Development and Validation of HPLC-DAD Method for Simultaneous Determination of Seven Food Additives and Caffeine in Powdered Drinks




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Chromatographic Method
2.3. Sample Preparation
2.4. Box–Behnken Design and Data Analysis
2.5. System Suitability Test
2.6. Method Validation
3. Results
3.1. Data Acquisition for the Responses
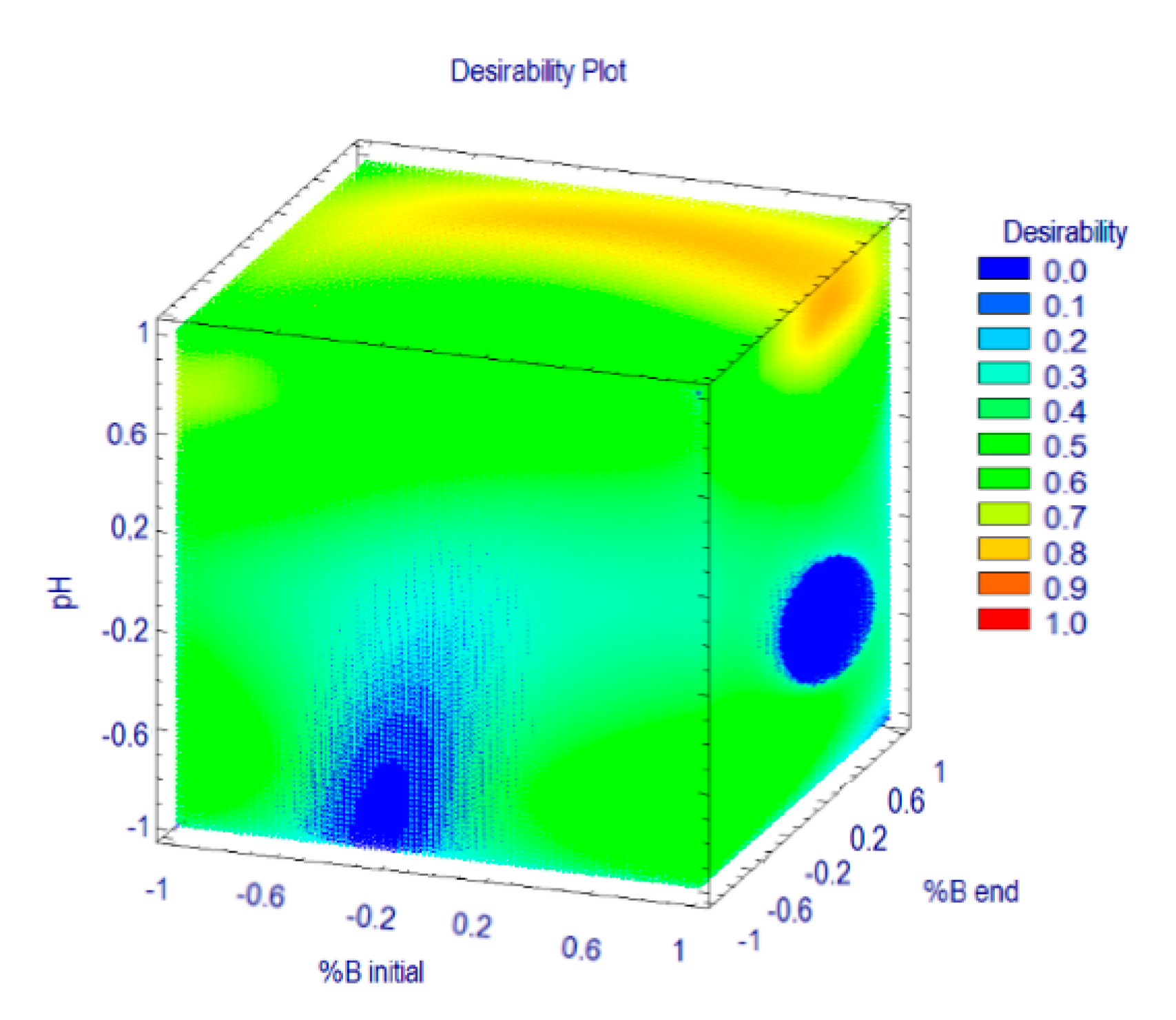
3.2. Optimization of the Separation Method
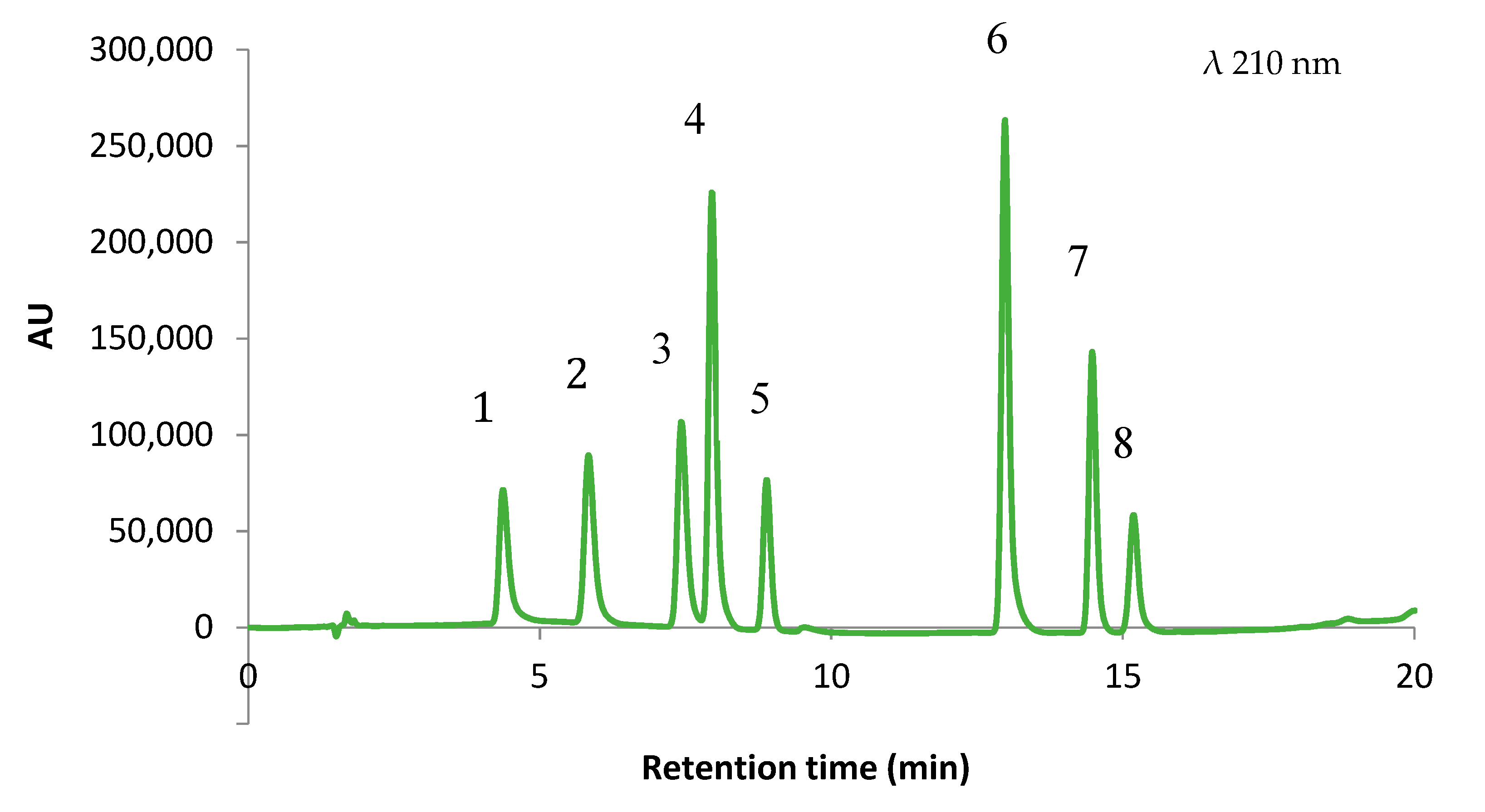
3.3. Validation of the Separation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zea, L.P.; Yusof, Y.A.; Aziz, M.G.; Ling, C.N.; Amin, N.A.M. Compressibility and dissolution characteristics of mixed fruit tablets made from guava and pitaya fruit powders. Powder Technol. 2013, 247, 112–119. [Google Scholar] [CrossRef]
- Fernandes, F.A.N.; Rodrigues, S.; Law, C.L.; Mujumdar, A.S. Drying of Exotic Tropical Fruits: A Comprehensive Review. Food Bioprocess Technol. 2011, 4, 163–185. [Google Scholar] [CrossRef]
- IFDA. Indonesian Food and Drug Authority Regulation No. 31/2018 Concerning Food Labelling; Indonesian Food and Drug Authority: Jakarta, Indonesia, 2018.
