
Impact of Particles on Pulmonary Endothelial Cells
by
 , , and
, , and
Marina Almeida-Silva
1 ,
,
Jéssica Cardoso
1,
Catarina Alemão
1,
Sara Santos
1,
Ana Monteiro
1,2 ,
,
Vítor Manteigas
1,2 and
and
Ana Marques-Ramos
1,* 1
HTRC-Health & Technology Research Center, ESTeSL—Escola Superior de Tecnologia da Saúde, Instituto Politécnico de Lisboa, 1990-096 Lisbon, Portugal
2
Centro de Ciências e Tecnologias Nucleares (C2TN), Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10, ao Km 139.7, 2695-066 Bobadela-Loures, Portugal
*
Author to whom correspondence should be addressed.
Toxics 2022, 10(6), 312; https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060312
Submission received: 22 March 2022
/
Revised: 3 June 2022
/
Accepted: 7 June 2022
/
Published: 9 June 2022
(This article belongs to the Special Issue Modelling & Impacts Assessments of Air Quality)
Abstract
:According to the WHO, air quality affects around 40 million people, contributing to around 21,000 premature deaths per year. Severe respiratory diseases, such as asthma and chronic obstructive pulmonary disorder, can be promoted by air pollution, which has already been documented; this is one of the reasons why air quality is a very relevant factor for human health and well-being. Aerosols are an aggregation of solid or liquid particles dispersed in the air and can be found in the form of dust or fumes. Aerosols can be easily inhaled or absorbed by the skin, which can lead to adverse health effects according to their sizes that range from the nanometre to the millimetre scale. Based on the PRISMA methodology and using the Rayyan QCRI platform, it was possible to assess more than four hundred research articles. This systematic review study aimed to understand the impact of particles on pulmonary endothelial cells, namely particulate matter in different sizes, cigarette smoke, diesel exhaust particles and carbon black. The main conclusions were that particles induce multiple health effects on endothelial cells, namely endothelial dysfunction, which can lead to apoptosis and necrosis, and it may also cause necroptosis in lung structure.
1. Introduction
Air quality has a significant influence in human health and well-being [1], and it has been recognised that air pollution contributes to severe respiratory diseases such as asthma and chronic obstructive pulmonary disorder (COPD) [2]. An aerosol is an aggregation of solid or liquid particles dispersed in the air, which can be found in the form of dust or fumes. Due to particle sizes ranging from the nanometre to the millimetre scale, these particles can be easily inhaled or absorbed by the skin, reaching the cells of the pulmonary alveoli and finally entering the bloodstream, which can lead to health adverse effects [3,4]. These include proinflammatory responses in vivo, such as the increased production of reactive oxygen species (ROS), the altered transcription of genes related to inflammation, the polarisation of macrophages and the overproduction of proinflammatory molecules [5]. Ultimately, be exposed to aerosol induces cardiovascular and respiratory diseases that, in some cases, might be fatal [1].
Air pollutants such as particulate matter (PM), polycyclic aromatic hydrocarbons (PAHs), carbon monoxide (CO), gaseous mixtures of nitrogen dioxide and volatile organic compounds can lead to severe lung damage and respiratory diseases such as asthma, bronchitis and chronic obstructive pulmonary disorder [2]. It is now known that the guidelines for air quality are not met in the 115 largest cities in the European Union (EU), which affects around 40 million people, ultimately leading to 21,000 premature deaths per year [6]. In Southeast Asia, it was estimated that in 2016, air pollution resulted in about 2.4 million premature deaths [6]. In other regions, in 2012, the number of deaths was also high: 194,000 deaths in the Eastern Mediterranean, 1.1 million in the Western Pacific and 211,000 deaths in Sub-Saharan Africa [7]. Accordingly, the study of the molecular mechanisms behind the effects of air pollutants on human health is mandatory. Among the most common and studied air pollutants are PM, cigarette smoke (CS), diesel exhaust particles (DEPs) and carbon black (CB).
Exposure to PM, specifically the particles derived from combustion, has increased significantly in Western countries in the last 250 years due to globalisation developments and industrialisation, such as increased traffic and shipping activities. This has led to the presence of substantial quantities of coarse (aerodynamic diameter < 10 μm—PM10), fine (aerodynamic diameter < 2.5 μm—PM2.5) and ultrafine (aerodynamic diameter < 0.1 μm—PM0.1) particles in the near-surface atmosphere [8]. Epidemiological and experimental studies have proven that exposure to atmospheric PM, especially fine particulate matter with sizes of 2.5 µm (PM2.5) or below, contributes to 3.3 million premature deaths per year worldwide [9], which are caused by respiratory and cardiovascular diseases such as ischemic heart disease, COPD, acute lower respiratory illness and lung cancer [5]. Studies have also shown that PM accelerates inflammation-mediated thrombosis [9]. It has been estimated that exposure to high concentrations of PM2.5 affects approximately 92% of the world’s population in areas with adverse air quality [1].
Nearly 1.25 billion people smoke cigarettes daily worldwide [10]. CS is a highly complex mixture that contains more than 4000 compounds, such as nicotine, a component of the tar phase that is the addictive substance of cigarette smoke [11]. Environmental tobacco smoke is generated from the combination of sidestream smoke (85%), smoke emanated from the burning part of a cigarette which contains a higher concentration of toxic gaseous constituents, and a small fraction of exhaled mainstream smoke (15%), which is smoke that is drawn through the tobacco into an active smoker’s mouth, which contains 8% tar and 92% gaseous components [11]. CS is linked to the most common causes of death in the elderly and contributes to the morbidity and disability rates associated with many chronic illnesses that are common in this age group [12].
Ambient air pollution by DEPs is a major cause of cardiovascular and metabolic mortality worldwide [13]. Diesel is a complex mixture of solid and liquid material with respirable size that can penetrate deep into human lungs [14]. DEPs combine approximately 20% ambient PM and belong to the fine (PM2.5) and ultrafine particle fractions (PM0.1); exposure to DEPs can impair endothelial and fibrinolytic functions, promote blood thrombogenicity and increase the gravity of cardiac ischemia in humans [13].
CB is a fine material that is used as a black pigment and is produced by the thermal decomposition of liquid or gaseous hydrocarbons under controlled conditions, which mainly consist of elemental carbon, with the rest being hydrogen, sulphur, oxygen, organic carbon, extractable organic materials and ash [15]. CB has high chemical stability and electrical conductivity and is a conductive additive [16]. Exposure to CB occurs in workers in industries that produce rubber and fortify vehicle tires and other rubber products, and it is used in coatings, plastics, inks or paints, ceramics, paper, battery production, carbon electrode production and in metallurgical processes such as carburisation [15]. In terms of the effects of CB on cells, it has been demonstrated that it strongly induces the production of reactive oxygen species (ROS) [17]. Additionally, studies on mice regarding gestation with CB through intratracheal instillation showed a change in testicular structure and reduced daily sperm production [18].
It has long been suggested in studies, in vitro and in vivo, that PM, CS, DEPs and CB can affect the endothelium and its cells, inducing lung injury and inflammation via endothelial dysfunction [13,19,20,21].
The endothelium is a layer that covers the interior of the walls of arteries, capillaries, veins and the lymphatic system [19]. This monolayer plays an active role by regulating the environment, responding to external stresses and also presenting itself as an endocrine and metabolic tissue [22]. Endothelial cells (ECs) guarantee permeability for the passage of blood, hormone fluids, macromolecules and platelets through arteries, veins, arterioles and venules and controls blood flow and vascular relaxation and constriction [23,24,25]. ECs secrete mediators and signalling molecules that regulate blood clotting to prevent platelet adhesion and aggregation in conditions of homeostasis [19]. The structure, characterisation and function of ECs vary according to their location [19]. Their structural heterogeneity includes differences in size, thickness, the position of the nuclei [19], changes in the genetic expression and in the production of the extracellular matrix and in the properties of the cell surface [25]. The function of pulmonary endothelial cells (PECs) is not very different from vascular endothelial cells; they regulate blood flow but also control the passage of liquid and macromolecules between the interstitial space, the vessels and the smooth muscle cells [26,27,28]. The role of PECs in haemostasis regulation can have opposite effects, as it displays anticoagulant, antiplatelet and fibrinolytic properties, whereas it promotes contrasting responses in the case of injury [29]. It also regulates the synthesis and metabolism of vasoactive compounds, which are regulators of pulmonary lung tone, such as endothelin-1 (ET-1) and nitrogen monoxide (NO) [30].
Based on the evidence that air particles are easily inhaled and deposited on the human respiratory tract, and knowing that their effects are poorly understood, we investigated the impact of the following pollutants on PECs: PM, CS, DEPs and CB.
