
Layered Double Hydroxide/Nanocarbon Composites as Heterogeneous Catalysts: A Review



1
ICGM, Université de Montpellier, CNRS, ENSCM, 34296 Montpellier, France
2
GIR-QUESCAT, Departamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad de Salamanca, 37008 Salamanca, Spain
*
Authors to whom correspondence should be addressed.
ChemEngineering 2022, 6(4), 45; https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040045
Submission received: 31 May 2022
/
Revised: 16 June 2022
/
Accepted: 20 June 2022
/
Published: 22 June 2022
(This article belongs to the Special Issue A Themed Issue in Honor of Prof. Dr. Vicente Rives)
Abstract
:The synthesis and applications of composites based on layered double hydroxides (LDHs) and nanocarbons have recently seen great development. On the one hand, LDHs are versatile 2D compounds that present a plethora of applications, from medicine to energy conversion, environmental remediation, and heterogeneous catalysis. On the other, nanocarbons present unique physical and chemical properties owing to their low-dimensional structure and sp2 hybridization of carbon atoms, which endows them with excellent charge carrier mobility, outstanding mechanical strength, and high thermal conductivity. Many reviews described the applications of LDH/nanocarbon composites in the areas of energy and photo- and electro-catalysis, but there is still scarce literature on their latest applications as heterogeneous catalysts in chemical synthesis and conversion, which is the object of this review. First, the properties of the LDHs and of the different types of carbon materials involved as building blocks of the composites are summarized. Then, the synthesis methods of the composites are described, emphasizing the parameters allowing their properties to be controlled. This highlights their great adaptability and easier implementation. Afterwards, the application of LDH/carbon composites as catalysts for C–C bond formation, higher alcohol synthesis (HAS), oxidation, and hydrogenation reactions is reported and discussed in depth.
1. Introduction
Nanocomposites combining LDHs and carbon-based materials as building blocks have attracted a great deal of interest lately, particularly as electrocatalysts and photocatalysts. Several excellent reviews related to the synthesis, characterization, and application of LDH/carbon nanocomposites highlight their remarkable properties and performance for energy storage and conversion, environmental protection, and pollution abatement [1,2,3,4,5,6,7]. Although less developed, the applications of LDH/carbon nanocomposites as nanofillers, non-enzymatic sensors, adsorbents for water remediation, and drug delivery systems have been also reported in several reviews [8,9,10,11]. Some of them point out the use of LDH/carbon nanocomposites as catalysts in acid–base and redox reactions [3,8,11,12]. However, there is now a remarkable number of publications reporting the high efficiency of LDH/carbon nanocomposites in these reactions.
This type of application was reported for the first time in 2005 [13,14]. MgAl-LDH/carbon nanofiber (CNF) composites were used in the base-catalyzed condensation of acetone, reaching specific activity four times higher than that of the unsupported MgAl-LDH. Moreover, impregnation of a MgAl-LDH/CNF composite with Pd led to a bifunctional catalyst achieving the single-stage synthesis of methylisobutylketone (MIBK) from acetone, with initial activity five times higher than the mechanical mixture of activated MgAl-LDH and Pd/CNF catalysts. Afterwards, various LDH/carbon nanocatalysts have been implemented, displaying highly successful performance compared to unsupported LDH-based catalysts for base-catalyzed C–C bond-forming reactions, as well as for oxidation and reduction reactions. Their higher efficiency than the single MgAl-LDH mainly results from the higher specific surface areas and porosity. Moreover, interactions between LDH nanosheets or metal nanoparticles (NPs) obtained from LDH precursors and nanocarbons improve electron transfer and avoid aggregation of the LDH or metal NPs. The LDH/carbon composites also exhibit high thermal and chemical stabilities. They generally retain the intrinsic properties of the individual LDH and carbon components with additional synergistic effects.
The hierarchical nanocomposites derived from a multitude of LDH compositions and 0D carbon dots (CD), 1D nanofibers (CF), single-walled carbon nanotubes (SWCNT) and multi-walled carbon nanotubes (MWCNT), and 2D graphene-like compounds give rise to a large family of materials.
The main characteristics of each type of carbon material have been extensively described [15,16,17,18,19]. Graphene, particularly its oxidized or reduced forms, i.e., graphene oxide (GO) or reduced graphene oxide (rGO), is currently the most widely used nanocarbon component in LDH/carbon nanocatalysts. The use of nitrogen-doped graphene is also now emerging. The success of the graphene-like supports for the design of LDH-containing composites is partly based on their 2D structural compatibility, which allows intercalation. The resulting sandwich-type structures are rarely used as catalysts, where disordered arrangements with more accessible and dispersed active sites are desirable [20,21]. GO and rGO are more likely chosen for their electronic and thermal conductivities, their ability to enhance the dispersion and avoid aggregation of the LDH, and to enhance the mechanical and chemical stability of the composites. CNF and CNT appear as the second type of nanocarbons used. More recently, carbon dots (CD) and nitrogen-doped carbon dots (NCD) have been considered promising to develop composites with improved basic properties, fast electron transfer through strong metal–CD interaction, and high mechanical resistance [20,21,22].
This review highlights the research implemented on the LDH/nanocarbon composites for base-catalyzed C–C bond formation, oxidation, hydrogenation reactions, and HAS. Firstly, the properties of the different types of carbon materials involved as building blocks of the composites will be summarized. Then, the synthesis methods of the LDH/carbon nanocomposites will be described. Afterwards, application of LDH/carbon composites as catalysts in the previously cited reactions will be reported. Finally, the main features emerging from the large survey of the literature considered in the review will be summarized and discussed. In addition, several possible future search directions will be indicated.
2. LDHs and Carbon Materials
2.1. LDHs
LDHs are built up of brucite (Mg(OH)2)-like layers, which consist of magnesium ions surrounded by six hydroxyl groups in an octahedral geometry where the divalent metal (M2+) is isomorphically substituted by a trivalent one (M3+). The excess of positive charge is balanced by intercalated anions coexisting with water molecules. The general formula of LDH can be written as [M2+1−xM3+x(OH)2]x+ (Am−x/m)x− · nH2O, where M2+ and M3+ are di- and trivalent cations, respectively, and Am− are the interlayer anions. The molar ratio M3+/(M2+ + M3+) generally ranges from 0.2 to 0.33, although some different M2+/M3+ molar ratios have also been reported [23]. LDHs exhibit remarkable versatility for the preparation of base- and metal-supported catalysts due to their variety of compositions and their activity either in the lamellar form or as layered double oxides (LDO) obtained by thermal decomposition. Moreover, they are highly suitable as precursors of metal-supported or multifunctional catalysts, with large specific surface areas and peculiar metal–support interactions [23,24,25,26]. However, LDHs are prone to particle aggregation, dissolution in liquid media, and sheet stacking, which causes low dispersion of metal NPs and hinders reactants’ accessibility to the active sites. This contributes to reducing the efficiency of the LDH-derived catalysts. Many of these drawbacks have been prevented by the dispersion of LDH-based catalysts on various supports. Among them, nanocarbons are increasingly used due to their complementary properties with LDHs.
2.2. Carbon Materials
Despite exhibiting the same general properties, i.e., electron conductivity, high mechanical strength, high thermal conductivity, and chemical inertness, each nanocarbon has specific properties. For catalytic applications, the nanocarbons should have a high surface area, suitable pore size, high graphitization degree, and strong interfacial coupling. In particular, 2D graphene-like materials, 1D carbon nanotubes (CNT), carbon fibers (CF) and nanofibers (CNF), and 0D carbon dots (CD) have been considered as components of the LDH/nanocarbons [15]. These nanocarbon components are very versatile for surface modification and functionalization, which is necessary to ensure interfacial coupling with LDHs.
Graphene-like materials, i.e., graphene (G), graphene oxide (GO), and reduced graphene oxide (rGO), are the most reported materials involved in LDH/nanocarbon composites. G consists of a single layer of sp2 and sp3-hybridized carbon atoms organized in a 2D hexagonal lattice. The synthesis of graphene-like materials involves either bottom-up methods starting from carbon molecules or top-down methods using a carbon source, generally graphite. For catalytic applications, chemical synthesis is most suitable since it provides a reactive surface with high density of functional groups. Hummers’ method and its modified methods are the most common routes for graphite oxidation [27]. The obtained GO can be easily exfoliated. Through a subsequent reduction step with a chemical agent or by ultrasonication or thermal or hydrothermal treatment, GO sheets are transformed into reduced graphene oxide (rGO).
A CNT can be considered as a rolled-up graphene sheet to form cylindrical molecules. They are classified as single-walled carbon nanotubes (SWCNT) and multi-walled carbon nanotubes (MWCNT) according to the number of rolled-up graphene layers forming the tubular nanostructure. CNTs present high strength, electrical and thermal conductivity and stability, and a high surface area, which makes them very attractive for catalytic applications.
CFs and CNFs exhibit structures and properties closely related and similar to those of CNTs. However, the geometry of CNFs, formed by regularly stacked truncated conical graphene layers, is different to that of CNTs [19]. CNFs can be defined as linear filaments formed of sp2 carbon atoms, giving a flexible, highly graphitic structure with lengths from several nanometers to microns. Similar to CNTs, CNFs present a high surface area and chemically active end planes, which facilitate its functionalization.
Carbon dots (CD), including carbon quantum dots CQD, graphene quantum dots (GQD) and N-doped carbon dots (NCD), and the CD/inorganic nanocomponents, are emerging as cutting-edge materials for the development of advanced catalysts. CD exhibit a size below 10 nm, whose structure is composed of sp2 and sp3 hybridized carbon atoms in the core and the outer part, respectively [28]. A variety of top-down and bottom-up syntheses have been developed for producing CD with different characteristics.
3. Preparation Methods
Among the various preparation methods of LDH/carbon nanocatalysts, two stand out: self-assembly of LDHs and carbon components and coprecipitation of LDHs on the carbon component acting as a growth substrate [1,3,8,10,11]. The self-assembly method is scarcely utilized, and an overwhelming majority of LDH/carbon nanocomposites are prepared by coprecipitation, which can be easily adapted to the nature of the carbon material and the design of various structures.
3.1. Self-Assembly Method
The self-assembly method, particularly employed in the case of GO, is based on the electrostatic interaction between the negatively and positively charged surfaces of the GO and LDH, respectively. It leads to different nanocomposite structures depending on the previous treatment of the LDH mixed with the GO nanosheets exfoliated by ultrasonication. When the LDH is exfoliated in water, the resulting LDH/GO composite is poorly homogeneous, exhibiting LDH and GO domains both separated and in intimate contact [20]. On the contrary, LDH nanosheets exfoliated in dimethyl formamide (DMF), when restacking is avoided during drying, leads to a nanocomposite with ultrathin LDH nanosheets highly dispersed on GO, boosting the catalytic performance [32].
A nanostructured Fe3O4@GO@Zn-Ni-Fe-LDH magnetic composite was developed by Zeynizadeh et al. combining three components: magnetic Fe3O4 nanoparticles (MNP), GO, and ZnNiFe-LDH [33]. Fe3O4 MNP allow the recovery and reusability of the nanocatalyst, while their immobilization on GO prevents aggregation. The overall synthetic process of the Fe3O4@GO@Zn-Ni-Fe-LDH composite started with the oxidation of graphite to GO (Hummers’ procedure), followed by the coprecipitation of Fe3O4 on GO exfoliated by sonication to give the Fe3O4@GO component. Then, the Fe3O4@GO constituent was assembled with a previously prepared ZnNiFe-LDH, which led to a nanocomposite where LDH was disposed in plane with the GO constituent, generating a highly porous material.