- IFDA. Indonesian Food and Drug Authority Regulation No. 37/2013 Concerning the Maximum Limits Allowed for Colorants; Indonesian Food and Drug Authority: Jakarta, Indonesia, 2013.
- IFDA. Indonesian Food and Drug Authority Regulation No. 4/2014 Concerning the Maximum Limits Allowed for Sweeteners; Indonesian Food and Drug Authority: Jakarta, Indonesia, 2014.
- IFDA. Indonesian Food and Drug Authority Regulation No. 36/2013 Concerning the Maximum Limits Allowed for Preservatives; Indonesian Food and Drug Authority: Jakarta, Indonesia, 2013.
- IFDA. Indonesian Food and Drug Authority Regulation Regulation No.22/2016 Concerning Maximum Limits Allowed for Flavouring Agents; Indonesian Food and Drug Authority: Jakarta, Indonesia, 2016.
- Fujiyoshi, T.; Ikami, T.; Kikukawa, K.; Kobayashi, M.; Takai, R.; Kozaki, D.; Yamamoto, A. Direct quantitation of the preservatives benzoic and sorbic acid in processed foods using derivative spectrophotometry combined with micro dialysis. Food Chem. 2018, 240, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, H.; Liu, Z.; Chen, Y.; Liu, Q.; Ding, Y.; Yu, R. Rapid and Sensitive Detection of Multi-Class Food Additives in Beverages for Quality Control by Using HPLC-DAD and Chemometrics Methods. Food Anal. Methods 2019, 12, 381–393. [Google Scholar] [CrossRef]
- Tighrine, A.; Amir, Y.; Alfaro, P.; Mamou, M.; Nerín, C. Simultaneous extraction and analysis of preservatives and artificial sweeteners in juices by salting out liquid-liquid extraction method prior to ultra-high performance liquid chromatography. Food Chem. 2019, 277, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Molognoni, L.; Valese, A.C.; Lorenzetti, A.; Daguer, H.; De Dea Lindner, J. Development of a LC–MS/MS method for the simultaneous determination of sorbic acid, natamycin and tylosin in Dulce de leche. Food Chem. 2016, 211, 748–756. [Google Scholar] [CrossRef]
- Purba, N.B.R.; Rohman, A.; Martono, S. The optimization of HPLC for quantitative analysis of acid orange 7 and Sudan II in cosmetic products using box behnken design. Int. J. Appl. Pharm. 2019, 11, 130–137. [Google Scholar] [CrossRef]
- Amini, T.; Hashemi, P. Preconcentration and GC–MS determination of caffeine in tea and coffee using homogeneous liquid–liquid microextraction based on solvents volume ratio alteration. J. Chromatogr. B 2018, 1092, 252–257. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Sato, K.; Abe, Y.; Sugimoto, N.; Akiyama, H. Quantification of acesulfame potassium in processed foods by quantitative 1H NMR. Talanta 2015, 131, 712–718. [Google Scholar] [CrossRef]
- Dos Santos, T.C.; Zocolo, G.J.; Morales, D.A.; de Aragão Umbuzeiro, G.; Zanoni, M.V.B. Assessment of the breakdown products of solar/UV induced photolytic degradation of food dye tartrazine. Food Chem. Toxicol. 2014, 68, 307–315. [Google Scholar] [CrossRef]