2. Materials and Methods
This study includes data published between 1 January 2000 and 6 March 2021 regarding studies about the effects of aerosols on endothelial cells, following the PRISMA methodology. The search terms used for each platform are described in Table 1.
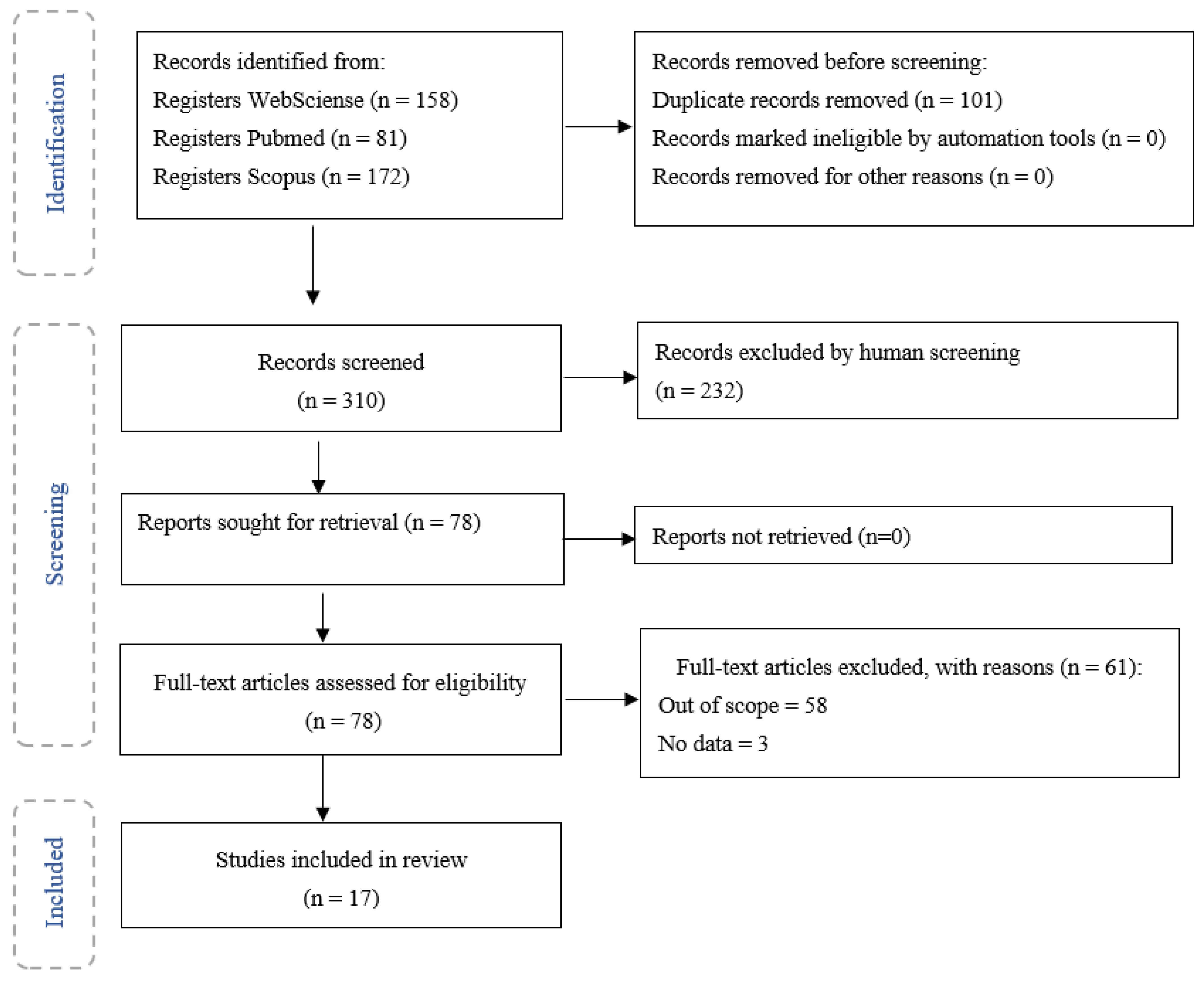
This search led to four hundred and eleven research articles in all databases, which were uploaded to an online and free platform named Rayyan QCRI. Through the application of the inclusion and exclusion criteria (Table 2), it was possible to select seventy-eight articles, of which seventeen were selected as eligible.
The diagram in Figure 1 describes the different phases of the selection of papers and the papers that were obtained in the final phase.
3. Results
From the seventy-eight articles obtained, seventeen were selected and analysed, being characterised according to the type of pollutant and its effects/impact on endothelial cells. The following air pollutants were found: CS, PM, including PM1, PM2.5, PM10, and fine and ultrafine nanoparticles, DEPs and CB (see Table 3). A total of nine articles assessed the effects of such pollutants on endothelial cells, through in vitro studies, using the following cells: lung endothelial cells in five articles, human pulmonary microvascular endothelial cells in three articles, and one article used rat microvascular cell monolayers. Three articles used in vivo methods, two with mice and one with humans. Finally, six articles that used both methods were analysed, with mice subjects used in (1) in vivo studies and (2) lung endothelial cells, lung microvascular endothelial cells, human pulmonary microvascular endothelial cells (HPMVECs), human pulmonary endothelial cells and pulmonary vascular endothelial cells in in vitro studies.
4. Discussion
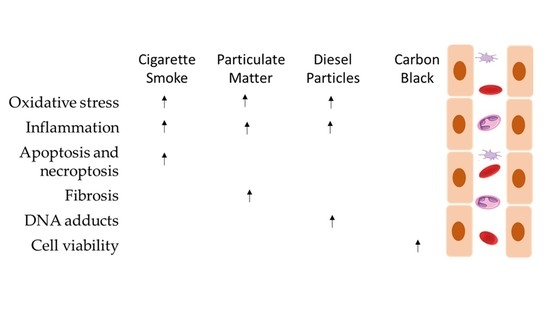
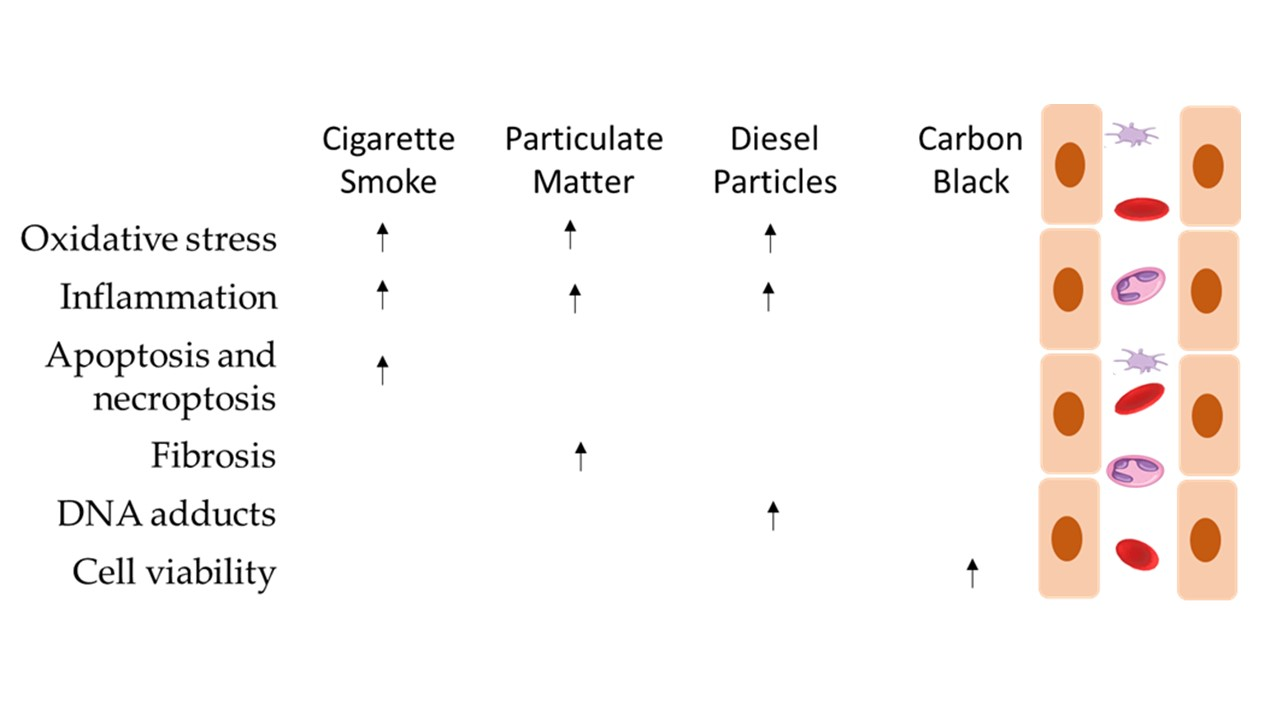
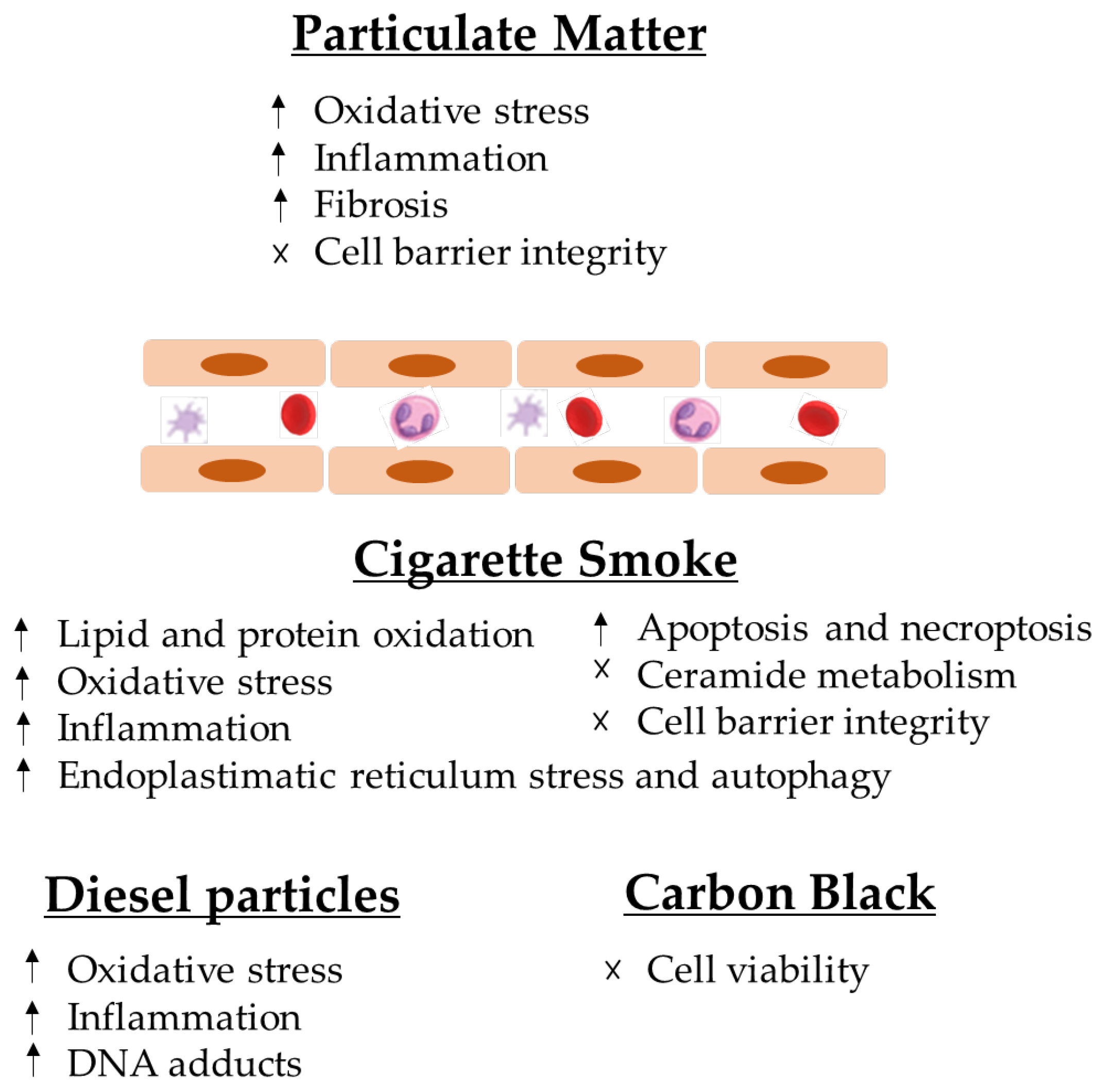
The analysed articles led the authors to conclude that particles induce multiple health effects on endothelial cells, namely endothelial dysfunction, which can cause apoptosis and necrosis, and it may also cause necroptosis in the lung structure (Figure 2).
4.1. Particulate Matter
Particulate matter (PM) can cause respiratory and cardiovascular diseases such as chronic obstructive pulmonary disease, ischemic heart disease, COPD, acute lower respiratory illness and lung cancer [5]. It affects around 40 million people, ultimately leading to 21,000 premature deaths per year [6]. Exposure to PM comes from combustion and can be presented in different sizes.
The results show that PM induces the disruption of the lung endothelial barrier through the induction of ROS and concomitant oxidative stress [31,37], with the activation of the p38 MAPK and HSP27 signalling pathways [37]. In particular, it was observed that the PM-induced phosphorylation of HSP27, an important protein in regulating actin polymerisation [47], leads to a contractile cytoskeletal arrangement [37] and that actin filament reorganisation was dependent on ROS production and p38 MAPK activation [37]. Accordingly, in order to protect endothelial integrity against PM exposure, it is necessary to restrain the activation of the p38–HSP27 pathway through the inhibition of oxidative stress [37]. Similarly, Karki, P. et al. [32] revealed that the effects of PM on EC barrier dysfunction were dependent on the production of ROS, which induced the production of truncated oxidised phospholipids (Tr-OxPLs). These events resulted in the destabilisation of cell junctions, as Tr-OxPLs induce the phosphorylation of the adherent junction VE-cadherin, which results in its internalisation with concomitant rescue from the plasma membrane [32]. Accordingly, the uncontrolled production of ROS seems to play a critical role in the PM-mediated effects on ECs. This event is caused by different mechanisms such as the activation of pro-oxidant enzymes and mitochondrial damages, and it constitutes a recognised mechanism of inflammation and cellular damage [32].
