1. Introduction
The C
Si
H
molecule has been the subject of very few literature studies, despite being a bicyclobutane/butadiene analog of fundamental interest. Holme et al. [
1] predicted the existence of over a dozen cyclic C
Si
H
stationary points at the Hartree-Fock (HF) level, assigning the lowest-energy isomer as the three-membered ring silylsilacyclopropenylidene. Inclusion of correlation energy using third-order many-body perturbation theory (MBPT(3)) left the isomer ordering unchanged, though it is worth noting that a modest 6-31G(d) basis set was employed. Holme remarked that a three-membered-ring isomer seemed an “unlikely choice” due to the enhanced strain destabilization as compared with competing four-membered-ring structures. Structural compression is energetically favorable when electron-correlation stabilization dominates over strain destabilization. If this is the case for C
Si
H
, one might expect that the maximally compressed isomer, a bridged bicyclobutane analog, should be the global minimum. One such isomer was considered by Holme et al., but it was predicted to be over 10 kcal/mol higher in energy than silylsilacyclopropenylidene.
In the first experimental study on C
Si
H
, Maier et al. reported infrared (IR) measurements supplemented by density functional theory (DFT) vibrational simulations [
2]. In their workup a mixture of silane structures was thermalized by pulsed flash pyrolysis at 1500 K, and the resulting mixture was rapidly frozen out in an Ar matrix at 10 K before IR characterization. Signatures of silylsilacyclopropenylidene were detected and upon further irradiation the corresponding ring-opening photolysis products were observed (i.e., (silylethynyl)silylene and ethynylsilylsilylene). Accompanying calculations were performed using the BLYP generalized-gradient approximation (GGA) exchange correlation functional (XCF) and assigned silylsilacyclopropenylidene as the global minimum, in agreement with the prediction of Holme et al. Due to this apparent agreement between theory and experiment, this assignment of the lowest-energy isomer of C
Si
H
has stood for over 20 years.
The purpose of the current study is to apply more sophisticated computational methods to investigate the delicate balance between electron-correlation stabilization and strain destabilization in C
Si
H
. Ground-state DFT methods, and their extension to excited states via time-dependent (TD) DFT [
3,
4,
5], are the most widely-used approaches for describing electron correlation in atoms, molecules, and solids [
6]. While superior to the HF self-consistent field (SCF) theory, the popular BLYP and B3LYP approaches have major shortcomings, which often result in qualitatively incorrect predictions [
7]. In light of recent advancements, the default choice of XCF needs to be augmented, updated, or supplanted entirely [
8]. Since it is unclear which of the hundreds of existing functionals perform best overall, several popular functionals were chosen from the BLYP [
9,
10], PBE [
11,
12], and Minnesota functional families [
13,
14,
15,
16] for evaluation in the context of the current application.
The quality of XCFs can be ranked as being on one of five rungs of Jacob’s ladder, with the fifth rung approaching the “heaven” of chemical accuracy [
17]. Here the term “chemical accuracy” takes the colloquial meaning that a method is predictive to within 1 kcal/mol. Functionals including only a local density approximation to the correlation functional [
18] represent the first rung, while those incorporating GGA correlation represent the second rung. After density gradients, Laplacians can be included in the mix, resulting in meta-GGA functionals, or the third rung of Jacob’s ladder [
19]. Hybrid functionals are on the fourth rung of Jacob’s ladder, often referred to as hyper-GGAs, while the fifth rung of Jacob’s ladder is represented here by double-hybrid (DH) DFT methods [
20,
21], which semiempirically blend DFT and wave-function theory. Only functionals from the highest three rungs will be considered here.