CD/LDH catalysts have also been generally prepared by self-assembly, mixing LDH and CD aqueous solutions without particular pretreatments in the preformed constituents [34]. In other cases, the LDH nanosheets are stabilized with an organic polymer to avoid particle aggregation and favor water dispersibility and CD dispersion on the LDH sheets. For instance, Yang et al. developed a method for the synthesis of an LDH@PDA@PNIPAM@Pd/CD nanocatalyst where polydopamine (PDA) was deposited on the MgAl-LDH nanosheets, dispersed by ultrasonic treatment through the mussel chemical method [22]. Afterwards, sulfhydryl-terminated poly(N-isopropylacrylamide) (PNIPAM) polymer was grafted via Michael addition. Finally, the LDH@PDA@PNIPAM@Pd/CD nanocatalyst was obtained by loading Pd and CD on the polymer brush-modified MgAl-LDH nanosheets dispersed by sonication (Figure 1a). This preparation favored both the formation of small and uniform CD and Ag NPs. The different components (LDH and CD) in the composite promoted electron transfer and migration but stabilized and improved the dispersion of Pd NPs.
3.2. Coprecipitation Method
Coprecipitation of LDH on the carbon support is facilitated by the strong adsorption ability of the metal cations in the solution with the negatively charged carbon surface. This gives rise to highly dispersed LDH nucleation sites.
Except for CD and amorphous carbon, other nanocarbons, such as G, CF, and CNT, need to be oxidized before the reaction due to their chemical inertness and hydrophobicity. Oxygenated functional groups are introduced through different procedures. GO is produced via oxidative exfoliation of graphite using the Hummers or modified Hummers method [27]. The functionalized surface of GO allows the nucleation and growth of LDH by electrostatic forces between GO and the LDH cation precursors [20,21,32,33,37,38,39,40]. Treatment of GO with citric acid (CA) leads to hierarchical LDH/GO composites containing completely exfoliated GO, supporting small LDH particles vertically oriented, due to the complexation effect of citrate on the LDH nucleation sites [35,39,41]. The preparation of nanocomposites based on CA-assisted synthesis is schematized in Figure 1b, with several key features at each main step favoring the formation of nanosheet array-like structures. Addition of the mixed base solution up to pH 10 to the CA-GO suspension induced deprotonation of the –COOH groups of CA (pKa1 = 3.13, pKa2 = 4.76, pKa3 = 5.40) and a decrease in the negative surface charge of GO. This led to hydrogen bonding between the carboxyl groups of the citrate species and the OH groups of GO. The dispersibility of GO was enhanced due to electrostatic repulsion among the negatively charged CA-functionalized sheets. When the mixed metal cations and the alkaline solutions were simultaneously added into the CA-GO suspension, the metal cations were electrostatically bonded to the citrate species, leading to highly dispersed nucleation sites and LDH sheets. The LDH nuclei presented faster growth in the a,b than in the c direction, resulting in plate-like LDH crystals, oriented vertically and covering both sides of the GO surface, preventing their restacking.
Functionalization of CNT goes from oxidation by sonication or reflux in a strong acid solution to the adsorption of organic molecules or coating with a polymer, e.g., polyacrylic acid (PAA), introducing carboxyl groups [42,43,44,45,46,47]. CNT functionalization followed by the addition of an organic linker efficiently immobilized the LDH cation precursors on the surface. For instance, L-cysteine (L-cys) has been added to functionalized PAA-CNT. The LDH was then coprecipitated on the L-cys-PAA-CNT materials, leading to the nanocomposite. L-cys contained on the surface of PAA-CNTs can interact with the cations, forming the LDH, acting as a bridging linker between LDH and CNT [47].
The presence of L-cyst led to the coprecipitation of a highly dispersed LDH phase on the CNT and inhibited the crystal growth of the LDH compared to single-functionalized PAA-CNT. This strategy can be applied to obtain highly dispersed metallic NPs derived from the LDH over CNT. In contrast, studies involving either CNF or CF report simple oxidation by refluxing in concentrated nitric acid [14,48].
Coprecipitation of LDH on the nanocarbon has been performed following different approaches. The metal precursor salts of the LDH can be added to the carbon material (GO, functionalized CNT, CD) with subsequent addition of the alkaline solution [20,37,38,47,49]. When LDH is precipitated on CNT, the particles nucleate and grow within the open framework of CNT, as well as separated domains. Due to the different geometry of the two components (1D vs. 2D), the contact area between the particles is relatively small.
Intimate contact has been found between LDH and GO particles in LDH/rGO nanocatalysts, where the LDH nanoplatelets are generally disposed in plane with the rGO surface, except for high LDH loadings, where the nanoplates grow perpendicularly. Separate aggregated LDH domains can be also observed depending on the loading and preparation route.
Previously oxidized CNF have also been used as a substrate for the precipitation and growth of LDH through incipient impregnation of the Mg2+ and Al3+ salt precursors and an alkaline solution, followed by hydrothermal treatment (50 °C for 24 h). The LDH platelets presented a lateral size of ca. 20 nm supported on the CNF, with a loading of 11 wt% [50,51,52].
In many other cases, the nanocarbon was first dispersed in the alkaline solution and the metal salt solution was subsequently added [39,40,42,44,53]. It led to well-dispersed and non-aggregated LDH nanosheets over the nanocarbons, particularly when GO contained citrate complexing groups, where nanoarray-like structures were observed. The different hierarchical structures induced by the order of addition of the metal cation salts and the alkaline solutions account for different supersaturation levels during synthesis.
The simultaneous addition of the metal-containing salt solution and the alkaline solution at constant pH into a suspension containing the well-dispersed nanocarbon (GO, CF, CNT) has been also reported [33,43,48]. In such cases, the carbon support was completely wrapped by well-dispersed LDH nanoflakes oriented vertically. Contrarily, a single-drop method, where the base solution was added dropwise to a mixture of GO and Ru, Co, and Al chloride salt solution, led to nanocomposites with parallel orientation of the LDH platelets on GO (Ru/LDH-GO-P) [54]. The high local supersaturation level in the former case favored the nucleation and perpendicular growth of the LDH platelets. Contrarily, in the single-drop method, the low supersaturation favored growth and led to parallel orientation of the LDH platelets onto GO with higher interaction.
LDH can also be coprecipitated on GO or N- and S-doped GO using urea or thiourea as a hydrolysis agent under hydrothermal conditions. Rohani et al. reported a catalyst with hollow inner and mesoporous hierarchically flower-like outer structures using a template-free method (Figure 1c) [36]. The preparation process was based on the synthesis of CoAl-LDH@GO hollow spheres previously reported involving Ostwald ripening by coprecipitation of the Co and Al metal salts with urea and ammonium fluoride within a GO suspension in ethanol [55]. The obtained hollow CoAl-LDH@GO was then dispersed in thiourea as source of N and S dopants and hydrothermally treated (180 °C for 12 h) to obtain CoAl-LDH@N,S-G hollow spheres. Finally, the Pd NPs were introduced to obtain CoAl-LDH@N,S-G@Pd hollow spheres. During the process, most of the Pd (II) was reduced to Pd (0) due both to the presence of reductive Co (II) in the LDH and of the electron rich N,S–G. SEM images show a spherical flower-like architecture of the assembly of CoAl-LDH and G with a dense outer part and a hollow inner part (Figure 1c) of the hierarchical CoAl-LDH@N,S-G@Pd. HRTEM confirmed the presence of (012) planes of CoAl-LDH and a good combination between Pd NP and G sheets of ~2 nm thickness. These structural features, creating high surface areas, also improved the dispersion of Pd NPs, electronically enriched due to the interaction with the basic LDH support.
The slow urea hydrolysis maintains low supersaturation during the coprecipitation and a lower nucleation than growth rate, leading to LDH materials with large crystallites and a homogeneous particle size distribution [56,57,58]. Urea hydrolysis also gives rise to GO reduction to G [32].
The influence of the synthesis route on the final structure and features of the composites was noticed in the work of Álvarez et al., which studied two series of MgAl-LDH/rGO with 0.5 ≤ LDH/rGO ≤ 10 mass ratio by the direct coprecipitation or self-assembly of MgAl-LDH and rGO. The LDH/rGO samples were dried either in static air at 80 °C or by freeze drying (Table 2, entry 7) [20].
The LDH:rGO ratio, the preparation, and the drying methods influenced the crystallinity of the composites, which was higher in the self-assembled than in the coprecipitated and freeze-dried samples. Intimate contact between LDH and rGO components occurred in all samples. However, coprecipitation led to more homogeneous LDH/rGO composites, with LDH nanoplatelets markedly smaller than those obtained by self-assembly. Moreover, drying at 80 °C developed more ordered structures by hydrogen bonding between both LDH and rGO building blocks than freeze drying, where restacking of LDH and rGO layers was avoided. The self-assembled samples exhibited, in general, a heterogeneous structure regardless of the drying method.
Differently from coprecipitation or self-assembly methods, LDH/CF composites have been prepared through the reconstruction of an LDO dispersed on pyrolyzed poly(vinyl alcohol) (PVA)/poly(ethylene oxide) (PEO) fibers obtained by electrospinning. In this case, MgAl-LDH precursors were directly introduced throughout the microstructured fiber matrix, whose confined space limited the LDH crystal growth, although this can also reduce their accessibility. The PVA/PEO/MgAl fibers were obtained from a PVA/PEO/MgAl precursor aqueous solution following three consecutive steps: (i) PVA/PEO/MgAl fiber templating by electrospinning of the precursor solution; (ii) carbonization of the PVA/PEO/MgAl fibers at 450 °C, leading to Mg(Al)O mixed oxide supported on carbon fiber; (iii) hydration in the gas phase leading to the reconstruction of Mg(Al)O into MgAl-LDH and the formation of the LDH/CF composite (Table 2, entry 3) [59]. Pyrolysis and rehydration resulted in the formation of agglomerated LDH particles both over the surface and within the network formed by the fused CNF. HRTEM and XRD analyses confirmed the formation of a crystalline layer of approximately 10–20 nm thickness containing LDH crystallites of approximately 4–8 nm (5–10 layers).
4. Applications of LDH/Nanocarbon Composites in Heterogeneous Catalysis
4.1. Base and Multi-Step Reactions
A series of publications reported the use of MgAl-LDH NPs deposited on various nanocarbons (CNF, MWCNT, or GO) as solid base catalysts for the self-condensation of acetone, the single-stage synthesis of MIBK, the Claisen–Schmidt condensation of acetophenone and benzaldehyde, the Knoevenagel reaction, and the transesterification of glycerol with diethyl carbonate [13,14,20,37,44,59,60,61].
The preparation method, activation procedure, and experimental conditions used in the different reactions considered in this review are summarized in Table 2.