Endothelial cells are the main tight-junction-associated cellular barriers for pulmonary gas exchange, and normally, they are impermeable to albumin and leukocytes. The place of adhesion between leukocytes and endothelial cells is determined by the presence of molecules such as VCAM-1, a very important marker for leukocytes, including PMNs [34]. The data collected showed that PMNs treated with PM2.5 are able to bind sVCAM-1 and that PM2.5 exposure induces PMN infiltration with increased VCAM-1 [34]. Furthermore, it was demonstrated that short-term exposure to PM2.5 increases neutrophil count in the peripheral blood and the neutrophil to WBC ratio, which indicated that PMNs participate in PM2.5-induced inflammatory-lung injury. This hypothesis was confirmed when PM2.5-induced lung injury was reversed by the depletion of circulatory PMNs before PM2.5 exposure [34]. Furthermore, as PM2.5 deposits in the alveolar space and ECs, it causes lung oedema, morphological changes and cell barrier disruption [34]. When it comes to ultrafine PM, mainly PM1, the decrease in cells’ viability depends on the concentration and exposure time, and it has been demonstrated that PM1 exposure induces the release of the proinflammatory cytokines IL-6 and TNF-α by surrounding epithelial cells and macrophages [36]. This resulted in the upregulation of ICAM-1 on endothelial cells through the activation of TNF-α/NF-κB signalling pathways, which promotes the adhesion of monocytes to ECs, a key event in the initiation of atherosclerosis [36]. Similarly, Mo et al. [38] demonstrated that UFP exposure (10 and 20 μg/mL) resulted in IL-6 induction through the activation of p38 and ERK1/2 MAPKs. In this study, the authors also demonstrated that these effects are caused by the induction of ROS, which occurs in a NAPDPH-oxidase-dependent manner [38].
By using tri-co-cultured systems consisting of alveolar epithelial cells, ECs and macrophages, Wang et al. [33] demonstrated that PM2.5 exposure induces a potent inflammatory response. In this study, EA.hy926 endothelial cells were not exposed to particulate matter directly, but different doses were chosen to study the mechanistic effects on both the cardiovascular and pulmonary system. Based on this methodology, it was possible to determine that the basolateral medium LDH that represents the cytotoxicity of EA.hy926, after PM2.5 exposure, did not induce significant EA.hy926 cytotoxicity, but the LDH levels were elevated meaningfully in high doses of PM2.5-treated apical co-cultures [33]. No reason was found for the different levels of LDH [48], but is known that its release into the extracellular space is a result of compromised or damaged cell membrane [48]. However, Bengalli et al. [39] found that exposure to PM10 increased IL-1β, a cytokine responsible for inflammatory responses on the cell [49], in ECs on the basolateral compartment; yet, no release in the apical compartment was observed, and it was not possible to find an explanation for that. The ABB (air–blood barrier) treated with CuO did not show a response in the basolateral compartment; this could be due to the integrity of the ABB which can inhibit the passage of CuO in the direction of the endothelial cells, while it enables to rise IL-6 and IL-8 release from the apical compartment [39]. Bengalli et al. [39] suggested that an explanation for CuO being free in the cytoplasm might be its higher oxidative potential compared with TiO2, and this can cause an oxidative degradation of lipids in vesicles, leaving CuO free. Exposure to PM10 and nanoparticles was shown to induce modulation in proinflammatory proteins and ECs [39].
