receptor) little hinders the drug’s efficacy, which also explains the lack of cross-resistance [73,85]. The phase I trial of NKP1339 was used as a dose-escalation study to assess its use for the treatment of advanced solid tumors. The study included patient tolerability as well as pharmacodynamic and pharmacokinetic studies of the drug (Niiki Pharma Inc. and Intezyne Technologies Inc., 2017). The trial (NCT0145297) was completed in 2016 and was deemed successful [46]. NKP1339 was also shown to be particularly effective against neuroendocrine tumors (and exhibited limited side effects in trial participants [46,73].
7. Current Developments in Ruthenium Anticancer Agents and Future Perspectives
The complexes in these four case-studies have paved the way for future research, much of which is now being completed by not only by well-established researchers but also by internationally recognized new and upcoming leaders in the field. Pushing the boundaries, new ruthenium anticancer complexes show a wider diversity of coordination spheres and a vast range of ligands that include, but are not limited to, extended intercalating aromatic units, polypyridyl rings, fluorescent derivatives, bio-active molecules, ferrocifen analogues, and carbenes, all of which exploit donor atoms such as C, N, O, S and P.
Even more interesting is observing the escalation of novel approaches regarding targeting strategies and MoA involved in the anticancer activity of Ru complexes, all of which are fueled by our increased understanding of the complexes’ fates at the cellular level. Disruption of protein-protein interactions, [91] enzymatic inhibition [92,93], and redox modulation [94], as well as chromatin [95] and histone [96,97] targeting, are only a few examples of cellular events being used as means for antiproliferative activity, with in-cell catalysis taking advantage of well-established reactions in the chemistry of materials field [77,78]. Crucially, the understanding of metal anticancer complexes as efficient multi-targeting agents is starting to have a positive outlook, if not yet for funding bodies, then at least from an academic perspective. Hence, the attempts to locate a single drug target may yet develop into the search for a majoritarian cellular event in a field that seems to move at a highly fast pace.
Such advances and new trends in thought should not forget how far NKP1339, NAMI-A, RM175, and RAPTA-C have advanced the field, and how critical these complexes have been in the development of the field. As a community, we may benefit from more closely examining these four compounds to learn, amongst other things, that small structural changes generate large variations in the MoA at the cellular level [98]. Although structure activity relationships are not easily established, current and further developments of analytical and cellular techniques will provide more investigative tools and, subsequently, a better understanding of cellular behavior. The key for the new generation of anticancer complexes may well be just round the corner.
Funding
This research received no external funding.
Acknowledgments
Economic support from the School of Pharmacy and Institute of Clinical Sciences at the University of Birmingham are acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Florea, A.M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Invest. 1991, 87, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalt. Trans. 2006, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, J.; Fandzloch, M.; Łakomska, I. The reduction of ruthenium(III) complexes with triazolopyrimidine ligands by ascorbic acid and mechanistic insight into their action in anticancer therapy. Inorg. Chim. Acta 2019, 484, 305–310. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougan, S.J.; Habtemariam, A.; McHale, S.E.; Parsons, S.; Sadler, P.J. Catalytic organometallic anticancer complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11628–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Goschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre- dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; Murdoch, P.D.S.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Masi, A.; Peacock, A.F.A.; Habtemariam, A.; Sadler, P.J.; Sava, G. In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model. J. Inorg. Biochem. 