4.1.1. Self-Condensation of Acetone and Single-Stage Synthesis of MIBK
The pioneering work of Winter et al. reports catalysts based on MgAl-LDH particles of controlled size supported on CNF to improve the number of accessible active sites and the LDH’s mechanical strength [13,14]. The MgAl-LDH/CNF composite was used as a base catalyst for the self-condensation of acetone to diacetone alcohol (DAA), the single-stage synthesis of MIBK, and the condensation of citral and acetone to pseudoionone. The MgAl-LDH/CNF nanocatalysts were activated at 500 °C, followed by rehydration under decarbonated water-saturated N2 flow, providing highly efficient base sites [50,51,52,70,71,72].
The small lateral dimension (21 nm) of the LDH platelets in the LDH/CNF catalysts increased the number of active sites situated at the edges, improving by approximately four times the catalytic activity in the self-condensation of acetone if compared with the unsupported LDH. The number of basic sites correlated well with the initial specific activity, confirming the influence of reducing the size of the LDH platelets. Selectivity to DAA was higher than 98%. Reactivation of the used catalyst by heat treatment and rehydration removed the adsorbed side products and restored the activity without leaching of the LDH phase.
Winter et al. also reported the preparation of a multifunctional Pd-MgAl-LDH/CNF catalyst, containing acid, base, and hydrogenating sites required for the single-stage synthesis of MIBK from acetone [14]. The as-synthesized LDH/CNF impregnated with Pd(acac)2 was then heated at 500 °C and reduced at 250 °C, and was rehydrated to obtain Brønsted basic sites, favoring the condensation of acetone with respect to Lewis basic sites (Table 2, entry 2).
The initial activity of the activated Pd-LDH/CNF was five times higher than that of a physical mixture of LDH and Pd/CNF. The formation of DAA was faster than the dehydration of DAA to mesityl oxide (MO), which was almost not observed, being rapidly hydrogenated to MIBK. The sum of selectivities into DAA, MO, and MIBK was higher than 99%.
These were the first reports on LDH/CNF-supported catalysts that confirmed that the catalytically active sites are situated at the edges of the LDH platelets and their amount greatly increases when the platelets are well-dispersed on a support.
Commercially available MWCNT were used by Celaya-Sanfiz et al. as supports for LDHs [44]. MWCNT have several advantages over CNF, particularly a higher aspect ratio, smaller diameter, and enhanced mechanical strength.
Coprecipitation of MgAl-LDH into a dispersion of previously oxidized MWCNT led to the MWCNT/MgAl-LDH nanocomposite with LDH loading ranging from 33 to 83 wt%. Activation was performed by calcination and rehydration in the liquid phase. The catalysts presented LDH particles disposed within the open structure of the MWCNT, decreasing the specific surface area from 230 m2 g−1 of bare MWCNT to 119–85 m2 g−1 depending on the LDH loading. The LDH crystallite sizes of the composites were slightly smaller than in the bulk LDH, and, apparently, they decreased with the increasing MWCNT content. However, larger LDH platelets were observed after hydration (ca. 62 nm) than for the as-synthesized MWCNT/LDH (ca. 30 nm). Álvarez et al. reported a similar trend for hydrated MgAl-LDH/CNF, assigned to dissolution/recrystallization during hydration in the liquid phase [60]. The basicity of the MWCNT/LDH ranged from 1.6 to 2.4 mmol gLDH−1 of CO2 desorbed for the composites with LDH content of 33–83 wt%, which is higher than the values reported previously in LDH/CNF samples, obtained by CO2 adsorption, with lower LDH loadings (11 to 20 wt%) [14,59,60].
The self-condensation of acetone at 0 °C with the hydrated MWCNT/LDH composites led to DAA only. There was an almost threefold increase in the initial reaction rate (V0) from ca. 71 to ca. 196 mmolDAA gLDH h−1 when going from the mere activated LDH to the MWCNT/LDH catalyst containing 83 wt% LDH. V0 decreased greatly with the MWCNT loading up to 9.8 mmolDAA gLDH h−1 at 33 wt% LDH. This value can be compared to V0 of 542 mmolDAA gLDH h−1 reported by Winter et al. for an LDH loading of 11 wt% [14]. Notably, in this latter case, the V0 of the LDH/CNF catalyst was fourfold higher than that of bulk LDH. Compared to LDH/CNF, the catalytic improvement of MWCNT/LDH is less important despite its higher LDH content. This accounts for the presence of carboxylated carbonaceous fragments generated during the MWCNT oxidation, which inhibits the active sites.
Álvarez et al. were the first to investigate the LDH/graphene-like composites as acid–base catalysts for C–C bond reactions, particularly the self-condensation of acetone [20]. Two series of MgAl-LDH/rGO with 0.5 ≤ LDH/rGO ≤ 10 mass ratio were prepared by either direct coprecipitation or self-assembly. In addition, both series of LDH/rGO samples were dried either in static air at 80 °C or by freeze drying (Table 2 entry 7).
After activation at 450 °C, all composites exhibited the XRD pattern of rGO at low LDH loading and of Mg(Al)O mixed oxide at high LDH loading; meanwhile, HRTEM images revealed similar morphologies and structural features to the non-calcined parent samples. The surface areas of the activated composites with LDH:rGO = 10 were in the range of 170–240 m2 g−1.
Only DAA was obtained with all the activated LDH/rGO catalysts, with a clear influence of the LDH/rGO mass ratio on the activity, from 19.8 to 104.7 mmol gLDH−1 for LDH/rGO mass ratios of 0.5 and 10, respectively. Only when LDH/rGO ≥5, the activity was higher than that of the bulk LDH (51.2 mmol gLDH−1), with a maximum at LDH/rGO = 10, achieving the best compromise between the number and accessibility of sites. The composite prepared by coprecipitation and freeze drying exhibited remarkable activity, leading to a DAA amount of 104.7 mmol gLDH−1 after 8 h, compared to around 70–80 mmol gLDH−1 for LDH/rGO prepared by self-assembly and either dried in air or freeze-dried. This can be related to the reduced lateral dimension of the LDH platelets (30–40 nm) and the disordered structure of the composite, with poorly stacked rGO and LDH sheets in weak interaction, which enhanced the accessibility to the active sites. Contrarily, those samples with lower LDH content exhibited poor activity significantly lower than that of the bulk calcined LDH. This was attributed to their low number of active sites and to the layer-by-layer hybridization between the LDH layers and the rGO sheets, hindering accessibility to the active sites.
Adsorption of CO2 on both series of samples with amounts from 63 to 157 µmol gcat−1 for LDH/rGO-0.5 and from 361 to 407 µmol gcat−1 for LDH/rGO-10 confirmed that the improved catalytic activity accounted for the LDH loading. Moreover, a clear correlation was found between the global rate of reaction and the number of stronger basic sites of the LDH/rGO series.
The basicity, determined by the adsorption of CO2, and the catalytic activity in the self-condensation reaction of acetone for the different MgAl-LDH/nanocarbon catalysts are compared in Table 3. All the reactions were performed at 0 °C with a similar LDH/acetone weight ratio of ~1.10 for CNF and MWCNT supports, and of 0.36 for rGO.
The number of basic sites varied in a large range, pointing to the influence of the loading and particle size of the LDH component and the structure of the carbonaceous support, which led to different accessibilities to the active sites in the composites. The unexpectedly different number of basic sites found between the two LDH/CNF samples with the same LDH loading and particle size probably resulted from the different conditions of CO2 adsorption performed at 0 and 25 °C, respectively. The higher number of basic sites of MWCNT/LDH compared to LDH/rGO having similar LDH loading (80–90 wt%) and particle size (~50 nm) typically accounts for the lower accessibility to the sites in the case of rGO due to its intimate contact with the LDH sheets. The LDH particles are, on the contrary, well exposed within the open framework of the MWCNT, acting as a supporting scaffold. The reaction rates are difficult to confirm as the values were calculated over very different times, but a global tendency can be underlined regarding the efficiency of the supports as CNF > MWCNT > rGO. However, the absence of a correlation between the basic properties and the catalytic activity among the different materials confirms that accessibility to the sites is a determining parameter.
4.1.2. Claisen–Schmidt Reaction (Synthesis of Chalcone)
LDH/rGO composites were investigated in the Claisen–Schmidt condensation of acetophenone and benzaldehyde to chalcone [37]. A wide range of basic catalysts were used for this reaction, but selectivity to chalcone is generally low because of the side reactions, such as Michael addition [73,74,75,76,77,78,79]. A series of catalysts with 0.5 ≤ MgAl-LDH/rGO ≤ 20 mass ratio was prepared by coprecipitation and drying in air at 80 °C, as previously reported (Table 2, entry 7) [20].
The number of basic sites of the activated catalysts, evaluated by the amount of adsorbed CO2, increased from 52 to 265 µmol gcat−1 when the LDH content increased. For LDH loadings higher than 80 wt%, the amount adsorbed was similar to or higher than in the bulk activated LDH (128 to 265 µmol gcat−1 compared to 129 µmol gcat−1, respectively), all exhibiting similar specific surface areas (~280 m2 g−1). Thus, dispersion of the LDH nanoparticles on the rGO surface increased the number and accessibility toward highly basic O2- sites. Accordingly, conversion reached 100% after 4 h with activated LDH/rGO catalysts when the condensation of acetophenone and benzaldehyde was carried out at 40 °C in the presence of either polar protic (methanol, MeOH), polar aprotic (acetonitrile, ACN), or non-polar (toluene) solvents or neat conditions. Bare activated LDH needed 8 h to complete the reaction.
The LDH/rGO ratio and the nature of the solvent greatly influenced the distribution of products. Chalcone, with yields in the range from 65 to 100%, was the main product using ACN, toluene, and in neat conditions. The weak acidity of ACN poisoned the stronger basic sites of the catalyst, inhibiting the Michael addition. Meanwhile, in solventless conditions, a Michael addition product was formed with ca. 20% selectivity along with c,t-chalcone (ca. 80% selectivity) due to the stronger basic sites, which initiated a side reaction.
With MeOH only, c,t-chalcone with yields in the range from 20% to 75% and an aldol product were formed. The Michael addition was totally inhibited due to the acidity of MeOH being slightly higher than that of ACN. Toluene led to the higher yield of c-chalcone and the Michael addition product, which was likely related to its very weak acidity and the different adsorption of the reactants on the catalyst surface.
4.1.3. Knoevenagel and One-Pot Oxidative Knoevenagel Reactions
Recently, Ce-containing MgAl-LDH-GO composites bearing different GO loading (5–25 wt%) were tested as catalysts in the Knoevenagel condensation of benzaldehyde with dimethyl malonate to diethyl benzylidene malonate (DBM) and subsequently to cinnamic acid (CNA), and the one-pot cascade oxidation–Knoevenagel condensation of benzyl alcohol (BA) and benzoyl acetonitrile to yield 2-benzoyl-3-phenylacrylonitrile (BPA) [61]. The Ce-MgAl-LDH/GO composite prepared by coprecipitation (Table 2, entry 9) presented the typical structural features corresponding to both LDH and GO. The acidity and basicity measurements showed a synergistic interaction between the Ce-containing MgAl-LDH and GO rather than a simple additive effect. Thus, while Ce-MgAl-LDH solid showed a mainly basic and GO a mainly acidic character, the composite presented both basic and acidic sites. Their numbers passed through a maximum for 15 wt% of GO (2.98 mmolacrylic acid g−1 and 0.46 mmolpyridine g−1, basic and acid sites, respectively).