In another study by Wang. et al. [37], an increase in Akt phosphorylation in endothelial cells due to the presence of NaSH was also reported, being dependent on the dose and the time of the exposure. This phosphorylation could be inhibited through the inhibition of PI3K in the presence of its inhibitors, LY 294002, leading Wang, T. et al. [37] to suggest that H2S stimulates Akt activation and that NaSH prevents not only vascular hyperpermeability through the system that combines ROS cleaning and Akt activation, but also endothelial integrity, thus suggesting that H2S may attenuate pulmonary inflammation caused by PM, although the associated mechanisms are still unclear [37].
Wang et al. [33] also showed that some inhaled PM2.5 can cross the epithelial barrier to the endothelial cells, causing damage in the cells, which leads to morbidity and mortality caused by cardiovascular diseases.
Ma et al. [35] reported that transforming growth factor-β (TGF-β) is decisive in the processes of development, injury and restoration in the lung tissue of mice. In this article, it was shown that exposure to PM2.5 leads to an activation of the TGF-β1 receptor that plays a fundamental role in the pathological process of pulmonary fibrosis, promoting ENDMT in the MHC cells, increasing the levels of Col1 and Acta2 and reducing the expression of CD31 and CDH5 after 48 h. After quantitative reverse transcription PCR (RT-qPCR) analysis, the lncRNA Gm16410 gained attention, because it is known that lncRNA Gm16410 is a pseudo-repressor gene of STAT interaction and it has been described that STAT3 and TGF-β1 form a closed loop, which can raise fibrosis in lung cancer cells [38].
4.1.1. Cigarette Smoke
It is established that CS interferes with the capacity of airway epithelial cells to uphold repair processes and also can lead to modifications in the airway structures and functions that characterise COPD [10]. It can cause detrimental cell responses, inducing tissue damage around the alveoli and airways, with the subjacent development of various lung diseases [50], such as lung cancer and asthma (9). From a mechanistic point of view, it has been demonstrated that CS increases the risk of lipid and protein oxidation and deregulates the normal ceramide metabolism, endoplasmic reticulum (ER) stress and cell death [50] (Figure 2). Furthermore, it has been reported that it can induce xenobiotic and redox-regulating genes and numerous oncogenes, whereas it blocks the expression genes that regulate airway inflammation as well as diverse tumour-suppressor genes [10] (Figure 2).
One of the molecular effects of e-smoking is the reduction in NO levels [40], and it is known that NO loss is a key event for endothelial dysfunction [51,52]. In this regard, it has been demonstrated that HPMVEC subjected to serum from regular e-cigarette smokers produce ROS and increase ICAM-1 expression, demonstrating endothelial activation and inflammation, which together represent the first step in signalling events that are conducive to the pathogenesis of numerous vascular disorders [40]. It seems that the activation of NADPH oxidase 2 is the origin of ROS production, as a decrease in ROS production following the pre-treatment of HPMVECs with the NADPH oxidase 2 inhibitor—apocynin—was observed [40]. Furthermore, it is known that NO reacts with ROS to create peroxynitrite, which triggers a cellular response that can lead to apoptosis and necrosis [48,49]. Indeed, it is known that CS induces the death of epithelial and endothelial lung cells through apoptosis [53,54]. Actually, both cultured and mouse-lung ECs exposed to CS exhibit pronounced apoptosis, which is accompanied by emphysema [42]. The mechanisms that seem to induce these effects are the activation of endoplasmic reticulum (ER) stress and autophagy with increased eIF2α signalling. Additionally, in those settings, FAK signalling, a prosurvival pathway, is inhibited through oxidative stress [42]. It is noteworthy to emphasise that the author points out that the induction of the unfolded protein response (UPR) (a response to ER stress) is not an injury but rather a compensatory mechanism to protect from lung EC injury through the induction of the apoptotic death of dysfunctional cells [42]. This suggestion was based on the observation that the evolution of lung EC apoptosis and emphysema was associated with the impairment of eIF2α signalling, which occurs when UPR is diminished [42].
The mechanism by which CS induces cell death seems not to be restricted to apoptosis, as Mizumura, K. et al. [41,53] revealed that chronic CS also induces the mitophagy of lung structural cells that ultimately leads to necroptosis, a caspase-independent form of regulated cell death [41,53]. This event occurs through the induction of acid sphingomyelinase and the accumulation of the ceramide C16-Cer in a PINK1-dependent manner [41]. It is known that both apoptosis and necroptosis might contribute to the pathogenesis of COPD, particularly to its emphysema phenotype.
Epithelial and ECs form an important barrier in the alveolus, forming a semi-permeable interface for gas exchange that prevents alveolar and interstitial oedema formation [55,56]. A study by Schweitzer, K.S. et al. [43] demonstrated that CS exposure induces the disruption of the endothelial cell barrier. From a mechanistic point of view, CS induces the upregulation of neutral sphingomyelinase-mediated ceramide and p38 MAPK and JNK activation in addition to oxidative stress and Rho activation. Neutral sphingomyelinase activation is responsible for the observed morphological changes upon CS exposure, as it induces actin cytoskeletal rearrangement, junctional protein zonula occludens-1 loss and intercellular gap formation [43]. Schweitzer et al. [43] referred to the importance of the development of CS-induced emphysema because it is believed that oxidative stress and ceramides, which also have a role in regulating apoptosis [57], are involved in the emphysema pathogenesis. This mechanism may make endothelial cells more sensible to effects of subsequent injuries associated with ARDS which can evolve to chronic inflammatory changes associated with COPD [43].
4.1.2. Diesel Particles
As cited before, DEPs are a mixture of solid and liquid material that easily penetrate human lungs [14], being able to damage endothelial and fibrinolytic functions, stimulate blood thrombogenicity and intensify the gravity of cardiac ischemia in humans [58]. Regarding diesel particles, which are an ensemble of compounds, extremely dependent on fuel and engine, and have physical–chemical characteristics, studies about their effects often use standard reference materials (SRMs), and according to the results, DEPs are constituted of ultrafine particles and a tendency toward aggregation [44]. Regarding the DEPs tested by Bengalli et al. (2017) [45], SRMs were developed under controlled conditions (SRM, 1650b), being related to the emission of a heavy engine and a light engine (SRM, 2975). The chemical speciation of the EuroIV (light duty engine—types of vehicles that are under the emission limit, defined in Directive 98/69/EC) revealed a common PAH composition with elevated levels of phenanthrene, pyrene, dibenzo(a,h) anthracene and benzo[a]anthracene [44]. A series of events that linked UFP exposure to inflammation and oxidative stress with the consequential indirect activation of the vascular endothelium [44] and the release of VEGF from BEAS-2B [44] has been demonstrated. The risk of exposure on endothelial cells by PAHs is related to the fact that due to its activation, it can affect DNA, forming DNA adducts, leading to deletions, translocations, fusions or aneuploidy [59] through the formation of phenols, catechols, diol-epoxides, quinones, o-quinones and radical cations, which occurs during that formation [60,61,62].
The availability of IL-6R in BEAS-2B supernatants was assessed, revealing its overexpression after diesel exposure [45]. With the exposure of EURO IV, an increased expression of ICAM-1 and VCAM-1 was also visible, and also, the release of proinflammatory mediators (IL-6), which was also reported by Bengalli et al. [45]. The diesel-induced overexpression of ICAM-1 and VCAM-1 was successfully inhibited by interfering with IL-6/gp130 binding by the addition of an IL-6 antibody to epithelial cell media to reveal the important role of IL-6 released by epithelial cells in inducing the observed effects on endothelial cells [45].