2010, 104, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Habtemariam, A.; van der Geer, E.; Fernández, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18269–18274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, N.P.E.; Sadler, P.J. Exploration of the medical periodic table: Towards new targets. Chem. Commun. 2013, 49, 5106–5131. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.J.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Westhorpe, A.; Romero, M.J.; Habtemariam, A.; Gallevo, C.R.; Bark, Y.; Menezes, N.; Sadler, P.J.; Sharma, R.A. Radiosensitisation of human colorectal cancer cells by ruthenium(II) arene anticancer complexes. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Parkinson, J.; Parsons, S.; Coxall, R.; Gould, R.O.; Sadler, P.J. Organometallic ruthenium(II) diamine anticancer complexes: Arene-nucleobase stacking and stereospecific hydrogen-bonding in guanine adducts. J. Am. Chem. Soc. 2002, 124, 3064–3082. [Google Scholar] [CrossRef] [PubMed]
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In vitro and in vivo activity and cross resistance profiles of novel ruthenium(II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tong, T.; Fan, W.; Fan, F.; Antinore, M.J.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; Rajasekaran, B.; et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene 2002, 21, 8696–8704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 2, 1396–1397. [Google Scholar] [CrossRef]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA): Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Van Beijnum, J.R.; Casini, A.; Nazarov, A.A.; Wagnières, G.; Van Den Bergh, H.; Dyson, P.J.; Griffioen, A.W. Organometallic ruthenium(II) arene compounds with antiangiogenic activity. J. Med. Chem. 2011, 54, 3895–3902. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Else, R.; Reid, E.; Zeitlin, B.; Aird, R.; Muir, M.; Dodds, M.; Fiebig, H.; Sadler, P.J.; Jodrell, D.I. Anti-tumour activity in non-small cell lung cancer models and toxicity profiles for novel ruthenium(II) based organo-metallic compounds. Biochem. Pharmacol. 2006, 71, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Berndsen, R.H.; Dubois, M.; Müller, C.; Schibli, R.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. In vivo anti-tumor activity of the organometallic ruthenium(II)-arene complex [Ru(η6-p-cymene)Cl2(pta)] (RAPTA-C) in human ovarian and colorectal carcinomas. Chem. Sci. 2014, 5, 4742–4748. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Risse, J.; Ang, W.H.; Schmitt, F.; Zava, O.; Ruggi, A.; Groessl, M.; Scopelitti, R.; Juillerat-Jeanneret, L.; Hartinger, C.G.; et al. Anthracene-tethered ruthenium(II) arene complexes as tools to visualize the cellular localization of putative organometallic anticancer compounds. Inorg. Chem. 2012, 51, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Furrer, M.A.; Schmitt, F.; Wiederkehr, M.; Juillerat-Jeanneret, L.; Therrien, B. Cellular delivery of pyrenyl-arene ruthenium complexes by a water-soluble arene ruthenium metalla-cage. Dalt. Trans. 2012, 41, 7201–7211. [Google Scholar] [CrossRef] [PubMed]
- Kilpin, K.J.; Clavel, C.M.; Edafe, F.; Dyson, P.J. Naphthalimide-tagged ruthenium–arene anticancer complexes: Combining coordination with intercalation. Organometallics 2012, 31, 7031–7039. [Google Scholar] [CrossRef]
- Bailly, C.; Braña, M.; Waring, M.J. Sequence-Selective Intercalation of Antitumour Bis-Naphthalimides into DNA. Eur. J. Biochem. 2018, 240, 195–208. [Google Scholar] [CrossRef]
- Ang, W.H.; Parker, L.J.; De Luca, A.; Juillerat-Jeanneret, L.; Morton, C.J.; Lo, B.M.; Parker, M.W.; Dyson, P.J. Rational design of an organometallic glutathione transferase inhibitor. Angew. Chemi. Int. Ed. 2009, 48, 3854–3857. [Google Scholar] [CrossRef] [PubMed]
- Chakree, K.; Ovatlarnporn, C.; Dyson, P.J.; Ratanaphan, A. Altered dna binding and amplification of human breast cancer suppressor gene BRCA1 induced by a novel antitumor compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. Int. J. Mol. Sci. 2012, 13, 13183–13202. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Biondi, I.; Dyson, P.J.; Bhattacharyya, A. A bifunctional organometallic ruthenium drug with multiple modes of inducing apoptosis. J. Biol. Inorg. Chem. 2011, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, H.U.; Movassaghi, S.; Morrow, S.J.; Kubanik, M.; Hartinger, C.G. Metallomic study on the metabolism of RAPTA-C and cisplatin in cell culture medium and its impact on cell accumulation. Metallomics 2018, 10, 455–462. [Google Scholar] [PubMed]