The conversion of the mere solids Ce-MgAl-LDH and GO (8% and 5% after 5 h, respectively) was rather low compared to the composites. The catalytic activity, in terms of aldehyde conversion, of the composites increased with the GO content up to an optimum for 15 wt% (24% and 76% after 5 h and 24 h, respectively), in line with the acidity and basicity results. Then, it decreased for higher GO content. The yield to CNA, with a maximum of ca. 45% after 24 h of reaction for the catalyst containing 15 wt% of GO, followed the same trend.
The ratio between the basic and acid sites, associated with the LDH and GO components, respectively, was a key factor determining the product distribution, since the basic sites are involved in the proton abstraction from the α-position of diethyl malonate to undergo nucleophilic addition to the carbonyl group of the benzaldehyde yielding DBM. However, selectivity to DBM decreased with the basicity. This is assigned to the favored decarbethoxylation of DBM by the basic sites, first leading to the ethyl cinnamate ester intermediate. At the same time, conversion of the latter into CNA occurs by the acid sites. Indeed, it was observed that the pure GO phase (essentially acid) led to high selectivity to CNA (99%), indicating a key role in its production in the composites. Again, the catalyst with 15 wt% GO presented the highest values of both total acidic and basic sites, but also showed the highest basic-to-acidic site ratio.
In the case of the cascade reaction, the conversion of BA was higher for the Ce-MgAl LDH-GO composites compared to the LDH and GO materials alone, the latter being completely inactive. BA conversion decreased with the increase in the GO concentration, which was attributed to a decrease in the number of accessible redox active sites needed in the first step of the reaction due to GO restacking at higher content. However, with lower GO content, the accessibility to the redox sites was increased by separation of the LDH particles, which favored the oxidation of BA.
4.1.4. Transesterification of Glycerol
The Mg(Al)O mixed oxide and meixnerite-like compound (OH-MgAl-LDH), obtained upon rehydration in the liquid or gas phase of Mg(Al)O, presented high efficiency in the base-catalyzed transesterification of glycerol with diethyl carbonate (DEC), leading to glycerol carbonate [80,81]. This reaction was also studied using MgAl-LDH/CNF with LDH nanoparticles of controlled size (20 nm), prepared following the protocol of Winter et al., with LDH content of ca. 12 wt% and activated either by calcination at 500 °C (LDH-CNFc) or by calcination followed by rehydration in the liquid phase (LDH-CNFrl) or gas phase (LDH-CNFrg) (Table 2, entries 5 and 6) [60].
The LDH-CNFc was the most efficient catalyst, showing a V0 of 53 mmol Gly g−1HT h−1 better than LDH-CNFrl and LDH-CNFrg (3 and 7 mmol Gly g−1HT h−1, respectively) and by far higher than bare calcined LDH (0.18 mmol Gly g−1HT h−1). Accordingly, total glycerol conversion was obtained after 1.5 h for LDH-CNFc but after 3 h for LDH-CNFrg and 8 h for LDH-CNFrl; meanwhile, the calcined LDH (LDHc) needed 22 h for total conversion. The nearly four times higher number of basic sites in LDH-CNFc than in the bulk catalyst (LDHc) cannot account for its 300 times higher initial reaction rate. This indicates different adsorption of the reactants depending on the different polarity of the catalysts. The heat-treated CNF is less polar than the bulk LDH and this improves DEC adsorption and the reaction rate. Likewise, the different behavior of the rehydrated samples can be explained by their different content of physisorbed and interlayer water.
The LDH-CNFc catalysts exhibited good stability, with slight decrease in glycerol conversion after three runs, without leaching of the active phase.
The transesterification of glycerol with DEC was also studied with catalysts formed by MgAl-LDH NP supported on CNF prepared by electrospinning [59]. The loading of LDH in the composite was ca. 20 wt%.
Glycerol transesterification with MgAl-LDH/CF was threefold higher than that of bare MgAl-LDH (5.6 × 10−3 vs. 1.7 × 10−3 mmol Gly g−1 HT s−1, respectively). This is consistent with their different numbers of basic sites located at the edges of the platelets. However, the activity of MgAl-LDH/CF (5.6 × 10−3 mmol Gly g−1 HT h−1 after 10 h) was by far lower than that of the MgAl-LDH/CNFrg catalyst (7 mmol Gly g−1 HT h−1 after 1 h) reported by Álvarez et al., both studies being performed in similar conditions [60]. The different behavior of MgAl-LDH/CF and MgAl-LDH/CNFrg catalysts both rehydrated in the gas phase can be related to their physico-chemical properties. LDH loading in MgAl-LDH/CF was slightly higher than in MgAl-LDH/CNFrg (20 vs. 12 wt%) and the mean crystallite size was also smaller in the former catalyst, two features able to improve the catalytic efficiency. However, the specific surface area was significantly higher for MgAl-LDH/CNFrg (164 m2 g−1) than for MgAl-LDH/CF (15 m2 g−1) due to the different textures of the CNF, being of lower porosity when prepared by electrospinning. Consistently, the amount of adsorbed CO2 was higher by a factor of 1.8 in MgAl-LDH/CNFrg (0.37 mmol g−1LDH) than in MgAl-LDH/CF (0.21 mmol g−1 LDH) and in both cases higher than in the bulk rehydrated MgAl-LDH (0.13 mmol g−1LDH). Therefore, despite the potentially higher number of active sites located at the edges of its smaller particles, MgAl-LDH/CF was found to be less active due to the lower accessibility of the particles embedded in the carbon fibers prepared by electrospinning. Moreover, covering of the LDH particles by coke provided by the carbonization process cannot be ruled out.
4.1.5. Ullman, Sonogashira, and Heck Reactions
Ahmed et al. investigated CuAl-LDH and CoAl-LDH supported on GO as catalysts for the classical Ullmann reaction [21]. The CuAl-LDH/GO composite (Cu:Al = 2:1; LDH:GO = 20:1) was prepared by coprecipitation of the CuAl-LDH onto an aqueous GO dispersion. Meanwhile, the CoAl-LDH/GO composite (Co:Al = 2:1; LDH:GO = 20:1) was prepared via a urea-mediated coprecipitation method (Table 2 entry 11). The LDH content was 97 wt% in both composites and the XRD patterns were typical of LDH, while GO was considered well exfoliated. Crystallite sizes (c direction) were around 19 and 43 nm for CuAl-LDH/GO and CoAl-LDH/GO, respectively. SEM images showed lateral particle sizes of 100 nm for CuAl-LDH. Consistently, they were larger (up to 8 µm) with a hexagonal shape for CoAl-LDH synthesized by the urea method. The specific surface area was higher for CuAl-LDH/GO (44 m2 g−1) than for CoAl-LDH/GO (17 m2 g−1). It increased by around 60–70% compared to the bare LDHs upon introduction of 3 wt% GO, with the creation of mesopores of 2–3 nm.
The catalysts’ activity in the condensation of iodobenzene to biphenyl was CuAl-LDH > CoAl-LDH > CuAl-LDH/GO > CoAl-LDH/GO. Remarkably, these results were obtained without the addition of a reducing agent, contrarily to usual operating conditions. The slightly lower activity of the LDH/GO catalysts compared to their bulk counterparts was attributed to a restacking of the GO layers in DMSO. In contrast, GO in the composite catalysts greatly improved their stability over reusability cycles. Thereby, the biphenyl yield dropped by around 30% after five reaction cycles with the LDH/GO catalysts, whereas it dropped by around 55% and 72% with CuAl-LDH and CoAl-LDH samples, respectively.
Wang et al. designed hierarchically structured nanoarray-like catalysts Pdx/rGO@CoAl-LDH for the Heck reaction using a lattice atomic-confined in situ reduction strategy of Pd2+ induced by the well-dispersed Co2+ atoms in the LDH layers [53]. CoAl-LDH provides both reductive Co2+ sites able to form highly dispersed Pd0 NP and basic sites able to increase their electron density. Moreover, graphene enhances electron conductivity at the same time, which provides high mechanical strength and a large surface area, inducing high dispersion of LDH nanoplates and improved adsorption of aromatic reactants via π–π interactions.
The catalysts presented a hierarchically structured nanoarray-like morphology with LDH nanoplates of ~65 × 7.5 nm grown perpendicular to both sides of the rGO layers. Pd NPs (<2 nm) were highly dispersed and preferentially located at the edges of the LDH nanoplatelets due to the interaction of the Pd precursor and the pending OH groups, suggesting strong metal–support interaction (SMSI).
Based on the time (t) required to reach the maximum conversion (C) and TOF values, hierarchical Pd0.6/rGO@CoAl-LDH (t = 20 min, C = 98.1%, TOF = 981 h-1) presented the highest catalytic activity in the Heck reaction between iodobenzene and styrene compared to non-hierarchical Pd0.6/rGO@CoAl-LDH-h (t = 50 min, C = 98.2%, TOF = 393 h-1) and control catalysts Pd0.92/CoAl-LDH (t = 60 min, C = 95.0%, TOF = 317 h-1) and Pd2 + 0.78/GO (t = 270 min, C = 98.1%, TOF = 73 h-1) (Figure 2a). The rGO layers largely contributed to the activity, and the hierarchical composite improved it due to its structured nanoarray-like morphology, which allowed a reduction in the Pd NP size and enhancement in their dispersion. It also provided more accessible active sites (Figure 2b).
When the Heck reaction was performed with the series of hierarchical Pdx/rGO@CoAl-LDH (x = 1.9, 1.2, 0.6, 0.33, 0.0098), the TOF increased from 846 h-1 to 2982 h-1 when Pd loading decreased from 1.9 to 0.33 wt%, in line with a concurrent increase in dispersion. The hierarchical Pd0.0098/rGO@CoAl-LDH catalyst with ultrasmall Pd NPs (1.3 nm) greatly dispersed exhibited a TOF value of 160,000 h-1 with 16.0% of iodobenzene conversion after 1 h, which is considerably higher than the values previously reported with Pd-based heterogeneous catalysts [53].
Moreover, the hierarchical composites showed a broad range of substrate applicability, having conversions higher than 94.3% within 40 min in reactions between varied substituted aryl halides with styrene and its derivatives. The catalysts could be reused in more than five runs without noticeable loss of activity.
Hierarchical flower-like CoAl-LDH@N,S-G@Pd catalysts were evaluated in the Sonogashira alkynylation reaction between a variety of aryl halides and phenylacetylene with optimized conditions (solvent, temperature, and base) [36]. The Pd and N,S-G loading was ~1.20 wt% and ~5 wt%, respectively. XPS analysis identified Co, Co2+ in CoAl-LDH structure, and Pd0, Pd2+ species in the CoAl-LDH@N,S-G@Pd composite.
Product yields higher than 90% were obtained in the coupling reaction of both electron-deficient and electron-rich aryl iodides with phenylacetylene, although completion of the reaction was reached after 3 h with the methyl substituent. The coupling was also effective with substituted aryl bromides and aryl chlorides, but longer reaction times were needed to achieve completion. This hierarchical catalyst presented activities comparable with other Pd-containing catalysts reported, with the advantages of a shorter reaction time, mild reaction conditions, high product yield, and smaller amount of Pd [36].