- Lindquist, E.; Yang, Y. Degradation of benzoic acid and its derivatives in subcritical water. J. Chromatogr. A 2011, 1218, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Guarino, C.; Fuselli, F.; Mantia, A.L.; Longo, L. Development of an RP-HPLC method for the simultaneous determination of benzoic acid, sorbic acid, natamycin and lysozyme in hard and pasta filata cheeses. Food Chem. 2011, 127, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, R.A.; Pena, M.T.; Fernández, P.; González, P.; Carro, A.M. Artificial sweeteners in beverages by ultra performance liquid chromatography with photodiode array and liquid chromatography tandem mass spectrometry. Food Control 2015, 47, 43–52. [Google Scholar] [CrossRef]
- Ding, M.; Liu, W.; Peng, J.; Liu, X.; Tang, Y. Simultaneous determination of seven preservatives in food by dispersive liquid-liquid microextraction coupled with gas chromatography-mass spectrometry. Food Chem. 2018, 269, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Viana, C.; Zemolin, G.M.; Dal Molin, T.R.; Gobo, L.; Ribeiro, S.M.; Leal, G.C.; Marcon, G.Z.; de Carvalho, L.M. Detection and determination of undeclared synthetic caffeine in weight loss formulations using HPLC-DAD and UHPLC-MS/MS. J. Pharm. Anal. 2018, 8, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Iverson, C.D.; Gu, X.; Lucy, C.A. The hydrophilicity vs. ion interaction selectivity plot revisited: The effect of mobile phase pH and buffer concentration on hydrophilic interaction liquid chromatography selectivity behavior. J. Chromatogr. A 2016, 1458, 82–89. [Google Scholar] [CrossRef]
- Lino, C.M.; Pena, A. Occurrence of caffeine, saccharin, benzoic acid and sorbic acid in soft drinks and nectars in Portugal and subsequent exposure assessment. Food Chem. 2010, 121, 503–508. [Google Scholar] [CrossRef]
- De Queiroz Pane, D.; Dias, C.B.; Meinhart, A.D.; Ballus, C.A.; Godoy, H.T. Evaluation of the sweetener content in diet/light/zero foods and drinks by HPLC-DAD. J. Food Sci. Technol. 2015, 52, 6900–6913. [Google Scholar] [CrossRef]
- Hadjmohammadi, M.; Sharifi, V. Simultaneous optimization of the resolution and analysis time of flavonoids in reverse phase liquid chromatography using Derringer’s desirability function. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2012, 880, 34–41. [Google Scholar] [CrossRef]
- Setyaningsih, W.; Saputro, I.E.; Carrera, C.A.; Palma, M.; García-Barroso, C. Fast Determination of Phenolic Compounds in Rice Grains by Ultraperformance Liquid Chromatography Coupled to Photodiode Array Detection: Method Development and Validation. J. Agric. Food Chem. 2019, 67, 3018–3027. [Google Scholar] [CrossRef]
- Islam, M.A.; Alam, M.R.; Hannan, M.O. Multiresponse optimization based on statistical response surface methodology and desirability function for the production of particleboard. Compos. Part B Eng. 2012, 43, 861–868. [Google Scholar] [CrossRef]