Electron correlation can be treated within a more hierarchical framework by using wave-function methods, which, unlike DFT, are systematically improvable to the exact solution of the Schrödinger equation. In the ground state, approximations based on the coupled-cluster (CC) hierarchy [
22,
23,
24,
25,
26,
27] are among the most rapidly convergent approaches, with the CCSD(T) method often referred to as the “gold standard” of quantum chemistry. While expensive, CCSD(T) energies extrapolated to the complete basis set (CBS) limit are widely considered benchmark-quality for molecules near their equilibrium geometries. In situations where degeneracy is commonly encountered (e.g., when crossing transition states, when accessing excited states, or when breaking or forming bonds), CCSD(T) can fail [
28], and in these cases one can instead turn to new generations of CC methods, such as the left-eigenstate completely renormalized (CR) CC method [
29,
30,
31,
32] called CR-CC(2,3). The CR-CC(2,3) approach provides an accurate description of single-bond-breaking processes [
33,
34], biradical and magnetic molecules [
35,
36], and transition states [
37,
38]. Excited states can be accessed in a straightforward manner using the equation of motion (EOM) CC formalism [
39,
40,
41,
42], resulting in the CR-EOMCC(2,3) approximation and its size-intensive variant
-CR-EOMCC(2,3) [
31,
43,
44,
45,
46]. Experience shows that reliable energetics are produced for ring-opening [
47] and bond-rearrangement processes [
48] when CR-CC(2,3) is applied on top of appropriate complete-active-space (CAS) SCF geometries. The steep
scalings of CCSD(T) and CR-CC methods prohibit their use for all but the smallest systems, but fortunately they are readily applicable to the title molecule.
The motivation for this work spans several fields. Mapping interstellar silicon reaction networks is one application [
49], as C
Si
H
may result from bimolecular collisions between the highly-abundant acetylene and its silicon-based analog disilyne. This bimolecular reaction and subsequent isomerization can also be considered as a model system for understanding preferred bonding configurations in coordinate complexes [
50], bulk silicon surface-adsorption [
51], and defect-inclusion processes [
52]. Many of the present modeling decisions were guided by prior benchmarking work on the structure and spectroscopy of hydrogen-free silicon carbide clusters [
53,
54]. This study continues along these lines, investigating the performance of various modern DFT methods by comparing against spectroscopic values and high-level computational results. The structure of the paper is as follows. The methods employed are detailed in
Section 2, the results and discussion are covered in
Section 3, while concluding remarks and final recommendations for density functional usage are offered in
Section 4.
2. Computational Methods
Electronic structure calculations were performed using the GAMESS [
55,
56] and Gaussian16 [
57] packages on the AFRL DSRC SGI Ice X Thunder and a local workstation. Visualizations were performed using the GaussView V6 [
58] and MacMolPlt V7.3 [
59] software packages. All DFT gradient calculations employed the tight
JANS = 2 grid in GAMESS and the ultrafine grid in Gaussian. Grimme’s D3(BJ) empirical dispersion correction was also employed [
60,
61,
62,
63].
We employed the basis sets DZP [
64], Def2-TZVP, and Def2-QZVP [
65]. Ahlrichs’ triple-
(
) and quadruple-
(
) basis sets were used in conjunction with the formula of Helgaker et al. [
66],
, for CBS extrapolation of the correlation energy, with the SCF component of CBS energies approximated at the HF/cc-pV5Z level. Geometry optimizations were performed at the level of theory indicated by the conventional double-forward slash notation (e.g., CCSD(T)//MBPT(2)) [
67,
68]. All MBPT(4) calculations correspond to the MBPT(4)-SDQ approximation, and all CR-CC(2,3) and CR-EOMCC(2,3) triples corrections correspond to the most complete “D” variant, which employs the exact Epstein-Nesbet-like denominator [
69]. Core orbitals were frozen in all wave-function calculations.