The efficiency of the CoAl-LDH@N,S-G@Pd composite can be ascribed to its mesoporous spherical flower-like architecture (Figure 1c) with a hollow structure, which facilitates the diffusion of the reactants to the Pd sites, also favored by the high dispersion and stabilization of the Pd NPs by the well-ordered Co(II) in the LDH sheets. Moreover, the π-π stacking of G with aromatic substrates makes the reactants more accessible to Pd sites, and the electron enrichment of Pd by the basic LDH and the N,S-G increases the rate of the oxidative addition step.
The absence of Pd leaching from CoAl-LDH@N,S-G@Pd showed that the reaction was purely heterogeneous. Moreover, the catalyst was highly recyclable. A decrease of only ~12% in the product yield in the model reaction was observed after six catalytic runs.
4.1.6. One-Pot and Cascade Reactions
A magnetic Fe3O4@GO@Zn-Ni-Fe-LDH system showed high efficiency in the one-pot Knoevenagel–Michael reaction between 4-hydroxycoumarin and a variety of benzaldehydes substituted with different electron-donating and electron-withdrawing groups [33]. The biscoumarin compounds were obtained in 87–95% yield within 30–40 min, showing the good activity of the catalyst. The condensation of 4-hydroxycoumarin and benzaldehyde with the Fe3O4@GO@Zn-Ni-Fe-LDH catalyst gave 95% yield of 3-3′-benzylidenebis(4-hydroxycoumarin) in 3 min, the highest performance compared with other catalytic systems such as CuO-CeO2 [82], SiO2-OSO3H NP [83], or phosphotungstic acid [84], which led to 93–94% yields in 12–30 min. GO provides a substrate for the formation of well-dispersed ZnNiFe-LDH platelets with acid–base properties and highly accessible sites required for the targeted reaction. After magnetic separation of Fe3O4@GO@Zn-Ni-Fe-LDH from the reaction media and washing, the catalyst can be reused up to five times, with a small loss in the yield of 3-3′-benzylidenebis(4-hydroxycoumarin) from 95% to 87%.
Zhang et al. have prepared nanocomposites based on RuCoAl-LDH and GO following either single-drop addition or simultaneous addition at constant pH for the cascade reaction between cinnamyl alcohol and ethyl cyanoacetate to produce cynnamilidene ethyl cyanoacetate [54].
The single-drop method led to the parallel orientation of the LDH platelets on the surface of GO (Ru/LDH-GO-P). Meanwhile, simultaneous addition led to the perpendicular orientation of the LDH platelets on GO (Ru/LDH-GO-V).
Ru/LDH-GO-P, with 99% yield of cinnamylidene ethyl cyanoacetate, was significantly more active than Ru/LDH-GO-V, with 48% yield (60 °C under O2; 3 h). Ru/LDH-GO-P presented the highest amount of weak and medium–strong basic sites. The higher catalytic efficiency of Ru/LDH-GO-P was also in line with its higher abundance of O22− and O2− species detected by O2-TPD, whose formation was promoted by the higher amount of Co3+ species and the higher specific surface area compared to Ru/LDH-GO-V. Two control catalysts, Ru/LDH-sd and Ru/LDH-cp, obtained by single-drop and coprecipitation methods, respectively, were poorly active (30% and 14% yield, respectively), showing that the presence of GO in the nanocomposites improves the dispersion of the active sites of the LDH nanosheets. The physical mixtures of the three components also were less active (<36% yield) than the corresponding nanocomposites. Ru/LDH-GO-P could be reused at least five times without an obvious decrease in the activity.
Interestingly, the Ru/LDH-GO-P catalyst showed high efficiency in the one-pot oxidation–Knoevenagel condensation reactions involving benzyl alcohols substituted with either an electron-withdrawing (NO2, Cl, Br) or an electron-donating group (CH3, CH3O) and active methylene groups (ethyl cyanoactate or malonitrile). Product yields were in the range of 91 to 99% within 2–3 h.
The different LDH/carbon nanocatalysts with hierarchical structures involved in the Ullmann, Sonogashira, Heck, and one-pot reactions are summarized in Table 2, entries 11–15.
It is noteworthy that the hierarchical nanocomposites combining transition metal-containing LDH, particularly CoAl-LDH, and Pd, with GO or rGO show remarkable efficiency for the achievement of a wide range of C–C coupling, such as Ullman, Sonogashira, and Heck reactions. Varying the LDH composition, e.g., RuCoAl-LDH, ZnNiFe-LDH, allows multifunctional catalysts that are able to perform one-pot Knoevenagel–Michael and oxidation–Knoevenagel cascade reactions. This behavior results from several main characteristics of these nanocomposites. The large surface area of the GO or rGO support favors the dispersion, and the number, strength, and accessibility of the most active sites of low coordination located at the edges of the LDH nanoplatelets. Moreover, it is possible to adjust the supersaturation rate during coprecipitation to obtain LDH nanosheets vertically or horizontally oriented on GO. Vertical orientation gives rise to the most efficient catalysts. Defects created by the reduction of GO to rGO during preparation of the nanocomposites are the preferential nucleation sites of the LDH crystallites and concur with their high dispersion. For CoAl-LDH and Pd systems, the presence of Co2+ species in the LDH facilitates the reduction of Pd2+, giving rise to highly dispersed Pd NPs of small size. Moreover, electron transfer occurs from both the LDH nanosheets with basic character and rGO to the Pd NPs. All these features contribute to enhancing the reactivity of Pd for oxidative addition. The specific surface area of the nanocomposites reaches up to 180 m2 g−1, accounting for the dispersion of the GO nanosheets due to the presence of LDH decreasing π-π interactions, generally responsible for their stacking. These properties make the Pd-containing nanocomposites more active than the classical catalysts in the C–C coupling reactions. The LDH-based nanocomposites can also lead to bi- and tri-functional catalysts required to achieve one-pot reactions and magnetic separation of the catalyst.
It is important to highlight the high robustness and stability of the LDH/carbon nanocomposites, which can be subjected to five or six cycles of regeneration with a decrease in activity not exceeding 30%.
4.2. Oxidation Reactions
LDH/nanocarbon composites are very attractive for a variety of oxidation reactions because the tunable composition of LDH allows the required catalytic active sites to be obtained, particularly efficient electron-deficient metal species. Moreover, the intrinsic LDH basicity can induce multifunctional composites. LDH/carbon nanocomposites are also designed for the oxidation of aromatic compounds due to their highly favorable adsorption on the carbon surface through π-π interaction closely to the dispersed active sites. The reactivity will also benefit from the balance between the hydrophobic and hydrophilic character induced by the carbon and LDH components, respectively, the large specific surface areas, and the strong interaction between the components. The preparation methods of the LDH/carbon nanocomposites and the conditions of the oxidation reactions considered are summarized in Table 2, entries 16–22.
4.2.1. Oxidation of Primary Alcohols
The development of highly active and selective catalysts for the solvent-free oxidation of primary alcohols is still challenging. Miao et al. reported for the first time the use of an LDH/graphene composite as a support for Au NPs for the selective oxidation of benzyl alcohol (BA) [40]. Its structure consisted of thin graphene sheets decorated by nanosized LDH particles grown parallel or perpendicular to the surface with an average size ~62 nm and Au NPs with an average particle size of 2.63 nm. Both the oxygenic functional groups and the defect sites on the surface of rGO acted as anchoring sites for the nucleation of LDH and Au NPs, with strong SMSI, high dispersion, and small size.
The BA conversion of Au/NiAl-LDH/rGO achieved in solvent-free conditions (140 °C; P O2: 2 bar) reached ca. 62% after 10 h, which was higher than that of Au/GO (7.1%), Au/NiAl-LDH (51.8%), and the physical mixture of Au/GO and Au/NiAl-LDH (38.2%). Selectivity toward the targeted benzylaldehyde of the Au/NiAl-LDH/rGO and Au/NiAl-LDH catalysts after 10 h reaching 65.2% and 63.3%, respectively, was higher than that of Au/GO (60.6%), showing that the over-oxidation of BA was prevented on less oxygenated rGO than GO. The main by-products were toluene, benzoic acid, and benzyl benzoate, the latter reaching up to 35% selectivity at higher BA conversion through the reaction of benzaldehyde with BA to form hemiacetyl, which was then oxidized. The optimum GO/NiAl-LDH mass ratio of 1:2.8 led to a 40% benzaldehyde yield.
Upon reusing Au/NiAl-LDH/rGO, the conversion of BA dropped by only 10%, with selectivity to benzaldehyde still reaching 68% after the third recycling step. This stability probably results from the strong anchoring of the Au NPs, which prevents their agglomeration, and from the presence of NiAl-LDH, preventing the agglomeration of rGO.
Wang et al. developed an approach aiming to replace noble metal-containing catalysts for the oxidation of BA using CoAl-LDH [32]. Co-based catalysts have demonstrated high efficiency in the oxidation of alcohols. However, the high Co loading required to reach high conversion leads to poorly dispersed active species and low TOF values. Furthermore, the addition of basic promotors is common to improve the activity. Co-containing LDH can provide both highly dispersed Co species and high basicity. Exfoliation of LDH is also an interesting method to improve the accessibility to the active sites. Wang et al. prepared CoAl-ELDH/GO composites achieving first the coprecipitation of CoAl-LDH using the urea method. An aqueous suspension of GO exfoliated by ultrasonication was then added into the suspension of CoAl-LDH nanosheets previously exfoliated in formamide (ELDH) (Table 2, entry 17). Both ultrathin ELDH nanosheets and GO nanosheets with apparent thickness in the range of 2.4–3.3 nm and 1.1–1.2 nm, respectively, were observed in the CoAl-ELDH/GO composite, with Co/Al = 1.6 and GO content of 15.3 wt%.
CoAl-ELDH/GO was significantly more active (92.2% conversion) than CoAl-LDH (37.3%), GO (10.7%), and their physical mixture (51.9%) in the BA oxidation (DMF; 120 °C; 4 h). The conversion reached with the CoAl-LDH and GO physical mixture corresponds to the sum of the values of the two components, showing that there is no synergetic effect.
Benzaldehyde was the main product of the reaction, with similar selectivity of 99.2% for CoAl-ELDH/GO and the bulk CoAl-LDH, while it decreased to 91.5% for GO with the formation of significant amounts of benzoic acid (5.9%) and benzyl benzoate (2.5%). These results revealed the predominant role of CoAl-LDH in the catalytic performance, while GO acted as a poorly active support. The positive effect of the dispersion of CoAl-LDH on GO is evidenced by the TOF value, being five times higher for CoAl-ELDH/GO (1.14 h−1) than for the bulk CoAl-LDH (0.23 h−1).
Investigation of the surface defects, local atomic arrangement, and electronic structure revealed that the Co-OOH and Co…Co distances remained unchanged but that the coordination number decreased significantly in CoAl-ELDH/GO in comparison to the bulk CoAl-LDH. This suggests that coordinatively unsaturated CoO6−x octahedra were formed in the ultrathin nanosheets of the composite with the generation of Co and O vacancies. The positron annihilation spectra (PAS) of CoAl-ELDH/GO showed the presence of larger amounts of Co vacancies and of negatively charged VCo-Co-OHδ− sites than in the bulk CoAl-LDH, with more lattice oxygen atoms exposed. Accordingly, both the strength and density of the basic sites of CoAl-ELDH/GO were improved with respect to bulk CoAl-LDH. The mobility of oxygen species was higher in the CoAl-ELDH/GO than in the bulk CoAl-LDH. This was consistent with the higher number of oxygen vacancies and surface O2− in the composite. The oxygen vacancies together with Co-OHδ− adjacent to Co vacancies (VCo-Co-OHδ−) were found by DFT calculation to be the sites of stronger BA adsorption via its OH group.