Meanwhile, by Klein et al. [8], it was demonstrated that DEPM exposure did not induce the expression of relevant markers for endothelial inflammation such as ICAM-1 or E-selectin, important for the adhesion of inflammatory cells and the induction of inflammation, leading to diseases such as myocardial infarction and atherosclerosis [8]. After exposure for 24 h and 48 h, the increase in the expression levels of the transcription factor NFkB (known to upregulate cytokines and the transcription of cellular adhesion molecules in the beginning of inflammation) and the chaperone HSP70 (a general marker for cellular stress) point towards a delayed response after acute exposure to DEPM [8]; these findings may indicate endothelial dysfunction and cardiovascular disease [8]. Nevertheless, this led to high nuclear translocation of the transcription factor Nrf2 and the expression of CYP1A1 mRNA in endothelial cells. The cellular viability or inflammatory statuses were not affected by the applied doses of DEPM [8]. In addition, in DEPM, the expression of HSP70 can be found, which was upregulated to respond to PAHs, such as B[a]P [8]. In addition, in the upregulation of CASP7 mRNA, after being directly exposed to DEPM, there was a delayed stress response in the endothelial cells that led to an upregulation of the marker genes and also indirect exposure to tetracultures in the endothelium after being exposed for 48 h [8].
In research by Bengalli et al. (2017) [45], it was demonstrated that cells that suffered indirect exposure to DEP had an overexpression of ICAM-1 and VCAM-1, and once directly exposed, IL-6R levels would also increase [45]. IL-6 is released on site in the case of inflammation [63]. IL-6 trans-signalling was also reported to increase the expression of molecules in endothelial cells [64]. Contributions to endothelial activation and proangiogenic processes are made by CAM-1 and ICAM-1, which can be induced by vascular endothelial growth factor (VEGF) and cytokines [65].
Through the interaction with endothelial cell membranes and FLT1, KDR or NRP1 receptors and the subsequent activation of RAS and MAPK kinases signalling cascades, VEGF is a powerful inducer of angiogenesis [45], which can have a very important role in cardiovascular diseases. In the study by Bengalli et al. [45], it was found that angiogenesis-related proteins in endothelial cells exposed to conditioned media and the modulation of the MAPK pathway was weak, so it was assumed that this was due to a low level of VEGF, and it is known that only higher levels can have effects on endothelial cells [45].
The study by Bengalli et al. (2017) [45] demonstrates that modulated genes which are oxidative-stress-responsive include NFE2L2 transcription factor and its targets TXNRD1 and HMOX1, enzymes that protect against oxidative stress. Amid genes with altered expression, some were related to the MAPK pathway, which influences signals from different stimuli and responds appropriately in cells with responses, such as cellular proliferation, differentiation, development, inflammatory responses and apoptosis [66]. By increasing the expression of phosphorylated active forms, specific Akt1/2 and ERK2, identified to respond to growth factor signals, it was possible to validate the pathway activation. It was also shown that these proteins are activated by ROS and are involved in the regulation of various processes including cytokine production, metabolism, proliferation, angiogenesis, growth and cell survival [45]. It has been reported that NF-kB is activated by Akt, a renowned factor inducing cytokine transcription, which through NF-kB and STAT3 regulation is able to directly modulate IL-6 levels. It was confirmed that lung epithelial cells exposed to diesel particles released IL-6 [45].
4.1.3. Carbon Black
Finally, only one study selected tried to identify the relation between carbon black and its effects on endothelial cells. As previously stated, CB is mainly used as a black pigment, and individuals’ exposure to it occurs in industries. Based on Dinmohammadi et al. [46], the interaction poorly contributes to prothrombotic and proinflammatory effects during the cells’ exposure, as shown by unclear evidence. As it can be seen in Table 2, Dinmohammadi et al. [46] did not find changes in FVIII, VWF, P-selectin and IL-8 release when cells were exposed for 24h to CB or MWCNTs at lower doses (under 10 µg/mL), while treatment with 100 µg/mL showed a decrease in cell viability.
Both FVIII and VWF are storage proteins in the endothelium which are released once stimulated by other exogenous or endogenous molecules [46], mediating platelet adhesion (Peyvandi et al., 2011), which binds to injured areas (Packham et al., 1984). FVIII has a role in the coagulation cascade [67] and is synthetised in endothelial cells [64]; the authors previously observed an increase in FVIII in cardiac microvascular endothelial cells once exposed to UFPs and measured the same protein in HPMECs once exposed to CB, but no results were visible. VWF is also a protein that can be found in endothelial cells, linked to haemostasis, by binding to FVIII [68]. The protein P-selectin can be also found on the surface of platelets and endothelial cells once it is activated [69], influencing the regulation of platelet function [70]. Finally, IL-8, which is a cytokine which is produced by different cells and is linked with inflammation [71], has been associated with a direct role in the survival, proliferation and angiogenesis of endothelial cells [72].
These findings showed that no changes occurred in Weibel–Palade bodies (WPBs), which store all these proteins [73], are involved in inflammation [68,74,75] as inflammation mediators and are also linked to coagulation factors [73]. Since there was no response from endothelial cells to the UFPs and no increases in these proteins, this study [46] concluded that exposure did not contribute to prothrombotic and proinflammatory effects.
The analysed articles lead us to conclude that particles induce multiple effects on endothelial cells, namely endothelial dysfunction, which can lead to apoptosis and necrosis, and it may also cause necroptosis in lung structures (Figure 2). Several studies have been published that have relevant results for the scientific community regarding exposure to atmospheric particles. Barosova et al. [76] studied the impact of nanomaterials in 3D lung co-cultures, realising that acute or prolonged exposure to nanomaterials may induce different responses [76]. Offer et al. [77] also used an air–liquid interface to assess lung cells’ exposure to particles and demonstrated the importance of the chemical PM composition in adverse health outcomes [77]. This evidence is in line with the most recent articles in this field [76,77,78,79].
5. Conclusions
The main conclusions of this systematic review are outlined in Figure 2.
Particles can cause inflammation in cells, the formation of paracellular gaps, cytoskeleton rearrangement, barrier dysfunction, emphysema and increase ICAM-1 and ROS generation, which contribute to the pathogenesis of many vascular disorders, acute respiratory distress syndrome and chronic obstructive pulmonary disease. Exposure to aerosols may contribute to asthma, cardiac arrhythmias, myocardial infarction and atherosclerosis.
Further investigations are suggested to create: (1) more knowledge about the impact of carbon black on endothelial cells; (2) as well as knowledge about the PM2.5 mechanism on apical and basolateral membranes; and (3) practical studies on inhibitory pathways to reduce the effects on endothelial cells from aerosols.
Several limitations were identified throughout this systematic review: (1) a high number of articles related to air pollution, but few specified the pollutant linked with the impacts on endothelial cells; (2) the authors had to exclude a massive number of articles since silica particles, nanoplastics or other type of pollutants that were not particles were used, as well as other types of cells; (3) the methodologies used in the different articles were not exactly the same, which means that the comparison and integration of the information have associated limitations, although we tried to account for these differences.