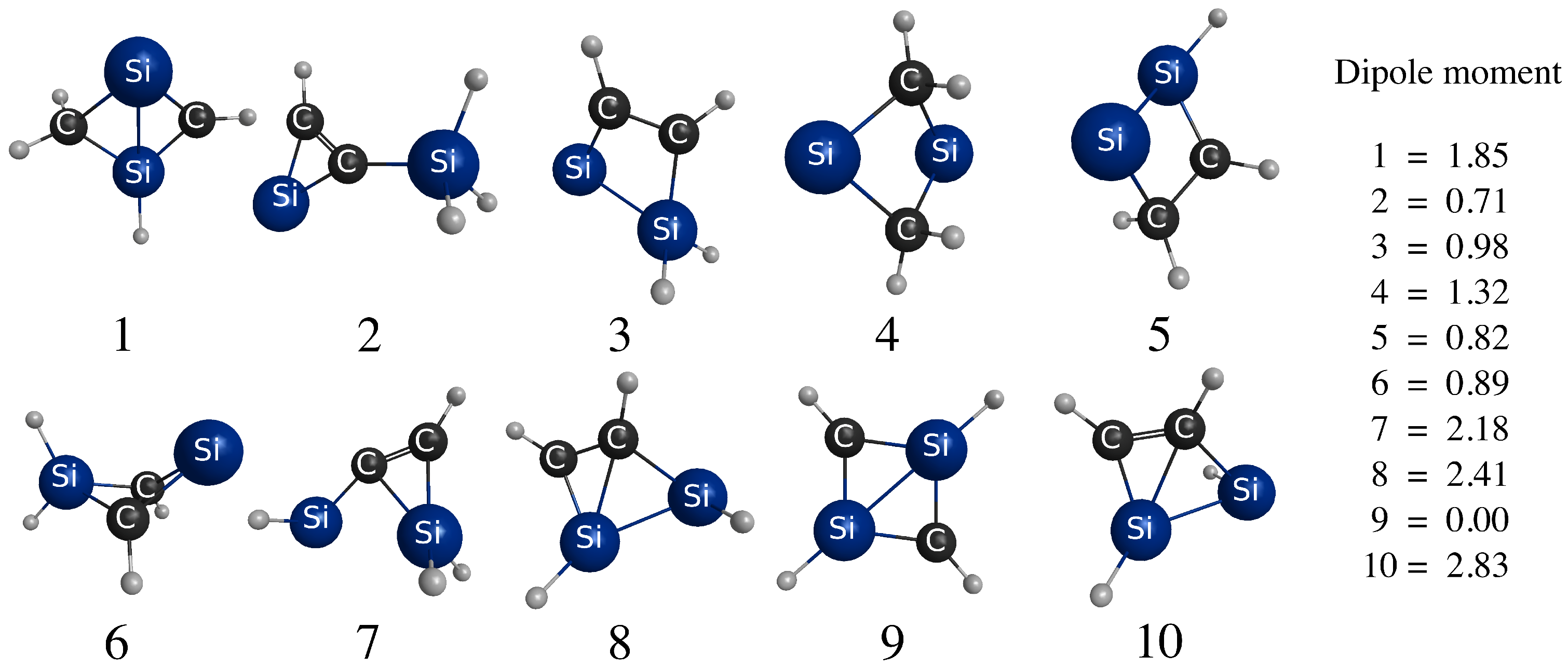
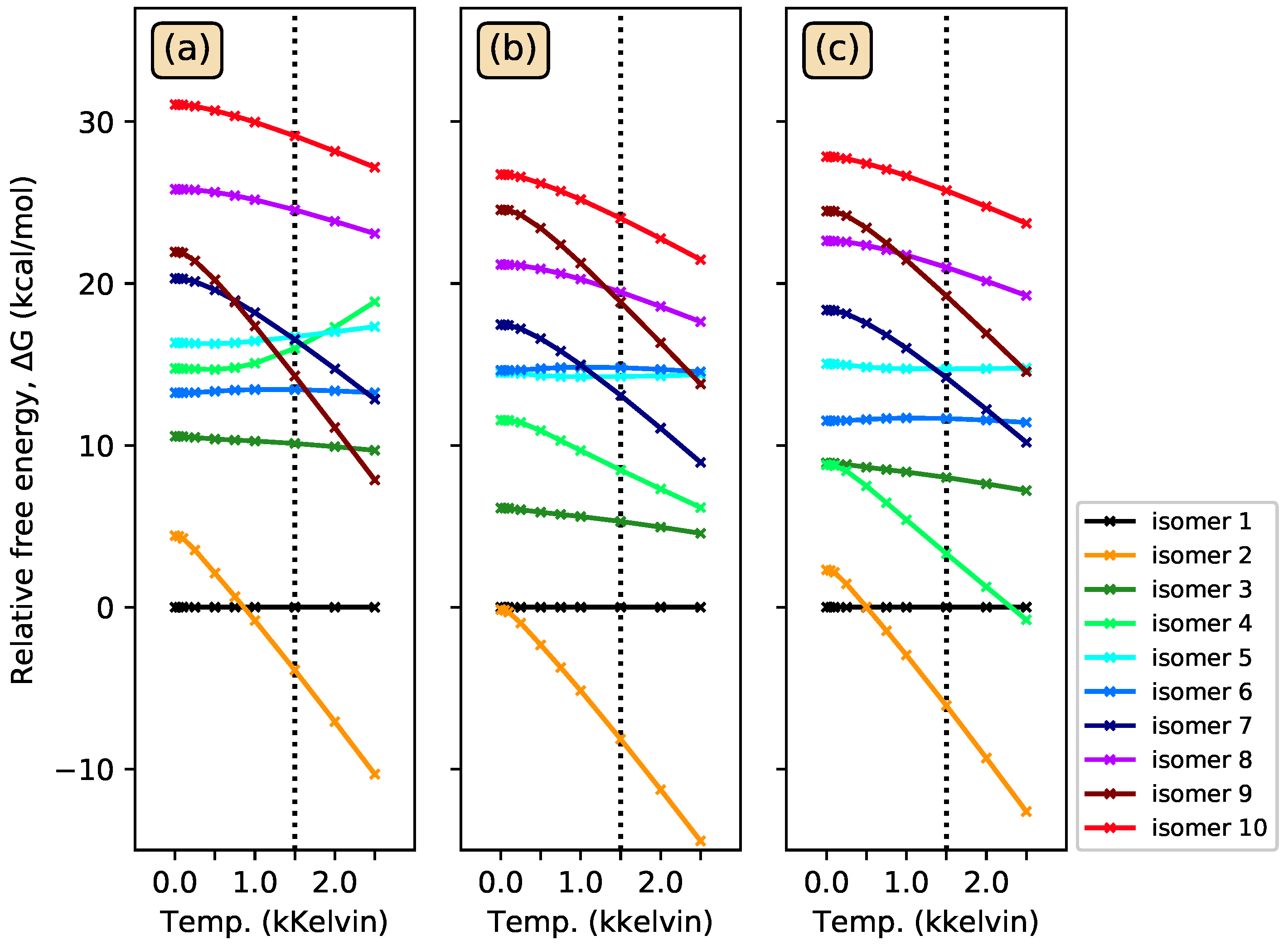
4. Conclusions
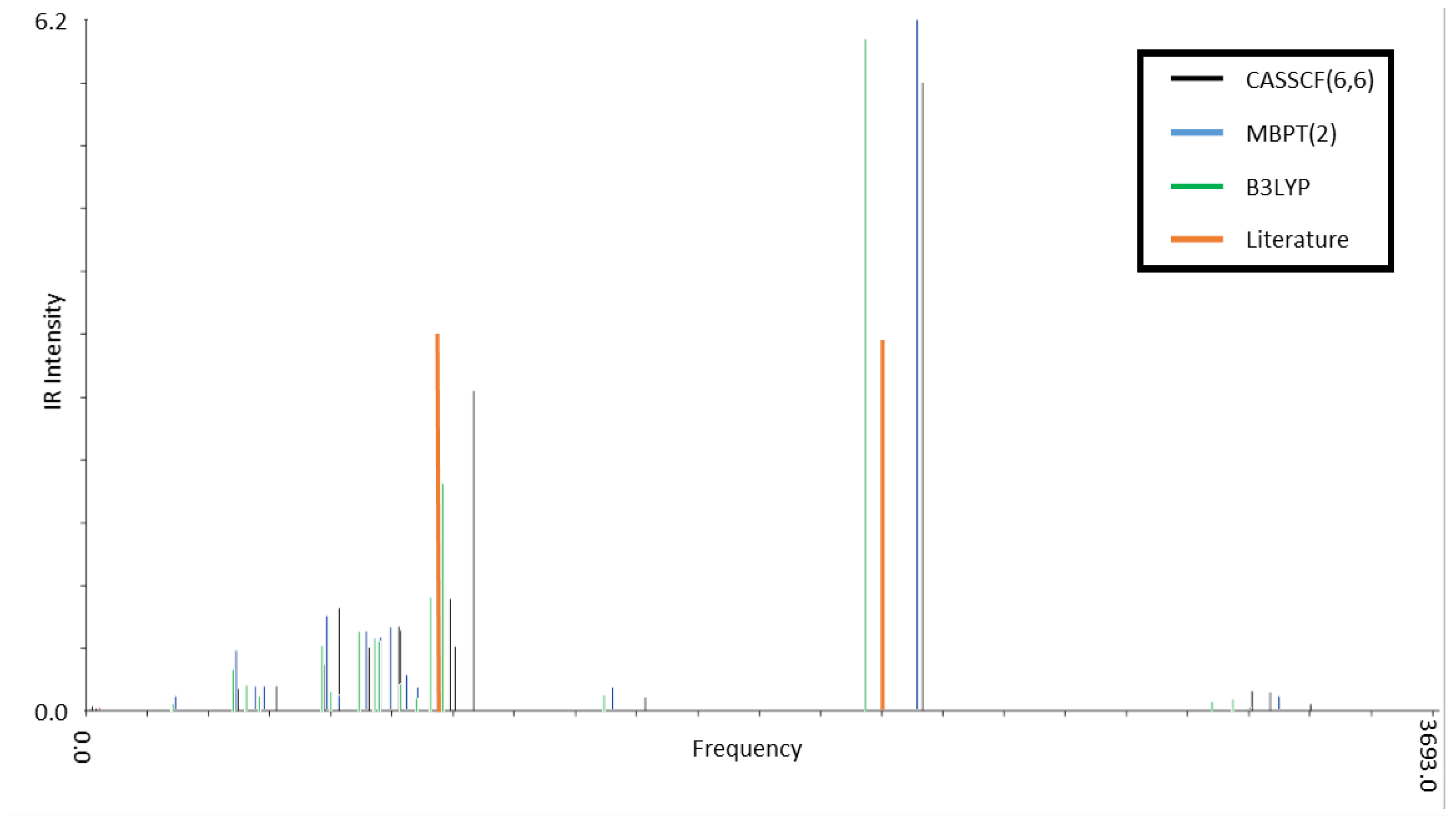
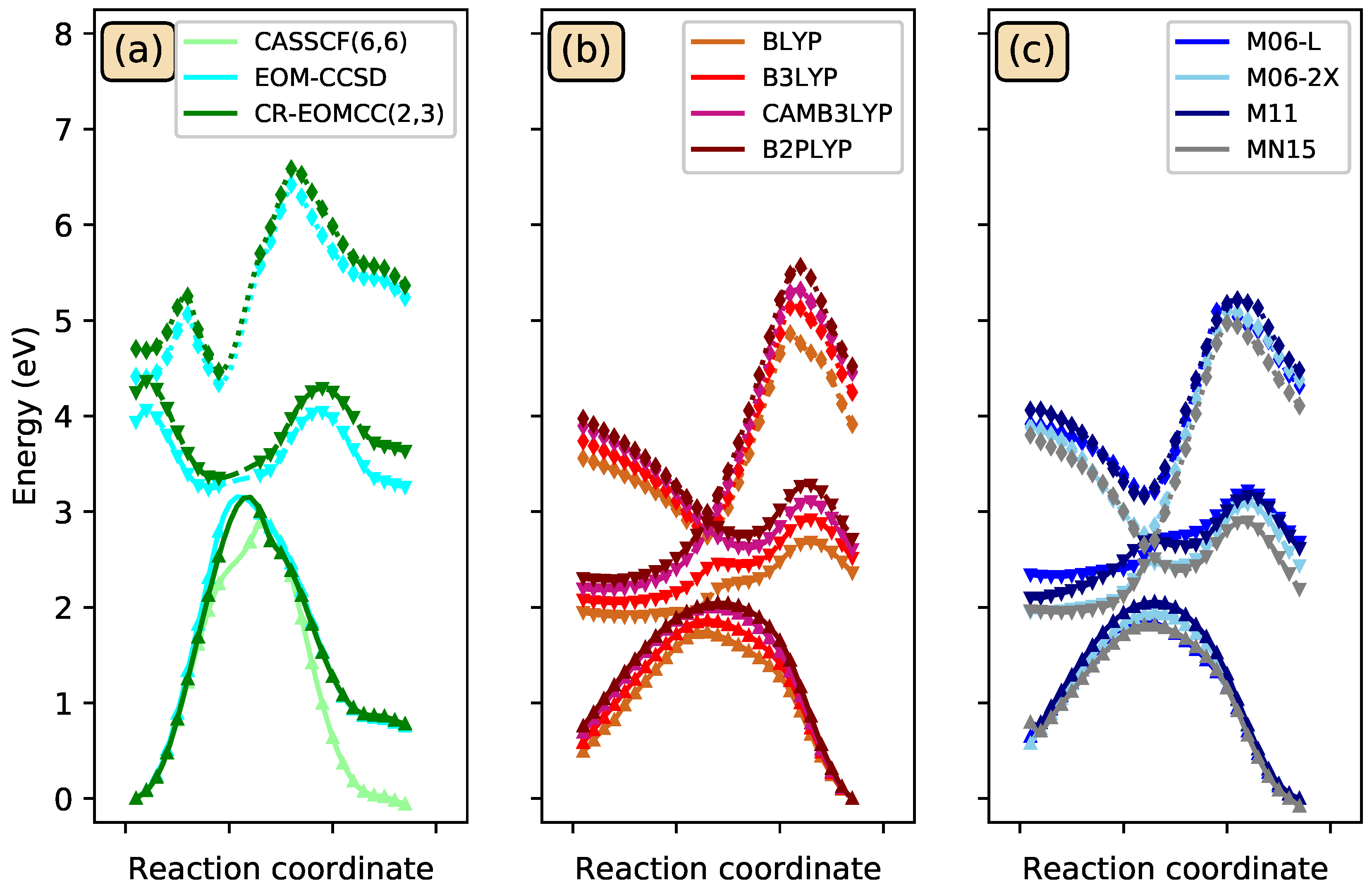
Ten CSiH isomers were characterized as PES minima and energy-ordered using high-level coupled-cluster calculations. Isomer 1, or 1,2-didehydro-1,3-disilabicyclo[1.1.0]butane, was assigned as the global minimum, in contrast to all existing literature on this system which favored isomer 2, or silylsilacyclopropenylidene. High-level coupled-cluster isomer energies were used to benchmark DFT methods, and BLYP and B3LYP incorrectly predicted isomer 2 as the global minimum, even when very large basis sets were employed. Many DFT methods struggled to order all ten isomer energies correctly with respect to benchmarks. Only the B2-PLYP+D3 DH-DFT method energy-ordered all isomers similarly to the benchmarks, while also producing an MUE within chemical accuracy. Among the pure and hybrid functionals tested, only M06-2X predicted the same energy ordering with respect to the benchmarks but its MUE was slightly above the chemical accuracy threshold.