The CoAl-ELDH/GO catalyst showed good stability over six runs. Furthermore, it was also highly active and selective in the oxidation of a wide range of other benzylic alcohols.
The authors proposed a mechanism with two possible pathways for the oxidation of BA under molecular oxygen on CoAl-ELDH/GO (Figure 2c). O2 initially adsorbed on the oxygen vacancies captured electrons from adjacent Co2+, giving activated O- and Co3+ species. The O-H group of the BA molecule simultaneously adsorbed on an oxygen vacancy (path I) or a VCo-Co-OHδ− site (path II) was activated, leading to H abstraction and the formation of an unstable metal-alkoxide species. Further, the α-Cδ+-Hδ− bond cleavage on an activated O− site accepting Hδ− led to the formation of benzaldehyde. The catalytic cycle was completed by the oxidation of the hydride by activated O−, with the concurrent reduction of Co3+-O− to Co2+-O− followed by the desorption of H2O.
CD-containing LDH-based nanocomposites are scarcely investigated in the literature. Notwithstanding, several recent papers indicate exciting outlooks for such composites, such as an N-doped CD/CoAl-LDH/g-C3N4 heterojunction photocatalyst and GdDy-LDH assembled with doxorubicin and folate–carbon dots, designed as cancer-targeted therapeutic agents or CD/LDH phosphors [85,86,87].
Interesting papers report nanocomposites with LDH and CD or N-doped CD (NCD) as supports of Au NPs and Ag NPs for heterogeneous catalysts. CD or NCD provide stabilizing and reducing ability, electro-donating capacity improving their basicity, and strong metal–carbon interaction favorable to the reduction of organic water pollutants and oxidation of alcohols [34,49].
Supported Au NPs on a NCD/MgAl-LDH composite (Au/NCD/MgAl-LDH) prepared by the coprecipitation of MgAl-LDH and NCD, and subsequent Au introduction by the deposition–reduction approach, gave rise to catalysts with improved basicity and metal–support interaction (Table 2, entry 18) [49]. Nitrogen atoms incorporated into carbon materials provided basic species inducing a Lewis basicity to the neighboring carbon atoms [88,89].
The Au/NCD/MgAl-LDH composite contained Au0 NPs of approximately 3.46 nm average size, uniformly dispersed and poorly aggregated, with a loading of 0.3 wt%. The highly dispersed NCD component on the surface of MgAl-LDH increased the surface density of stronger basic sites compared to the Au/MgAl-LDH sample. In addition, SMSI was produced in the Au/NCD/MgAl-LDH composite, indicated by BE values of Au0 species lower than for the Au/MgAl-LDH. This is due to the strong coordination of electron-donating N atoms in NCD and Au0 NPs.
The BA oxidation with the Au/NCD/MgAl-LDH composite was conducted without a solvent and the addition of bases in the reaction media, as previously reported for an Au/NiAl-LDH/rGO composite [40].
MgAl-LDH and NCD/MgAl-LDH were poorly active in BA oxidation, leading to conversion of 4.2% and 5.8%, respectively, after 4 h. The conversion increased upon introduction of Au on MgAl-LDH (38.2%) and further with the introduction of NCD (47.3%) with the Au/NCD/MgAl-LDH composite at similar Au loading compared to Au/MgAl-LDH (0.82% and 0.91%, respectively). This suggests that the improvement of basicity promotes the activity of the composite. Consistently, the initial TOF based on surface Au atoms after 0.5 h of reaction greatly increased from 8591 h−1 with Au/MgAl-LDH up to 20175 h−1 with Au/NCD/MgAl-LDH. The role of the basicity was also confirmed when the catalytic results were compared with those previously obtained with the Au/NiAl-LDH/rGO composite [40]. Conversion of BA was indeed 49 and 35% with Au/NCD/MgAl-LDH and Au/NiAl-LDH/RGO, respectively, after 5 h.
Benzaldehyde was the main reaction product, with benzoic acid and benzyl benzoate as by-products. The conversion increased with the Au loading, with a concurrent slight decrease in benzaldehyde selectivity.
Similar selectivity to benzaldehyde of approximately 80%, with benzyl benzoate as the main by-product, was obtained with Au/NiAl-LDH/rGO [40]. However, it must be underlined that Au/NiAl-LDH/rGO exhibited a specific surface area of 172.5 m2 g−1 and Au NP size of 2.63 nm, while they were 61 m2 g−1 and 3.46 nm, respectively, for Au/NCD/MgAl-LDH. These structural features were more favorable to the former catalyst. Moreover, Au/NCD/MgAl-LDH was more active despite the softer reaction conditions used, i.e., 120 °C reaction temperature vs. 140 °C for Au/NiAl-LDH/rGO. The presence of NCD and of MgAl-LDH instead of NiAl-LDH contributes to improving the basicity of Au/NCD/MgAl-LDH in comparison to Au/NiAl-LDH/rGO and, therefore, the catalytic activity in the oxidation of BA.
BA conversion with Au/NCD/MgAl-LDH decreased only by 6.0% after five consecutive runs, instead of 12.4% with Au/MgAl-LDH, showing the high stability of the former composite, whose Au leaching was around 1.0%.
Shan et al. developed a method aiming to obtain highly thermodynamically stable Pickering emulsion using an amphiphilic nanocomposite based on LDH and CNT components. The selective oxidation of BA was studied using an Ru-based LDH-CNT catalyst as a solid emulsifier [42].
LDH-CNT composites were prepared by the coprecipitation of MgAl-LDH on the acid-treated CNT (Table 2 entry 19). The Ru/MgAl-LDH-CNT catalyst was obtained by wet impregnation of the MgAl-LDH-CNT support with RuCl3·3H2O and reduced at 400 °C under H2 flow. The collapsed LDH structure obtained after reduction was recovered after redispersion for 3 h in a water–oil interface (H2O:toluene = 1:2), giving a homogeneous emulsion.
The Ru content was ca. 0.7 wt% in the Ru/LDH-CNT composite, Ru/LDH, and Ru/CNT, with particle sizes of 2–3 nm uniformly dispersed on the surfaces. They also showed similar specific surface areas of ~90 m2 g−1. However, the catalytic activities of the three materials revealed different behaviors for the selective oxidation of BA to benzaldehyde. The selectivity was in all cases 99.9%, but conversion of 92% was reached by Ru/LDH-CNT, compared to 52% and less than 5% for Ru/CNT and Ru/LDH, respectively, after 5 h of reaction. These results can be attributed to the different capacities of LDH, CNT, and LDH-CNT to stabilize water–toluene emulsions. LDH-CNT presented a 100% stabilized volume fraction with the smallest emulsion droplets (30–150 µm) much lower than those of CNT and unstable hydrophilic LDH nanosheets at the water–oil interface (100–300 µm). This resulted in an increased emulsion interfacial surface area, where the Pickering interfacial catalysis (PIC) process took place, leading to the high catalytic activity observed with Ru/LDH-CNT in the aerobic oxidation of BA. The advantage of the PIC process is obvious when one considers that 54% conversion of BA is obtained over Ru/LDH-CNT with toluene as the solvent.
With LDH at 85 °C, coalescence of the droplets at the water–oil interface occurred and the LDH was transferred to the aqueous phase. Therefore, Ru/LDH did not catalyze the oxidation of BA likely dissolved in toluene. CNT dispersed at the water–oil interface allowed PIC emulsion catalysis to occur, although partial coalescence of the droplets occurred. Ru/CNT (conversion 52%) was then found more active than Ru/LDH (conversion < 5%). LDH-CNT exhibited the higher thermostability at the water–oil interface due to the smaller size of the droplets and the restricted rotation of the emulsifier induced by the nanosheet-shaped structure, which led to higher conversion on Ru/LDH-CNT. The positive effect of LDH nanosheets for the PIC process was confirmed using a Ru/LDH-CNT catalyst whose LDH-CNT support was hydrothermally treated for 4 h instead of 16 h, leading to a smaller LDH particle size. The conversion of BA on this Ru/LDH-CNT reached 84% at 5 h, higher than that over Ru/CNT despite a lower emulsion volume (55% versus 77%). The three-phase contact angle of the LDH-CNT at the water–oil interface of 97°, instead of 110° for CNT, showed that the wettability of CNT is modulated in the presence of LDH, leading to a more stable emulsion, accounting for the higher stability of the Pickering emulsion with LDH-CNT.
Table 4 summarizes the reaction conditions and catalytic results of the considered nanocomposite catalysts. Benzaldehyde is always the main product of the reaction. Higher conversion of BA and selectivity to benzaldehyde are obtained in the presence of a solvent, DMF, or water–toluene, than in solvent-free conditions. In the latter, higher catalytic activity is obtained when the basicity of the catalyst is improved. The CoAl-ELDH/GO composite allows similar conversion and selectivity to the Ru-containing catalyst to be obtained in the PIC process, showing that a non-noble-metal-containing catalyst can be efficient in the oxidation reaction. It can be noted that the PIC process allows the performance of the reaction at a lower temperature. In the case of Au-containing catalysts, the high selectivity to benzyl benzoate probably results from the reaction conditions, particularly the high oxygen pressure and temperature, promoting the reaction of benzaldehyde and BA.
4.2.2. Oxidation of Alkylaromatics
Nanocomposites based on non-noble-metal-containing LDH and carbon compounds offer wide possibilities to obtain highly efficient catalysts for the selective oxidation of alkylaromatics able to fulfil sustainable chemistry requirements. Two main model reactions have been particularly investigated: the selective oxidation of ethylbenzene (EB) to acetophenone (AP) and the oxidation of styrene to styrene oxide (SO).
Among the selective oxidation of alkylaromatic compounds, that of EB to produce AP is relevant because AP is an important intermediate to produce esters, aldehydes, and pharmaceuticals. Cobalt-based metal oxides have been reported as efficient catalysts for the oxidation of EB. For example, hierarchical flower-like core–shell-structured CoZnAl-MMO supported on amorphous alumina microspheres or a flower-like Al2O3@CoCuAl-MMO catalyst exhibited high activity and selectivity toward AP [90,91].
CoCuAl-LDH on a graphene sheet nanocomposite prepared by coprecipitation of the LDH on GO (Table 2 entry 20) was also tested in EB oxidation [38].
The main products of the oxidation of EB at 120 °C using tert-butyl hydroperoxide (TBHP) as an oxidant were AP, BA, and 1-phenylethanol (1-PA). The catalytic activity of the CoCuAl-LDH/graphene nanocomposites depended on the graphene/LDH mass ratio. A mass ratio of 0.4 led to a maximum EB conversion of 96.8% and AP selectivity of 95.4%. GO and graphene were poorly active, with conversion not exceeding 13.5%, while pristine CoCuAl-LDH exhibited significant conversion (68.8%) and selectivity to targeted AP (88.1%). Moreover, the catalyst could be recycled at least four times without significant loss of activity.