Author Contributions
Statement of each author’s contribution: M.A.-S.: Conceptualisation, Methodology and Writing—Reviewing and Editing. J.C., C.A. and S.S.: Writing—Original Draft Preparation. A.M.: Writing—Reviewing and Editing. V.M.: Writing—Reviewing and Editing. A.M.-R.: Supervision and Writing—Reviewing and Editing. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully acknowledge the FCT/MCTES national support through the UIDB/05608/2020 and UIDP/05608/2020.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
M. Almeida-Silva and A. Ramos also acknowledge Instituto Politécnico de Lisboa for funding the project PLASCONGEN (IPL/2021/PLASCONGEN_ESTeSL) under its IDI&CA Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nwanaji-Enwerem, J.C.; Bind, M.-A.; Dai, L.; Oulhote, Y.; Colicino, E.; Di, Q.; Just, A.C.; Hou, L.; Vokonas, P.; Coull, B.A.; et al. Editor’s Highlight: Modifying Role of Endothelial Function Gene Variants on the Association of Long-Term PM2.5 Exposure with Blood DNA Methylation Age: The VA Normative Aging Study. Toxicol. Sci. 2017, 158, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, S.; Pusadkar, V.; McDonald, J.; Mirpuri, J.; Azad, R.K.; Goven, A.; Lund, A.K. Traffic Generated Emissions Alter the Lung Microbiota by Promoting the Expansion of Proteobacteria in C57Bl/6 Mice Placed on a High-Fat Diet. Ecotoxicol. Environ. Saf. 2021, 213, 112035. [Google Scholar] [CrossRef] [PubMed]
- Marques-Ramos, A.; Candeias, M.M.; Menezes, J.; Lacerda, R.; Willcocks, M.; Teixeira, A.; Locker, N.; Romao, L. Cap-Independent Translation Ensures MTOR Expression and Function upon Protein Synthesis Inhibition. RNA 2017, 23, 1712–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlemann, M.; Zeidler, R.; Lang, S.; Mack, B.; Münz, M.; Gires, O. Carcinoma-Associated EIF3i Overexpression Facilitates MTOR-Dependent Growth Transformation. Mol. Carcinog. 2006, 45, 957–967. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, Y.; Gao, Y.; Li, C.; Zhou, L.; Qi, W.; Zhang, Y.; Ye, L. Effects of Different Components of PM2.5 on the Expression Levels of NF-ΚB Family Gene MRNA and Inflammatory Molecules in Human Macrophage. Int. J. Environ. Res. Public Health 2019, 16, 1408. [Google Scholar] [CrossRef] [Green Version]
- Alain, T.; Morita, M.; Fonseca, B.D.; Yanagiya, A.; Siddiqui, N.; Bhat, M.; Zammit, D.; Marcus, V.; Metrakos, P.; Voyer, L.-A.; et al. EIF4E/4E-BP Ratio Predicts the Efficacy of MTOR Targeted Therapies. Cancer Res. 2012, 72, 6468–6476. [Google Scholar] [CrossRef] [Green Version]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional Crosstalk between Endoplasmic Reticulum Stress and MTOR Signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.G.; Cambier, S.; Hennen, J.; Legay, S.; Serchi, T.; Nelissen, I.; Chary, A.; Moschini, E.; Krein, A.; Blömeke, B.; et al. Endothelial Responses of the Alveolar Barrier in Vitro in a Dose-Controlled Exposure to Diesel Exhaust Particulate Matter. Part. Fibre Toxicol. 2017, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Meliton, A.; Sitikov, A.; Tian, Y.; Ohmura, T.; Birukova, A.A. Microtubule Destabilization Caused by Particulate Matter Contributes to Lung Endothelial Barrier Dysfunction and Inflammation. Cell. Signal. 2019, 53, 246–255. [Google Scholar] [CrossRef]
- Spira, A.; Beane, J.; Shah, V.; Liu, G.; Schembri, F.; Yang, X.; Palma, J.; Brody, J.S. Effects of Cigarette Smoke on the Human Airway Epithelial Cell Transcriptome. Proc. Natl. Acad. Sci. USA 2004, 101, 10143–10148. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 1,3-Butadiene, Ethylene Oxide and Vinyl Halides (Vinyl Fluoride, Vinyl Chloride and Vinyl Bromide). IARC Monogr. Eval. Carcinog. Risks Hum. 2008, 97, 3–471. [Google Scholar]
- Das, S.K. Harmful Health Effects of Cigarette Smoking. Mol. Cell. Biochem. 2003, 253, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O.; Davids, L.M.; Marnewick, J.L. Diesel Exhaust Particles and Endothelial Cells Dysfunction: An Update. Toxicol. In Vitro 2016, 32, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Holz, M.K. MTOR Signaling for Biological Control and Cancer. J. Cell. Physiol. 2013, 228, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Long, C.M.; Nascarella, M.A.; Valberg, P.A. Carbon Black vs. Black Carbon and Other Airborne Materials Containing Elemental Carbon: Physical and Chemical Distinctions. Environ. Pollut. 2013, 181, 271–286. [Google Scholar] [CrossRef] [Green Version]
- Casanova, A.; Gomis-Berenguer, A.; Canizares, A.; Simon, P.; Calzada, D.; Ania, C.O. Carbon Black as Conductive Additive and Structural Director of Porous Carbon Gels. Materials 2020, 13, 217. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, N.R.; Pojana, G.; White, P.; Møller, P.; Cohn, C.A.; Korsholm, K.S.; Vogel, U.; Marcomini, A.; Loft, S.; Wallin, H. Genotoxicity, Cytotoxicity, and Reactive Oxygen Species Induced by Single-walled Carbon Nanotubes and C60 Fullerenes in the FE1-MutaTM Mouse Lung Epithelial Cells. Environ. Mol. Mutagen. 2008, 49, 476–487. [Google Scholar] [CrossRef]
- Yoshida, S.; Hiyoshi, K.; Oshio, S.; Takano, H.; Takeda, K.; Ichinose, T. Effects of Fetal Exposure to Carbon Nanoparticles on Reproductive Function in Male Offspring. Fertil. Steril. 2010, 93, 1695–1699. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Giebe, S.; Cockcroft, N.; Hewitt, K.; Brux, M.; Hofmann, A.; Morawietz, H.; Brunssen, C. Cigarette Smoke Extract Counteracts Atheroprotective Effects of High Laminar Flow on Endothelial Function. Redox Biol. 2017, 12, 776–786. [Google Scholar] [CrossRef]
- Vesterdal, L.K.; Folkmann, J.K.; Jacobsen, N.R.; Sheykhzade, M.; Wallin, H.; Loft, S.; Møller, P. Pulmonary Exposure to Carbon Black Nanoparticles and Vascular Effects. Part. Fibre Toxicol. 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garland, C.J.; Hiley, C.R.; Dora, K.A. EDHF: Spreading the Influence of the Endothelium: EDHF and Arterial Dilatation. Br. J. Pharmacol. 2011, 164, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Tailor, A.; Cooper, D.; Granger, D.N. Platelet–Vessel Wall Interactions in the Microcirculation. Microcirculation 2005, 12, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Mrowietz, C.; Lendlein, A.; Jung, F. Interaction of Human Umbilical Vein Endothelial Cells (HUVEC) with Platelets in Vitro: Influence of Platelet Concentration and Reactivity. Clin. Hemorheol. Microcirc. 2013, 55, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial Cell Control of Thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Rounds, S.I.S.; Lu, Q.; Harrington, E.O.; Newton, J.; Casserly, B. Pulmonary Endothelial Cell Signaling and Function. Trans. Am. Clin. Climatol. Assoc. 2008, 119, 155–167; discussion 167-9. [Google Scholar]
- Michiels, C. Endothelial Cell Functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Gonzales, J.N.; Verin, A.D. Pulmonary Vascular Endothelial Cells. In Endothelial Dysfunction—Old Concepts and New Challenges; Lenasi, H., Ed.; InTech: Vienna, Austria, 2018; ISBN 978-1-78984-253-1. [Google Scholar]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The Pulmonary Endothelium in Acute Respiratory Distress Syndrome: Insights and Therapeutic Opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, L.; Zaidi, S.R.; Sammani, S.; Siegler, J.; Moreno-Vinasco, L.; Mathew, B.; Natarajan, V.; Garcia, J.G.N. Hydrogen Sulfide Attenuates Particulate Matter-Induced Human Lung Endothelial Barrier Disruption via Combined Reactive Oxygen Species Scavenging and Akt Activation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Meliton, A.; Shah, A.; Tian, Y.; Ohmura, T.; Sarich, N.; Birukova, A.A.; Birukov, K.G. Role of Truncated Oxidized Phospholipids in Acute Endothelial Barrier Dysfunction Caused by Particulate Matter. PLoS ONE 2018, 13, e0206251. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, X.; Liu, X.; Zheng, J.; Chen, R.; Kan, H. Ambient Fine Particulate Matter Induce Toxicity in Lung Epithelial-Endothelial Co-Culture Models. Toxicol. Lett. 2019, 301, 133–145. [Google Scholar] [CrossRef]
- Cui, A.; Xiang, M.; Xu, M.; Lu, P.; Wang, S.; Zou, Y.; Qiao, K.; Jin, C.; Li, Y.; Lu, M.; et al. VCAM-1-Mediated Neutrophil Infiltration Exacerbates Ambient Fine Particle-Induced Lung Injury. Toxicol. Lett. 2019, 302, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Li, C.; Xu, J.; Ren, F.; Xu, X.; Liu, C.; Niu, B.; Li, F. LncRNA Gm16410 Regulates PM2.5-Induced Lung Endothelial-Mesenchymal Transition via the TGF-Β1/Smad3/p-Smad3 Pathway. Ecotoxicol. Environ. Saf. 2020, 205, 111327. [Google Scholar] [CrossRef]