- Weiss, A.; Ding, X.; van Beijnum, J.R.; Wong, I.; Wong, T.J.; Berndsen, R.H.; Dormond, O.; Dallinga, M.; Shen, L.; Schlingemann, R.O.; et al. Rapid optimization of drug combinations for the optimal angiostatic treatment of cancer. Angiogenesis 2015, 18, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Berndsen, R.H.; Ding, X.; Ho, C.M.; Dyson, P.J.; Van Den Bergh, H.; Griffioen, A.W.; Nowak-Sliwinska, P. A streamlined search technology for identification of synergistic drug combinations. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 1549–1560. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2003, 236, 209–233. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Artner, C.; Holtkamp, H.U.; Hartinger, C.G.; Meier-Menches, S.M. Characterizing activation mechanisms and binding preferences of ruthenium metallo-prodrugs by a competitive binding assay. J. Inorg. Biochem. 2017, 177, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, G.; Alessio, E.; Sava, G.; Pacor, S.; Coluccia, M.; Boccarelli, A. Water-soluble ruthenium(III)-dimethyl sulfoxide complexes: Chemical behaviour and pharmaceutical properties. Met. Based. Drugs 1993, 1, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P.J.; Sava, G. Metal-based antitumour drugs in the post genomic era. Dalt. Trans. 2006, 1929–1933. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Filippi, S.; Messori, L.; Piccioli, F.; Orioli, P.; Sava, G.; Ziche, M. Antiangiogenic properties of selected ruthenium(III) complexes that are nitric oxide scavengers. Br. J. Cancer 2003, 88, 1484–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oszajca, M.; Kuliś, E.; Stochel, G.; Brindell, M. Interaction of the NAMI-A complex with nitric oxide under physiological conditions. New J. Chem. 2014, 38, 3386–3394. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.; Zhao, Z.; Li, L.; Zheng, W.; Chen, T. Antiangiogenic ruthenium(II) benzimidazole complexes, structure-based activation of distinct signaling pathways. Metallomics 2015, 7, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases Marie. Microbiol. Mol. Biol. Rev. 2011, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Pelillo, C.; Chambery, A.; Sava, G. Influence of components of tumour microenvironment on the response of HCT-116 colorectal cancer to the ruthenium-based drug NAMI-A. J. Inorg. Biochem. 2017, 168, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Gasparoli, L.; Stefanini, M.; Ristori, M.; D’Amico, M.; Alessio, E.; Scaletti, F.; Becchetti, A.; Arcangeli, A.; Messori, L. NAMI-A is highly cytotoxic toward leukaemia cell lines: Evidence of inhibition of KCa 3.1 channels. Dalt. Trans. 2014, 43, 12150–12155. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stühmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grössinger, E.M.; Weiss, L.; Zierler, S.; Rebhandl, S.; Krenn, P.W.; Hinterseer, E.; Schmölzer, J.; Asslaber, D.; Hainzl, S.; Neureiter, D.; et al. Targeting proliferation of chronic lymphocytic leukemia (CLL) cells through KCa3.1 blockade. Leukemia 2014, 28, 954–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulk, E.; Ay, A.S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic dysregulation of KCa3.1 channels induces poor prognosis in lung cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATMand ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a12716. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Delfino, R.; Casarsa, C.; Sava, G. CDK1 Hyperphosphorylation Maintenance Drives the Time-course of G2-M Cell Cycle Arrest after Short Treatment with NAMI-A in Kb Cells. Anticancer. Agents Med. Chem. 2012, 12, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Bruno, M.; Boccarelli, A.; Coluccia, M.; Ribatti, D.; Bergamo, A.; Garbisa, S.; Sartor, L.; Sava, G. Inhibition of endothelial cell functions and of angiogenesis by the metastasis inhibitor NAMI-A. Br. J. Cancer 2002, 4, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, G.; Theocharis, A.D.; Karamanos, N.K. Roles of MMP in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.M.