- Vera Candioti, L.; De Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental design and multiple response optimization. Using the desirability function in analytical methods development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Xie, N.; Zhao, S.; Wu, Y.; Li, J.; Wang, Z. Simultaneous determination of ten preservatives in ten kinds of foods by micellar electrokinetic chromatography. Food Chem. 2015, 181, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Belguidoum, K.; Amira-Guebailia, H.; Boulmokh, Y.; Houache, O. HPLC coupled to UV–vis detection for quantitative determination of phenolic compounds and caffeine in different brands of coffee in the Algerian market. J. Taiwan Inst. Chem. Eng. 2014, 45, 1314–1320. [Google Scholar] [CrossRef]
- Gan, Z.; Sun, H.; Wang, R.; Feng, B. A novel solid-phase extraction for the concentration of sweeteners in water and analysis by ion-pair liquid chromatography–triple quadrupole mass spectrometry. J. Chromatogr. A 2013, 1274, 87–96. [Google Scholar] [CrossRef]
- Baş, D.; Boyacı, İ.H. Modeling and optimization I: Usability of response surface methodology. J. Food Eng. 2007, 78, 836–845. [Google Scholar] [CrossRef]
- Snyder, L.R.; Kirkland, J.J.; Dolan, J.W. Introduction to Modern Liquid Chromatography, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; ISBN 9780470508183. [Google Scholar]
- ISO. ISO/IEC 17025:2005 General Requirements for the Competence of Testing and Calibration Laboratories; ISO: Geneva, Switzerland, 2005; Volume 2005. [Google Scholar]
- ICH. Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: Geneva, Switzerland, 2005; Volume 2005. [Google Scholar]
- AOAC. Appendix F: Guidelines for Standard Method Performance Requirements; AOAC International: Rockville, MA, USA, 2016. [Google Scholar]
- Minten, J.; Adolfsson-Erici, M.; Björlenius, B.; Alsberg, T. A method for the analysis of sucralose with electrospray LC/MS in recipient waters and in sewage effluent subjected to tertiary treatment technologies. Int. J. Environ. Anal. Chem. 2011, 91, 357–366. [Google Scholar] [CrossRef]
- Ding, M.; Peng, J.; Ma, S.; Zhang, Y. An environment-friendly procedure for the high performance liquid chromatography determination of benzoic acid and sorbic acid in soy sauce. Food Chem. 2015, 183, 26–29. [Google Scholar] [CrossRef]
- Setyaningsih, W.; Saputro, I.E.; Carrera, C.A.; Palma, M.; Barroso, C.G. Multiresponse optimization of a UPLC method for the simultaneous determination of tryptophan and 15 tryptophan-derived compounds using a Box-Behnken design with a desirability function. Food Chem. 2017, 225, 1–9. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | −1 | 0 | +1 |
|---|---|---|---|
| x1, %B initial | 0 | 5 | 10 |
| x2, %B end | 60 | 80 | 100 |
| x3, pH | 3 | 5 | 7 |
| BBD Point | Resolution (Rs) | Analysis Time (min) | ||||||
|---|---|---|---|---|---|---|---|---|
| Rs1–2 | Rs2–3 | Rs3–4 | Rs4–5 | Rs5–6 | Rs6–7 | Rs7–8 | ||