- Tian, G.; Wang, J.; Lu, Z.; Wang, H.; Zhang, W.; Ding, W.; Zhang, F. Indirect Effect of PM1 on Endothelial Cells via Inducing the Release of Respiratory Inflammatory Cytokines. Toxicol. In Vitro 2019, 57, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chiang, E.T.; Moreno-Vinasco, L.; Lang, G.D.; Pendyala, S.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; Chillrud, S.N.; Natarajan, V.; et al. Particulate Matter Disrupts Human Lung Endothelial Barrier Integrity via ROS- and P38 MAPK-Dependent Pathways. Am. J. Respir. Cell Mol. Biol. 2010, 42, 442–449. [Google Scholar] [CrossRef]
- Mo, Y.; Wan, R.; Chien, S.; Tollerud, D.J.; Zhang, Q. Activation of Endothelial Cells after Exposure to Ambient Ultrafine Particles: The Role of NADPH Oxidase. Toxicol. Appl. Pharmacol. 2009, 236, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bengalli, R.; Mantecca, P.; Camatini, M.; Gualtieri, M. Effect of Nanoparticles and Environmental Particles on a Cocultures Model of the Air-Blood Barrier. BioMed Res. Int. 2013, 2013, 801214. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Tao, J.-Q.; Johncola, A.; Guo, W.; Caporale, A.; Langham, M.C.; Wehrli, F.W. Acute Exposure to E-Cigarettes Causes Inflammation and Pulmonary Endothelial Oxidative Stress in Nonsmoking, Healthy Young Subjects. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 317, L155–L166. [Google Scholar] [CrossRef]
- Mizumura, K.; Justice, M.J.; Schweitzer, K.S.; Krishnan, S.; Bronova, I.; Berdyshev, E.V.; Hubbard, W.C.; Pewzner-Jung, Y.; Futerman, A.H.; Choi, A.M.K.; et al. Sphingolipid Regulation of Lung Epithelial Cell Mitophagy and Necroptosis during Cigarette Smoke Exposure. FASEB J. 2018, 32, 1880–1890. [Google Scholar] [CrossRef] [Green Version]
- Sakhatskyy, P.; Miranda, G.A.G.; Newton, J.; Lee, C.G.; Choudhary, G.; Vang, A.; Rounds, S.; Lu, Q. Cigarette Smoke-Induced Lung Endothelial Apoptosis and Emphysema Are Associated with Impairment of FAK and EIF2α. Microvasc. Res. 2014, 94, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G.; Hubbard, W.C.; et al. Mechanisms of Lung Endothelial Barrier Disruption Induced by Cigarette Smoke: Role of Oxidative Stress and Ceramides. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef]
- Bengalli, R.; Zerboni, A.; Marchetti, S.; Longhin, E.; Priola, M.; Camatini, M.; Mantecca, P. In Vitro Pulmonary and Vascular Effects Induced by Different Diesel Exhaust Particles. Toxicol. Lett. 2019, 306, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bengalli, R.; Longhin, E.; Marchetti, S.; Proverbio, M.C.; Battaglia, C.; Camatini, M. The Role of IL-6 Released from Pulmonary Epithelial Cells in Diesel UFP-Induced Endothelial Activation. Environ. Pollut. 2017, 231, 1314–1321. [Google Scholar] [CrossRef]
- Dinmohammadi, H.; Pirdel, Z.; Salarilak, L.; Hoylaerts, M.; Nejatbakhsh, R.; Biglari, A.; Jacquemin, M.; Shahani, T. Pure Ultra-Fine Carbon Particles Do Not Exert pro-Coagulation and Inflammatory Effects on Microvascular Endothelial Cells. Environ. Sci. Pollut. Res. 2019, 26, 991–999. [Google Scholar] [CrossRef]
- Zuo, H.; Faiz, A.; van den Berge, M.; Mudiyanselage, S.N.H.R.; Borghuis, T.; Timens, W.; Nikolaev, V.O.; Burgess, J.K.; Schmidt, M. Cigarette Smoke Exposure Alters Phosphodiesterases in Human Structural Lung Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L59–L64. [Google Scholar] [CrossRef]
- Loscalzo, J. The Identification of Nitric Oxide as Endothelium-Derived Relaxing Factor. Circ. Res. 2013, 113, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric Oxide Activates Guanylate Cyclase and Increases Guanosine 3′:5′-Cyclic Monophosphate Levels in Various Tissue Preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-Dependent Necroptosis Contributes to the Pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide Upregulation Causes Pulmonary Cell Apoptosis and Emphysema-like Disease in Mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel Mechanisms Regulating Endothelial Barrier Function in the Pulmonary Microcirculation. J. Physiol. 2019, 597, 997–1021. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A. Functions of Ceramide in Coordinating Cellular Responses to Stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ren, X.; Liu, J.; Guo, X.; Jiang, X.; Zhang, D.; Huang, Y.; Zhang, J. HSP27 Phosphorylation Protects against Endothelial Barrier Dysfunction under Burn Serum Challenge. Biochem. Biophys. Res. Commun. 2015, 463, 377–383. [Google Scholar] [CrossRef]
- Hiebl, B.; Peters, S.; Gemeinhardt, O.; Niehues, S.M.; Jung, F. Impact of Serum in Cell Culture Media on in Vitro Lactate Dehydrogenase (LDH) Release Determination. J. Cell. Biotechnol. 2017, 3, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G.; Brough, D. Understanding the Mechanism of IL-1β Secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- van den Brule, S.; Rappe, M.; Ambroise, J.; Bouzin, C.; Dessy, C.; Paquot, A.; Muccioli, G.G.; Lison, D. Diesel Exhaust Particles Alter the Profile and Function of the Gut Microbiota upon Subchronic Oral Administration in Mice. Part. Fibre Toxicol. 2021, 18, 7. [Google Scholar] [CrossRef]
- Bai, H.; Wu, M.; Zhang, H.; Tang, G. Chronic Polycyclic Aromatic Hydrocarbon Exposure Causes DNA Damage and Genomic Instability in Lung Epithelial Cells. Oncotarget 2017, 8, 79034–79045. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Mangal, D.; Tacka, K.A.; Quinn, A.M.; Harvey, R.G.; Blair, I.A.; Penning, T.M. Evidence for the Aldo-Keto Reductase Pathway of Polycyclic Aromatic Trans-Dihydrodiol Activation in Human Lung A549 Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6846–6851. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Dalton, T.P. The Role of Cytochrome P450 Enzymes in Endogenous Signalling Pathways and Environmental Carcinogenesis. Nat. Rev. Cancer 2006, 6, 947–960. [Google Scholar] [CrossRef]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic Activation of Polycyclic Aromatic Hydrocarbons to Carcinogens by Cytochromes P450 1A1 And1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Moake, J.L. Factor VIII is Synthesized in Human Endothelial Cells, Packaged in Weibel-Palade Bodies and Secreted Bound to ULVWF Strings. PLoS ONE 2015, 10, e0140740. [Google Scholar] [CrossRef] [PubMed]
- Jośko, J.; Gwóźdź, B.; Jedrzejowska-Szypułka, H.; Hendryk, S. Vascular Endothelial Growth Factor (VEGF) and Its Effect on Angiogenesis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2000, 6, 1047–1052. [Google Scholar]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchini, M.; Mannucci, P.M. Past, Present and Future of Hemophilia: A Narrative Review. Orphanet J. Rare Dis. 2012, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandi, F.; Garagiola, I.; Baronciani, L. Role of von Willebrand Factor in the Haemostasis. Blood Transfus. 2011, 9, s3–s8. [Google Scholar] [CrossRef]
- Borsig, L. Selectins in Cancer Immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Théorêt, J.-F.; Yacoub, D.; Hachem, A.; Gillis, M.-A.; Merhi, Y. P-Selectin Ligation Induces Platelet Activation and Enhances Microaggregate and Thrombus Formation. Thromb. Res. 2011, 128, 243–250. [Google Scholar] [CrossRef]
- El Ayadi, A.; Herndon, D.N.; Finnerty, C.C. Biomarkers in Burn Patient Care. In Total Burn Care; Elsevier: Amsterdam, The Netherlands, 2018; pp. 232–235.e2. ISBN 978-0-323-47661-4. [Google Scholar]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Valentijn, K.M.; Sadler, J.E.; Valentijn, J.A.; Voorberg, J.; Eikenboom, J. Functional Architecture of Weibel-Palade Bodies. Blood 2011, 117, 5033–5043. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Packham, M.; Mustard, J. Platelet Adhesion. Prog. Hemost. Thromb. 1984, 7, 211–288. [Google Scholar] [PubMed]
- Barosova, H.; Karakocak, B.B.; Septiadi, D.; Petri-Fink, A.; Stone, V.; Rothen-Rutishauser, B. An In Vitro Lung System to Assess the Proinflammatory Hazard of Carbon Nanotube Aerosols. Int. J. Mol. Sci. 2022, 21, 5335. [Google Scholar] [CrossRef] [PubMed]
- Offer, S.; Hartner, E.; Di Bucchianico, S.; Bisig, C.; Bauer, S.; Pantzke, J.; Zimmermann, E.J.; Cao, X.; Binder, S.; Kuhn, E.; et al. Effect of Atmospheric Aging on Soot Particle Toxicity in Lung Cell Models at the Air–Liquid Interface: Differential Toxicological Impacts of Biogenic and Anthropogenic Secondary Organic Aerosols (SOAs). Environ. Health Perspect. 2022, 130, 027003. [Google Scholar] [CrossRef]
- Sherratt, S.C.R.R.; Dawoud, H.; Libby, P.; Bhatt, D.L.; Malinski, T.; Mason, R.P. Eicosapentaenoic acid (EPA) reduces inflammation and improves nitric oxide bioavailability in pulmonary endothelial cells following exposure to air pollution particles. J. Am. Coll. Cardiol. 2022, 79, 1758. [Google Scholar] [CrossRef]
- Eckhardt, C.M.; Baccarelli, A.A.; Wu, H. Environmental Exposures and Extracellular Vesicles: Indicators of Systemic Effects and Human Disease. Curr. Environ. Health Rep. 2022. [Google Scholar] [CrossRef]
Figure 1.