CoCuAl-LDH/graphene nanocomposites presented improved catalytic performance if compared to other types of catalysts previously reported, despite the different reaction conditions used in each case [38].
The higher activity shown by CoCuAl-LDH/graphene nanocomposites is assignable to: (i) the adsorption of EB on the graphene through π-π interaction in close proximity to Co2+ and Cu2+ active sites; (ii) the high dispersion of CoCuAl-LDH nanoplatelets of small size; (iii) promotion of THBP activation due to the strong interaction of graphene with Co2+ and Cu2+ sites; and (iv) the preferential adsorption of EB than water on the hydrophobic graphene surface.
ZnCr-LDH/CNT was the best-performing of the different carbonaceous composites studied by Zhao et al. in solvent-free aerobic EB oxidation with O2 at 130 °C, with conversion of 54.2% and AP selectivity of 93.7% [62]. The ZnCr-LDH/CNT nanocomposite, prepared as shown in Table 2, entry 21, presented LDH platelets with an average size of ~10 nm, highly dispersed on the surface of CNT (Figure 3a). Larger ZnCr-LDH particles (30–40 nm) were obtained over the graphene (G) surface, while aggregation occurred on active carbon (AC). These ZnCr-LDH/G and ZnCr-LDH/C composites yielded EB conversion of 45.1% and 40.0%, respectively, with AP selectivity of ca. 87% in both cases. The physical mixture of CNT and ZnCr-LDH (20 wt% CNT) leading to EB conversion of 28.5% was less active than ZnCr-LDH/CNT and the pristine ZnCr-LDH (37.9%), revealing a synergistic effect in the composite. The strong interaction between ZnCr-LDH particles and CNT was revealed by the largest positive shift in the BE of the Zn 2p and Cr 2p regions in ZnCr-LDH/CNT in comparison to the pristine ZnCr-LDH.
Interestingly, oxidation of EB over ZnCr-LDH/CNT using either O2, a small amount of TBHP (0.5 mL, 70% aqueous solution), or both oxidants (Figure 3b) led to contrasting results.
An induction period of around 4 h was observed using O2 that was, on the contrary, absent using TBHP, but EB conversion was very low, reaching only 7.1% with AP selectivity of 55.8% after 6 h. Using both O2 and TBHP, EB conversion of 50.8% with high AP selectivity of 93.6% was achieved after only 1 h. The addition of butylated hydroxytoluene, as a radical scavenger, inhibited the reaction performed under O2, suggesting that it obeyed a free radical process because free radicals generated from TBHP greatly enhanced the reactivity. The ZnCr-LDH/CNT catalyst was also found to be active for the oxidation of other alkylaromatics.
The high efficiency with a clear synergistic effect between the components of the ZnCr-LDH/CNT catalyst can be mainly related to the dispersion of the ZnCr-LDH nanoplatelets on the CNT surface, which increases the accessibility, the strength of the active sites, and their strong electronic interaction. Such features promote EB adsorption on CNT through π-π interaction and its contact with the adjacent Cr3+ sites interacting with O2.
The CoCuAl-LDH/graphene catalyst (G/LDH mass ratio of 0.4), with an AP yield of 92.35% using TBHP, is significantly more efficient than ZnCr-LDH/CNT as reported by Xie et al., leading to an AP yield of ca. 4% or 50.8% with TBHP or O2, respectively, which emphasizes the higher intrinsic efficiency of the CoCuAl-LDH/graphene catalyst [38,90]. This was also confirmed in the oxidation of cumene, 1,2,3,4-tetrahydronaphtalene, and diphenylmethane, with conversions of 84.8, 98.1, and 97.3%, respectively, with the CoCuAl-LDH/graphene nanocatalyst, and of 49.0, 53.6, and 63.8%, respectively, with ZnCr-LDH/CNT.
The different behavior of the CoCuAl-LDH/graphene and ZnCr-LDH/CNT catalysts mainly accounts for their different numbers of active sites. The total loading of Co2+ and Cu2+ indeed reached 11.2 wt% in CoCuAl-LDH/graphene, while Cr3+ loading was 0.87 wt% in ZnCr-LDH/CNT. Notably, the performance of the CoCuAl-LDH/graphene and ZnCr-LDH/CNT catalysts was better than that of a range of previously reported catalysts. The literature data suggest that higher EB conversions are generally obtained when using TBHP operating at a lower temperature (70–130 °C) but a larger reaction time (8–24 h) than with O2 (120–160 °C and 1–9 h) [38,90].
The epoxidation of styrene is of outstanding importance. For cleaner processes, oxidation of styrene with H2O2 or TBHP rather than with organic peracids is preferred, with higher selectivity to styrene oxide generally obtained with TBHP.
Shen et al. have reported a series of NiCo-LDH/rGO nanocomposites (NiCo-LDH/RGOx, x = 1–10 wt%), synthesized by a one-pot hydrothermal method, as catalysts using TBHP as an oxidant [63]. A GO colloid was dispersed in a C2H5OH/H2O solution, where a Ni and Co chloride salt solution (Ni/Co = 1) was added dropwise along with NH4Cl under stirring (pH adjusted to 9). The suspension was hydrothermally treated (120 °C for 12 h), producing both LDH crystallization and GO reduction.
LDH nanoplatelets were dispersed around rGO as nanoflower clusters (Figure 3c,d) and presented a smaller size (50–100 nm) than in the pristine NiCo-LDH (ca. 200 nm).
There was a significant influence of the rGO content on the catalytic activity of the reaction performed at 80 °C for 8 h. Styrene conversion increased from 88.6% to 95.1% when the rGO content increased from 0% to 5 wt%. Such behavior accounted for an increase in the number and strength of accessible Ni2+ and Co2+ active sites, leading to improved THBP binding (Figure 3e). This gave rise to MIII-oxo and MIII-peroxo species, able to attack the C=C bond of styrene, producing a peroxo metallocycle that was then decomposed to form styrene oxide (path 1). Benzaldehyde was also generated due to the cleavage of C=C bond of the styrene adsorbed on MII species.
The best SO yield (70.2%) was obtained with 5 wt% of rGO and decreased when the rGO content increased; meanwhile, selectivity to BAL remained almost similar around 27%, regardless of the rGO content. These catalytic results improve those ones previously reported with catalysts such as Fe-sal-CMK-3, NiO NP and Au/L-Fe3O4, giving SO yields of 32.4%, 10.8%, and 53.4%, respectively [92,93,94]. NiCo-LDH/RGO catalysts showed remarkable stability along five catalytic runs, with styrene conversion decreasing only by around 2% and SO selectivity remaining constant.
In summary, the previous results illustrate the great potential of the LDH/nanocarbon composites to perform the oxidation of various substrates through the tailoring of their composition. The LDH composition and/or the introduction of nitrogen-doped carbon dots (NCD) allow the adjustment of the basicity of the final nanocomposite. The ability to obtain Pickering emulsions highly stabilized with LDH/nanocarbon composites offers interesting outlooks that have not yet been explored in the hydrogenation of benzaldehyde. The LDH/nanocarbon catalysts are particularly suitable for the oxidation of molecules containing aromatic cycles due to the π-π interactions with the carbon component and the enhanced electron transfer.
4.3. Hydrogenation Reactions
Since 2012, Mg, Co, Ni, or Cu-based LDHs have been combined with carbon materials, i.e., amorphous carbon, GO, CNT, or CD, to obtain hierarchical structures acting as precursors of catalysts for a wide variety of hydrogenation reactions. They were involved in the reduction of organic dyes and nitroarenes, the selective hydrogenation of C=O or C=C conjugated bonds and of 5-hydroxymethylfurfural (HMF) to 2,5-dimethylfuran (DMF), the hydrodechlorination (HDC) of chlorobenzene, and the hydrogenolysis of glycerol [22,34,35,39,41,47,64,65,66,95,96,97,98,99].
The nature of the components and the preparation methods of the LDH/nanocarbon nanocomposites and the hydrogenation reactions considered are summarized in Table 2, entries 23–30.
4.3.1. Hydrogenation of Organic Dyes and Nitroarenes
Nitro derivatives have a highly pollutant nature, especially 4-nitrophenol (4-NP) (or p-nitrophenol), which is anthropogenic, carcinogenic, and toxic, with, in addition, high solubility and stability in water. Reduction of 4-NP to produce 4-aminophenol (4-AP) is an important issue because this latter is a valuable industrial raw material and pharmaceutical intermediate. Several transition metals, mainly Cu, Co, and Ni and their oxides, have been widely used for the reduction of 4-NP [99,100,101,102]. A wide variety of LDH/nanocarbon composites have been also explored as catalysts or precursors for the reduction of 4-NP and organic dyes.
CuxMg3−xAl-LDH/rGO (x = 0.5, 1, 1.5) and Cu3−xNixAl-LDH/rGO (x = 2.5, 2.0, 1.5) nanocomposites with nanoarray-like structures (Figure 1) were prepared according to the CA-assisted aqueous-phase coprecipitation method and studied for the reduction of 4-NP with an excess amount of NaBH4 [35,39,41].
The reduction of GO and the presence of abundant and smaller sp2 graphitic regions, with the creation of defects acting as nucleation sites, account for the vertical and uniform growth of the LDH in the nanoarray-like CuxMg3−xAl-LDH/rGO (x = 0.5, 1, 1.5) (named xCu-LDH/rGO) nanocomposites, reducing the strong interparticle interaction among LDH nanoplates (Table 2, entry 23). Thus, the hierarchical nanoarray-like morphology is in line with the higher specific surface area of the composites (204–215 m2 g−1) compared to Cu1Mg2Al-LDH (named CuAl-LDH) (102 m2 g−1).
Complete conversion in the reduction of 4-NP was achieved within 2.5 min with the xCu-LDH/rGO composites, in comparison to 11 min with CuAl-LDH, while GO and Mg3Al-LDH were inactive [39]. The active Cu-related species were more efficient in the hierarchical nanoarray-like structure. Both the apparent reaction rate constant (pseudo-first-order kinetics) and the TOF number increased with the Cu content and were higher than with pure Cu-LDH or previously reported catalysts [103,104,105]. Cu 2p XPS, XRD, and HRTEM analyses revealed that the active sites were Cu2+ ions belonging to LDH nanosheets and Cu+ formed by reduction upon dissociation of NaBH4 in water, which led to the in situ formation of Cu2O NPs on the LDH nanosheets. Indeed, the presence of spherical Cu2O NPs (~7 nm) was ascertained by HRTEM, with also the presence of interfaces between LDH domains supporting Cu2O NPs and single rGO layers. A synergistic Cu2O-Cu-LDH-rGO three-phase interface is then suggested, which can be considered as the most active catalytic domain due to the enhanced electron transfer. The xCu-LDH/rGO composites act as efficient Cu2O reservoirs without needing a pre-reduction step. These composites were also active for the reduction of various nitroarenes (2-NP, 3-NP, 2,4-dinitrotoluene, and 4-nitrobenzaldehyde).