| 1 | FOP | FOP | 1.2280 | 0.9515 | 0.6610 | 1.6360 | 0.7855 | 19.09 |
| 2 | 0.2555 | 1.1254 | 1.5789 | 1.5465 | 3.0378 | 2.7325 | 0.3874 | 21.18 |
| 3 | FOP | FOP | 1.5295 | 1.0145 | 0.5500 | 2.5180 | 1.0330 | 20.27 |
| 4 | 3.1026 | 1.2854 | 0.8438 | 2.6014 | FOP | 1.2804 | 0.8706 | 17.84 |
| 5 | 3.0046 | 1.2852 | 0.8660 | 2.6402 | FOP | 1.2772 | 0.8807 | 17.84 |
| 6 | FOP | 1.1549 | 1.6432 | 1.1495 | 3.1962 | 1.5370 | 0.9020 | 19.56 |
| 7 | 3.0033 | 1.9175 | 1.1825 | 3.2517 | 0.1641 | 1.1538 | FOP | 18.60 |
| 8 | 3.4380 | 1.2154 | 0.8799 | 2.4530 | FOP | 1.2758 | 0.9002 | 19.20 |
| 9 | 2.4517 | 0.9708 | 2.6548 | 1.7886 | 1.0280 | FOP | 0.9276 | 14.70 |
| 10 | FOP | FOP | 1.2460 | 1.4690 | 0.5300 | 2.3685 | 1.0175 | 22.76 |
| 11 | 3.2329 | 0.9794 | 2.1677 | 1.8457 | 1.2248 | 0.6605 | 0.9197 | 17.15 |
| 12 | 3.1698 | 1.3190 | 0.8337 | 2.5400 | FOP | 1.2681 | 0.8802 | 17.82 |
| 13 | 1.0052 | 1.4346 | 1.1752 | 1.3097 | 4.0203 | 2.2164 | 0.8106 | 16.67 |
| 14 | 0.6670 | 1.8968 | 1.7036 | 1.2585 | 3.4432 | 1.0378 | 1.1120 | 16.25 |
| 15 | FOP | FOP | 1.0430 | 1.2720 | 3.1895 | 3.1895 | 1.3860 | 25.40 |
| Model Term | Regression Coefficients | |||||||
|---|---|---|---|---|---|---|---|---|
| Rs1–2 | Rs2–3 | Rs3–4 | Rs4–5 | Rs5–6 | Rs6–7 | Rs7–8 | Analysis Time | |
| constant | 3.092 | 1.297 | 0.848 | 2.594 | 0.000 | 1.275 | 0.877 | 23.085 |
| x1, %B initial | −0.026 | 0.122 | 0.076 | 0.056 | 0.098 | 0.006 | −0.121 | −0.620 |
| x2,%B end | −0.043 | −0.051 | 0.384 | −0.335 | −0.008 | −0.627 | 0.134 | −3.039 |
| x3, pH | 0.241 | 0.701 | 0.132 | 0.070 | 1.096 | −0.274 | −0.126 | −0.617 |
| x1x1 | −0.020 | −0.067 | 0.442 | −0.140 | 0.044 | −0.246 | −0.084 | −0.672 |
| x1x2 | −0.087 | −0.178 | 0.046 | −0.214 | −0.097 | −0.135 | 0.227 | −0.774 |
| x1x3 | 0.251 | 0.070 | −0.188 | 0.154 | 0.201 | 0.132 | −0.027 | −0.025 |
| x2x2 | −0.041 | 0.041 | 0.432 | −0.119 | 0.553 | −0.257 | −0.107 | 0.208 |
| x2x3 | 0.103 | 0.193 | −0.015 | 0.008 | 0.733 | −0.035 | 0.331 | 0.085 |
| x3x3 | −2.821 | −0.582 | 0.109 | −1.218 | 2.030 | 1.131 | 0.147 | 0.366 |
| Model Validation | ||||||||
| R2 | 0.9743 | 0.9153 | 0.7414 | 0.9370 | 0.8794 | 0.9401 | 0.8679 | 0.9375 |
| R2(adjustedford.f) | 0.9281 | 0.7629 | 0.2758 | 0.8236 | 0.6624 | 0.8324 | 0.6301 | 0.8260 |
| SE | 0.4005 | 0.3234 | 0.4443 | 0.3008 | 0.8749 | 0.3461 | 0.1908 | 1.0064 |
| MAE | 0.1987 | 0.1477 | 0.1983 | 0.1392 | 0.3958 | 0.1577 | 0.0830 | 0.5456 |
| Analytes | SST (%CV) | Linear Equation | R2 | Limits (mg kg−1) | Recovery (%) | Intraday, %CV (n = 9) | Interday, %CV (n = 3 × 3) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t | Area | LOD | LOQ | 80% | 100% | 120% | t | Area | t | Area | |||
| ACE | 0.56 | 0.60 | y = 66,040.5x − 19359.2 | 0.9992 | 3.00 | 10.02 | 99.30 | 95.37 | 100.29 | 0.46 | 1.34 | 0.33 | 2.67 |
| BEN | 0.33 | 1.37 | y = 78,686.4x − 21997.5 | 0.9994 | 2.16 | 7.21 | 101.41 | 97.87 | 96.28 | 0.40 | 2.77 | 0.36 | 3.03 |