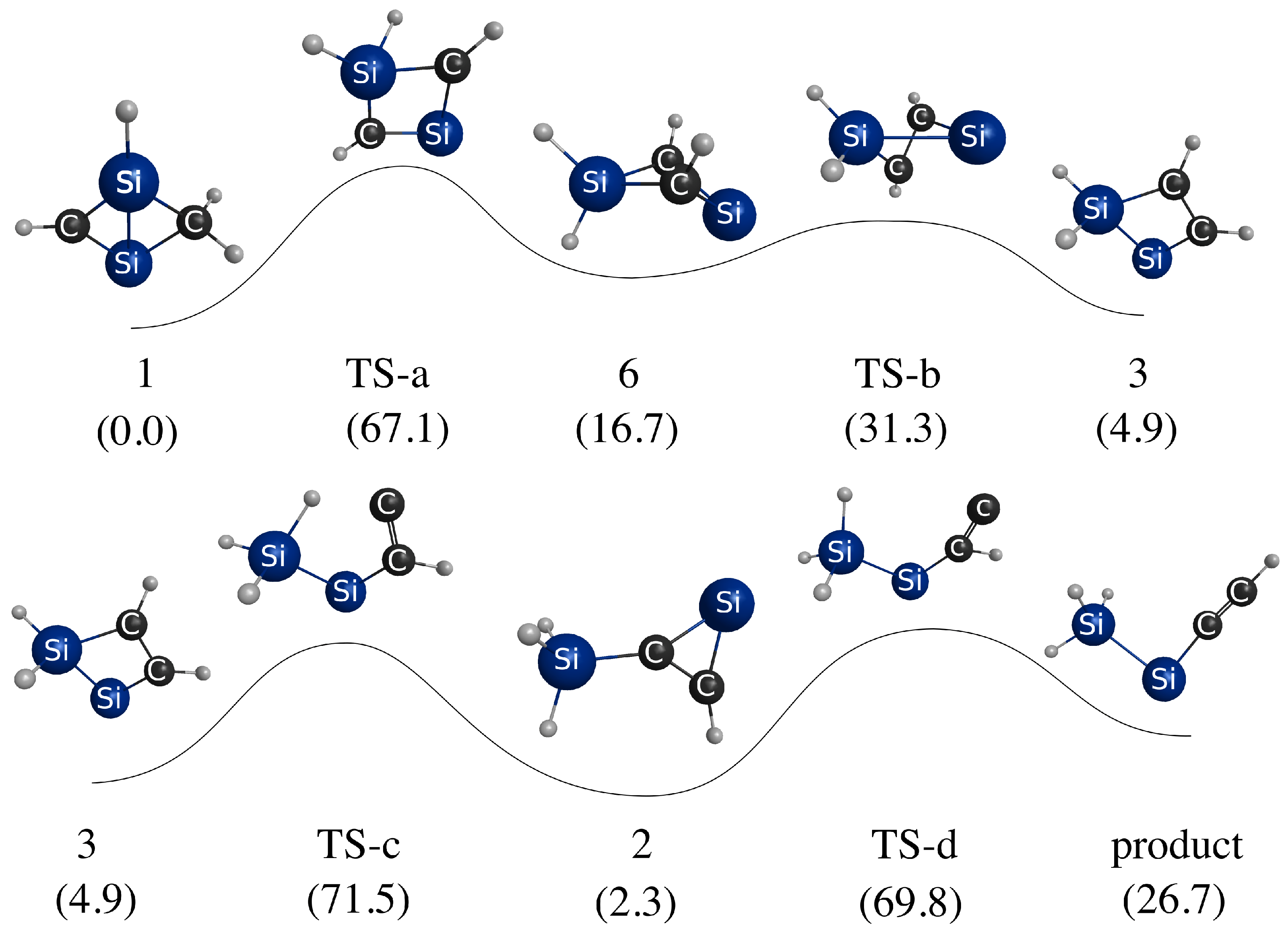
Assignment of isomer 1 as the global minimum required a reinterpretation of prior experimental data to reconcile with past assignments of isomer 2 as the global minimum. Computations helped show that the frequencies of isomers 1 and 2 overlap, which likely contributed to the broad bands previously observed in the IR difference spectrum. Some PES mapping was also performed to provided insight into the mechanism of photolysis of isomers 1 and 2 to form the observed products HSiCCSiH and HSiSiCCH. These simulations suggested that the ultraviolet frequencies used to irradiate the sample may initiate photolysis in both isomers 1 and 2, leading to the observed products.
Taken together, these simulations provide compelling evidence that the energetically similar isomers
1 and
2 were likely both present as reactants in the measurements reported in Ref. [
2]. In light of the present analysis, there exists no contradictory evidence in the literature to dismiss assignment of isomer
1 as the global minimum. In closing, isomer
1 has been identified as a prominent configuration on the global PES of a barrierless acetylene-disilyne collision, and its dipole moment is strong enough for electron binding. Thus, it may be of significance in interstellar silicon carbide reaction networks. This prospect will be investigated in a future study.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}