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sava, L.B.A.M.G.; Bergamo, A. Effects of the ruthenium-based drug NAMI-A on the roles played by TGF-β1 in the metastatic process. J. Biol. Inorg. Chem. 2015, 20, 1163–1173. [Google Scholar]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Pongratz, M.; Jakupec, M.A.; Heffeter, P.; Berger, W.; Lackinger, L.; Keppler, B.K.; Marian, B. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Parkinson, J.; Morris, R.E.; Sadler, P.J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: Novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Dickson, N.R.; Jones, S.F.; Burris, H.A.; Ramanathan, R.K.; Weiss, G.J.; Infante, J.R.; Bendell, J.C.; McCulloch, W.; Von Hoff, D.D. A phase I dose-escalation study of NKP-1339 in patients with advanced solid tumors refractory to treatment. J. Clin. Oncol. 2011, 29, 2607. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z. Increased oxidative stress as a selective anticancer therapy. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015, 6, 6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—Involves ER- and ROS-related effects in colon carcinoma cell lines. Invest. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Atil, B.; Kryeziu, K.; Groza, D.; Koellensperger, G.; Körner, W.; Jungwirth, U.; Mohr, T.; Keppler, B.K.; Berger, W. The ruthenium compound KP1339 potentiates the anticancer activity of sorafenib in vitro and in vivo. Eur. J. Cancer 2013, 49, 3366–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, T.M.; Rincon, M. Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009, 5, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001, 495, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta, Mol. Cell. Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-J.; Wang, W.; Huang, S.-Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.-M.D.; Ma, D.-L.; et al. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Casini, A.; Sava, G.; Dyson, P.J. Organometallic ruthenium-based antitumor compounds with novel modes of action. J. Organomet. Chem. 2011, 696, 989–998. [Google Scholar] [CrossRef]
- Mitrović, A.; Kljun, J.; Sosič, I.; Gobec, S.; Turel, I.; Kos, J. Clioquinol–ruthenium complex impairs tumour cell invasion by inhibiting cathepsin B activity. Dalt. Trans. 2016, 45, 16913–16921. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxidants Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Magistrato, A.; Riedel, T.; von Erlach, T.; Davey, C.A.; Dyson, P.J.; Rothlisberger, U. Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies. ChemMedChem 2016, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef]
- Wu, B.; Ong, M.S.; Groessl, M.; Adhireksan, Z.; Hartinger, C.G.; Dyson, P.J.; Davey, C.A. A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem. Eur. J. 2011, 17, 3562–3566. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
Figure 1.
RM175 (left) and RAPTA-C (right) are typical examples of 18-electron ruthenium arene “piano-stool” complexes, in which an η6-arene ring stabilises the 2+ oxidation state of the ruthenium metal centre.
Figure 1.
RM175 (left) and RAPTA-C (right) are typical examples of 18-electron ruthenium arene “piano-stool” complexes, in which an η6-arene ring stabilises the 2+ oxidation state of the ruthenium metal centre.

Figure 2.
NAMI-A (left) and KP1019/NKP1339 (right) are octahedral Ru(III) pro-drugs, which are hypothesized to undergo an “activation by reduction” mechanism inside cells to form more active Ru(II) species.
Figure 2.
NAMI-A (left) and KP1019/NKP1339 (right) are octahedral Ru(III) pro-drugs, which are hypothesized to undergo an “activation by reduction” mechanism inside cells to form more active Ru(II) species.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030031
AMA Style
Coverdale JPC, Laroiya-McCarron T, Romero-Canelón I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics. 2019; 7(3):31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030031
Chicago/Turabian StyleCoverdale, James P. C., Thaisa Laroiya-McCarron, and Isolda Romero-Canelón. 2019. "Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates?" Inorganics 7, no. 3: 31. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030031
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.