PRISMA methodology of selection of papers.

Figure 2.
Effects of particulate matter and, consequently, cigarette smoke, diesel particles and carbon black on lung endothelial cells.
Figure 2.
Effects of particulate matter and, consequently, cigarette smoke, diesel particles and carbon black on lung endothelial cells.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Description of search databases and search terms.
| Search Databases | Search Terms |
|---|---|
| Pubmed | “endothelial cells” [MeSH Terms] AND “lung” [MeSH Terms] AND (“particulate matter” [MeSH Terms] OR “particle*” [All Fields]) |
| Web of Science | “endothelial cells” AND “lung” AND (“particulate matter” OR “particle*”) |
| Scopus | “endothelial cells” AND “lung” AND (“particulate matter” OR “particle*”) |
Table 2.
Inclusion and exclusion criteria.
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Articles in English language | Articles in other languages |
| Articles published from 1 January 2000 to 6 March 2021 | Articles published prior to 2000 |
| Articles related to lung endothelial cells | Articles related to other cells |
| Articles related to particulate matter effects | Articles related to another organs |
| Articles on in vitro and/or in vivo studies | Articles related with other pollutants |
Table 3.
Data obtained from the chosen articles.
| Pollutant | Objective | Methodology | Results | Main Findings | |
|---|---|---|---|---|---|
| [31] | PM | The importance of the protective role of hydrogen sulphide (H2S) on PM-induced human lung EC barrier disruption and pulmonary inflammation. | (In vitro) HLMVECs. (In vivo) Mice. |
|
|
| [32] | PM | To investigate if the PM challenge triggers the production of bioactive Tr-OxPLs by pulmonary EC. | (In vitro) Human pulmonary endothelial cells. (In vivo) PM administered as suspension in saline to mice. |
|
|
| [33] | PM2.5 | The stabilisation of a suitable in vitro model to investigate PM2.5-mediated toxicity in vascular endothelial cells. | (In vitro) The Transwell culture method was used on A549 cells in apical chamber and EA.hy926 cells in the basolateral chamber to establish bi-culture, while tri-culture systems consisting of co-culture (A549 cells and THP-1-differentiated macrophages) in the apical chamber and also EA.hy926 cells in the basolateral chamber. |
|
|
| [34] | PM2.5 | To determine if polymorphonuclear leukocyte PMNs exacerbate PM2.5-induced lung in-jury and the role of vascular cell adhesion molecule 1 (VCAM-1) in this process. | (In vivo) The association between blood PMNs and ambient PM2.5 levels on the previous day was analysed. Neutropenia was achieved by injecting mice with PMN-specific antibodies. The inhibition of PMN infiltration was achieved by pre-treating PMNs with soluble VCAM-1. |
|
|
| [35] | PM2.5 | A simulation of the state of PM2.5 in a real environment and chronic exposure to PM2.5 in mice was used to construct a lung injury model to detect whether exposure to PM2.5 could trigger EndMT in the lungs. | (In vivo) Balb/c Mice. (In vitro) Pulmonary vascular endothelial cells (PVECs). The toxicity of PM2.5 investigated by determining its effect on the growth of MHC cells using the MTT assay. |
|
|
| [36] | PM1 | The examination of PM1-induced inflammatory response in the respiratory system and the indirect effect on endothelial cell function. | (In vitro) Transwell co-culture model of EA.hy926 cells with BEAS-2B cells and macrophages. |
|
|
| [37] | Fine/ultrafine PM | To analyse if PM, via enhanced oxidative stress, disrupts lung endothelial cells’ (LECs’) barrier integrity, thereby enhancing organ dysfunction. | (In vitro) Cultivated human pulmonary artery ECs on gold electrodes and assessed TER. The investigation of the involvement of ROS and mitogen-activated protein kinase (MAPK) signalling pathways. |
|
|
| [38] | Ultrafine particles | To test if exposure to UFPs leads to endothelial cell O2 U−generation via NADPH oxidase and results in activation of endothelial cells. | (In vivo) Mice. (In vitro) MPMVEC. |
|
|
| [39] | Nanoparticles/PM10 | To report the effects of metal oxide NPs (CuO and TiO2) and of PM on an in vitro model of the ABB constituted by the type II epithelial cell line (NCI-H441) and the endothelial one (HPMECST1.6R). | (In vitro) HPMECs. |
|
|
| [40] | E-cigarette smoke | The evaluation of the acute response to aerosol inhalation of e-cigarettes in terms of oxidative stress and indices of endothelial activation in human pulmonary microvascular endothelial cells (HPMVECs). | (In vivo) Healthy subjects were subjected to e-cig challenge. Serum was monitored for markers of inflammation (C-reactive protein (CRP) and soluble intercellular adhesion molecule (sICAM)) and nitric oxide metabolites (NOx). |
|
|
| [41] | Cigarette smoke | The investigation of the role of ceramide (Cer) in CS-induced lung cell mitophagy and cell death. | (In vitro) Cer and DHC treatments and aqueous CS extract (100%). (In vivo) DBA2/Jmice exposed for 5 h/d, 5 d/ wk, for 4 mo; PTEN-induced kinase 1 (Pink1)-/- mice exposed to room air or CS for 2 h/d, 5 d/wk, for 6 mo; CerS2-/- exposed to room air or CS for 2 h/d, 5 d/wk, for 1 mo. |
|
|
| [42] | Cigarette smoke | A comparation between Focal Adhesion Kinase (FAK) activation in the lungs of highly and mildly susceptible mice after exposure to CS for three weeks. | (In vitro) Rat lung microvascular endothelial cells (LMVECs). (In vivo) Male C57BL/6 and FAK mice exposed to room air or CS for 3 weeks at 6 h per day and 4 days per week. |
|
|
| [43] | Cigarette smoke | The investigation of the occurrence and mechanisms by which soluble components of mainstream CS disrupt the lung endothelial cell barrier function. | (In vitro) Cultured primary rat microvascular cell monolayers. |
|
|
| [44] | Diesel exhaust particles | The investigation of LECs’ primary response on bronchial cells BEAS-2B exposed to three different DEPs. | (In vitro) Microvascular lung HPMEC cells and BEAS-2B placed in aco-culture model and conditioned media. |
|
|
| [8] | Diesel exhaust particulate matter (DEPM) | Researching the response of ECs to DEPM, using a complex 3D tetraculture model of the alveolar barrier. | (In vitro) Aerosol exposure of DEPM; viability evaluated at 6, 24 and 48 h after exposure. |
|
|
| [45] | Diesel UFP | The investigation of the endothelial cells’ activation through epithelial-released mediators’ role. | (in vitro) HPMEC-ST1.6R (EC) treated with 40% of media (epithelial cells diluted in M199 medium), HPMEC-ST1.6R cells exposure lasted for 24 h. |
|
|
| [46] | Carbon black (CB) and multi-walled carbon nanotubes (MWCNTs) | The investigation of industrial CB and CNTs’ potential toxicity in endothelial cells by measuring Coagulation factor VIII (FVIII) and the Von-Willebrand factor (VWF). | (In vitro) MECs, 48 h after reaching confluence, were treated with culture medium of either MWCNTs or CB with a solution of 0.1, 1, 10, or 100 μg/mL in. |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Almeida-Silva, M.; Cardoso, J.; Alemão, C.; Santos, S.; Monteiro, A.; Manteigas, V.; Marques-Ramos, A. Impact of Particles on Pulmonary Endothelial Cells. Toxics 2022, 10, 312. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060312
AMA Style
Almeida-Silva M, Cardoso J, Alemão C, Santos S, Monteiro A, Manteigas V, Marques-Ramos A. Impact of Particles on Pulmonary Endothelial Cells. Toxics. 2022; 10(6):312. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060312
Chicago/Turabian StyleAlmeida-Silva, Marina, Jéssica Cardoso, Catarina Alemão, Sara Santos, Ana Monteiro, Vítor Manteigas, and Ana Marques-Ramos. 2022. "Impact of Particles on Pulmonary Endothelial Cells" Toxics 10, no. 6: 312. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10060312
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.