| SOR | 0.24 | 0.75 | y = 84,155.0x + 15085.0 | 0.9994 | 2.70 | 9.00 | 100.19 | 97.86 | 99.15 | 0.27 | 2.10 | 0.24 | 1.75 |
| SAC | 0.27 | 1.08 | y = 169,869.9x − 28700.4 | 0.9998 | 1.16 | 3.86 | 96.90 | 98.75 | 97.23 | 0.30 | 3.02 | 0.29 | 4.06 |
| TAR | 0.33 | 1.08 | y = 71,332.1x + 5665.2 | 0.9995 | 2.06 | 6.86 | 100.54 | 101.19 | 99.43 | 0.30 | 2.26 | 0.42 | 2.31 |
| CAF | 0.17 | 0.98 | y = 122,490.4x + 27542.7 | 0.9997 | 1.93 | 6.43 | 99.32 | 96.11 | 97.11 | 0.14 | 1.29 | 0.10 | 2.58 |
| SUN | 0.24 | 0.24 | y = 63,931.9x + 15471.2 | 0.9997 | 1.75 | 5.85 | 100.23 | 100.76 | 95.30 | 0.20 | 1.44 | 0.19 | 2.48 |
| ASP | 0.17 | 1.70 | y = 55,293.4x − 31270.7 | 0.9991 | 2.98 | 9.95 | 98.68 | 98.31 | 101.74 | 0.14 | 2.94 | 0.08 | 1.85 |
| (a) | ||||||||
| Sample | ACE | BEN | SOR | SAC | TAR | CAF | SUN | ASP |
| 01 | √ | - | - | - | √ | - | √ | √ |
| 02 | √ | - | - | - | √ | - | √ | √ |
| 03 | √ | - | - | - | √ | - | - | √ |
| 04 | - | √ | √ | √ | - | - | - | √ |
| 05 | √ | - | - | - | √ | - | - | √ |
| 06 | √ | - | - | - | √ | - | - | √ |
| 07 | - | - | - | - | - | - | - | √ |
| 08 | √ | - | - | - | - | - | - | √ |
| 09 | √ | - | - | - | √ | - | - | √ |
| (b) | ||||||||
| Sample | Concentration (mg kg−1) | |||||||
| ACE | BEN | SOR | SAC | TAR | CAF | SUN | ASP | |
| 01 | 181.97 ± 0.45 | ND | ND | ND | 61.72 ± 0.14 | ND | 5.33 ± 0.44 | 446.96 ± 0.44 |
| 02 | 140.86 ± 0.56 | ND | ND | ND | 18.80 ± 0.13 | ND | 8.11 ± 0.01 | 495.24 ± 0.38 |
| 03 | 185.71 ± 0.72 | ND | ND | ND | 12.77 ± 0.06 | ND | ND | 156.75 ± 0.14 |
| 04 | ND | 77.87 ± 0.53 | 42.50 ± 0.13 | 117.97 ± 0.45 | ND | ND | ND | 476.01 ± 0.85 |
| 05 | 204.68 ± 0.87 | ND | ND | ND | 15.66 ± 0.15 | ND | ND | 163.25 ± 0.28 |
| 06 | 491.17 ± 0.72 | ND | ND | ND | 12.04 ± 0.32 | 31.45 ± 0.20 | ND | 188.11 ± 0.96 |
| 07 | ND | ND | ND | ND | ND | 94.62 ± 0.46 | ND | 274.82 ± 0.17 |
| 08 | 70.68 ± 0.50 | ND | ND | ND | ND | 66.17 ± 0.64 | ND | 437.57 ± 0.27 |
| 09 | 173.10 ± 0.73 | ND | ND | ND | 10.51 ± 0.06 | 22.06 ± 0.38 | ND | 258.76 ± 0.62 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imanulkhan; Setyaningsih, W.; Rohman, A.; Palma, M. Development and Validation of HPLC-DAD Method for Simultaneous Determination of Seven Food Additives and Caffeine in Powdered Drinks. Foods 2020, 9, 1119. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9081119
Imanulkhan, Setyaningsih W, Rohman A, Palma M. Development and Validation of HPLC-DAD Method for Simultaneous Determination of Seven Food Additives and Caffeine in Powdered Drinks. Foods. 2020; 9(8):1119. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9081119
Chicago/Turabian StyleImanulkhan, Widiastuti Setyaningsih, Abdul Rohman, and Miguel Palma. 2020. "Development and Validation of HPLC-DAD Method for Simultaneous Determination of Seven Food Additives and Caffeine in Powdered Drinks" Foods 9, no. 8: 1119. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9081119