The high catalytic efficiency of the composites accounts for: (i) the electronic mobility of rGO in the Cu2O-Cu-LDH-rGO three-phase interface, which facilitates the electron transfer from BH4− to 4-NP mediated by small Cu2O NPs; (ii) the enhanced contact between the reactant and the active Cu2O centers due to the π–π stacking interaction between rGO and adsorbed 4-NP; (iii) the improved diffusion and transfer of 4-NP and BH4− to the catalytic sites due to the large specific surface area of the composites.
Considering the well-known influence of calcination to improve metal–support interaction, Dou et al. investigated the catalytic activity of the xCu@Cu2O/MgAlO-rGO hierarchical nanocomposites obtained by calcination (600 °C under N2 flow) of the CuxMg3−xAl-LDH/rGO precursors (x = 0.5, 1, 1.5) (Table 2, entry 24) [41].
The xCu@Cu2O/MgAlO-rGO nanocomposites contained Cu, Cu2O, and CuO phases according to the reduction of Cu2+ to Cu+ and Cu0 by rGO, and poorly crystallized MgAlO mixed oxide with highly dispersed Cu-based spherical-like particles. Their average size increased (12.8 to 40.3 nm) with the copper content, and they were located near the border between MgAlO and rGO layers. These structural features conferred upon the composite catalysts a mesoporous structure with high specific surface areas, decreasing from 200 to 157 m2 g−1 when the copper content increased.
FTIR suggested charge transfer between π electrons of rGO and the copper atom, weakening the Cu-O bonds and stabilizing Cu+. Catalysts 1.0Cu@Cu2O/MgAlO-rGO and 1.5Cu@Cu2O/MgAlO-rGO exhibited core–shell-like Cu@CuO NPs with Cu0 in the core and Cu2O in the outer shell, while 0.5Cu@Cu2O/MgAlO showed a mixture of Cu nanocrystallites and Cu2O phases. These results confirmed the in situ self-reduction of Cu2+ into Cu0 by rGO and Cu2O NPs located on the border between MgAlO and rGO, creating a Cu-Cu2O-MgAlO-rGO four-phase interface. These latter must greatly improve the catalytic activity, particularly due to the presence of well-dispersed and poorly aggregated core–shell-like Cu@Cu2O NPs in the MgAlO matrix, in close contact with rGO layers, and electron transfer from Cu or Cu2O to MgAlO and rGO (XPS).
Complete reduction of 4-NP to 4-AP occurred within 3, 1, and 1.2 min with 0.5Cu@Cu2O/MgAlO-rGO, 1.0Cu@Cu2O/MgAlO-rGO, and 1.5Cu@Cu2O/MgAlO-rGO catalyst, respectively. The 1.0CuMgAlO with CuO phases supported on MgAlO led to the reduction of 88% of 4-NP within 17 min. The dramatically higher activity of the xCu@Cu2O/MgAlO-rGO catalysts may be related to the four-phase synergistic effect and the presence of core–shell-like Cu@Cu2O NPs. The apparent rate constant (pseudo-first-order kinetics) was 2.03-fold higher for 1.0Cu@Cu2O/MgAlO-rGO and 1.5Cu@Cu2O/MgAlO-rGO than for 0.5Cu@Cu2O/MgAlO-rGO. This clearly showed the higher catalytic efficiency of the core–shell-like Cu@Cu2O NPs than the mixture of Cu and Cu2O NPs, in agreement with the core–shell metal–metal oxide interaction. Moreover, 1.0Cu@Cu2O/MgAlO-rGO, with the smaller core–shell-like Cu@Cu2O NPs, showed 1.2-fold higher activity than its non-calcined precursor. Therefore, calcination, which allows a shift from the three-phase to the four-phase interface system and the formation of core–shell-like Cu@Cu2O NPs interacting with MgAlO and rGO, creates more active species. In addition, 1.0Cu@Cu2O/MgAlO-rGO exhibited remarkable activity in 4-NP reduction comparable to that of several noble-metal-containing catalysts [103,106,107,108].
The peculiarity of the four-phase interface system is that the core–shell-like Cu@Cu2O NPs play a key role as efficient electron transport entities instead of Cu2O alone in the three-phase interface system [39]. Indeed, 4-NP anions and BH4− are both adsorbed on the surface of the catalyst, supporting the electrophilic Cu@Cu2O entities, while BH4− reacts with H2O to produce H2. Dissociation of H2 on Cu@Cu2O generates Cu-H species, reducing –NO2 groups to –NH2 groups through several sequential steps, including the formation of nitroso groups and hydroxylamine intermediates and the generation of 4-AP. The high dispersion of the active Cu@Cu2O species and the strong four-phase synergistic effect also serve to improve the catalytic activity. rGO facilitates both the electron transfer from BH4− to 4-NP and the adsorption of 4-nitrophenate anions through π-π interaction with the surface-active Cu@Cu2O NPs.
A very interesting proof-of-concept for the use of the −1.0Cu@Cu2O/MgAlO-rGO catalyst in a fixed bed reactor for the treatment of industrial effluents has been given by the reduction of 4-NP, methyl orange (MO), and a mixture of 4-NP and MO. Complete reduction of 4-NP to 4-AP and complete degradation of MO as an individual or mixture of substrates were obtained.
Bimetallic Cu-based catalysts, especially with Ni as a second transition metal, have been found more active than monometallic ones for the reduction of nitroarenes, particularly 4-NP [109,110,111,112]. Wei et al. have considered that bi-transition-metal-based Cu3−xNixAl-LDH/rGO nanocomposites may improve the catalytic activity compared to the previous CuxMg3−xAl-LDH/rGO [35]. Therefore, a series of Cu3−xNixAl-LDH/rGO (x = 2.5, 2, 1.5) composites was prepared via an AC-assisted aqueous-phase coprecipitation method (Table 2, entry 25). The samples exhibited the same structural and morphological features as the CuxMg3−xAl-LDH/rGO series, with highly uniform, dispersed nanosheets forming an array-like structure, high accessibility and surface area, and strong interaction between LDH and GO, which led to electron transfer. The BE values for Cu 2p3/2 and Ni 2p3/2 varied with the Cu2+ on Ni2+ molar ratio, suggesting an electronic interaction and synergistic effect.
The time to reach the entire conversion of 4-NP to 4-AP ranged as follows: Cu1Ni2Al-LDH/rGO (1.5 min) < Cu1.5Ni1.5Al-LDH/rGO (2 min) < Cu0.5Ni2.5Al-LDH/rGO (2.5 min). This shows that the activity depended on the composition. Cu1Ni2Al-LDH/rGO was more active than the single transition metal sample Cu1Mg2Al-LDH/rGO, reaching complete conversion in 2 min [39]. Because complete 4-NP conversion occurred in 3 min with Cu1Ni2Al-LDH and both Ni3Al-LDH and rGO were inactive, it was concluded that the Cu-related species were the active sites, whose efficiency was improved due to the synergistic effect with Ni2+. Moreover, the positive influence of the dispersed Cu species in the LDH nanosheets strongly interacting with rGO was reflected by the 1.3-fold higher kapp values (pseudo-first-order kinetics) obtained with the composites than with their corresponding Cu3−xNixAl-LDH.
The excess of NaBH4 benefited the formation of small Cu2O NPs, also directed by the isolation and stabilization effects of the Ni-OH groups of the LDH layers. XPS peaks in Cu1Ni2Al-LDH/rGO upon reduction with NaBH4 showed an increase in electron density around Ni and Cu core level compared to untreated Cu1Ni2Al-LDH/rGO. Moreover, 15.9% of total Cu in the LDH was in situ reduced to Cu2O species. Therefore, Cu2+ ions can be considered as a reservoir continuously providing highly active Cu2O NPs when an excess of NaBH4 is used in the reduction reaction, as already reported for the CuxMg3−xAl-LDH/rGO composites [39]. Moreover, in the reaction medium, the electron transfer from Cu to Ni (slightly more electronegative) improved the hydrophilicity of the surface of Cu2O in the Cu1Ni2Al-LDH/rGO composite when compared to the single transition metal Cu1Mg2Al-LDH/rGO. Cu2O NPs were more stabilized in the former hybrid due to the stronger interaction with the OH groups of LDH. All these features suggest that the three-phase interface Cu2O-Ni-OH-rGO between the Cu1Ni2Al-LDH domains and the single-layer rGO induces a strong synergistic effect, enhancing the catalytic activity.
Furthermore, 4-NP conversion of 90.3% after 10 catalytic cycles demonstrated the remarkable reusability of the Cu1Ni2Al-LDH/rGO catalyst, whose slight deactivation was mainly due to the increase in the average Cu2O NP size (from ~3.8 to ~9.8 nm).
The previous results emphasize the influence of the composites’ composition (CuxMg1−xAl-LDH/rGO and Cu3−xNixAl-LDH/rGO) and of their topotactic decomposition (xCu@Cu2O/MgAlO-rGO). The main physico-chemical characteristics, the performance in the reduction of 4-NP, and the recyclability of the most active catalysts in each series are compared in Table 5.
All the as-prepared composites exhibited ultrathin LDH nanosheets vertically grown on both sides of the rGO substrate, giving rise to large surface areas (>150 m2 g−1) and mesopores (2–15 nm). Their catalytic efficiency arose from the existence of three-phase interface domains formed upon in situ reduction with NaBH4 during the reaction process. They contained well-dispersed Cu2O NPs of small size (3–7 nm), continuously provided by the reduction of Cu2+ in the LDH layers, which acted as a reservoir of the active species. The Cu2O NPs supported on the LDH layers in close contact with the rGO layers benefited from significant electron transfer. These features and the π-π stacking effect between the catalytic surface and the aromatic cycle of the nitroarenes led to a synergistic effect. This was enhanced in the bi-transition-metal composites due to the higher isolation and stabilization effect induced by Ni-OH rather than Mg-OH species and to the electron transfer from Cu to Ni.
The topotactic decomposition of the CuxMg1−xAl-LDH/rGO precursors by calcination led to four-phase interfaces in the xCu@Cu2O/MgAlO-rGO nanocomposites with core–shell-like Cu@Cu2O NPs instead of Cu2O NPs in the non-calcined precursor. The former Cu species improved the synergistic effect and the catalytic performance due to faster electron transfer, with also the contribution of the larger surface area and mesoporosity. It is noteworthy that the reduction of the Cu2O species into well-dispersed Cu crystallites was observed by HRTEM in the CuxMg1−xAl-LDH/rGO precursors recycled 20 times, with only a slight decrease in activity.
Remarkably, calcination of the single-transition-metal-containing composites gave rise to catalysts (xCu@Cu2O/MgAlO-rGO) exhibiting rather similar performance to the non-calcined bi-transition-metal Cu3−xNixAl-LDH/rGO composites. Then, core–shell-like Cu@Cu2O NPs presented the same efficiency as regards electron transfer and the adsorption of reactants as materials with the presence of a second transition metal. This provides many possibilities to tune the catalytic properties.
The LDH/rGO composites presented significantly higher catalytic reduction efficiency for 4-NP than several recently reported catalysts [64,103,104,105,113,114].
Iqbal et al. have designed a multicomponent CD/Ag@MgAlCe-LDH catalyst combining Ag NPs and CD NPs with Ce-doped MgAl-LDH for the reduction of 4-NP and several organic dyes [34]. Ag NPs act as electron relays between electron donors and electron acceptor species, making them efficient redox catalysts [115]. CD acted as stabilizers and reducing agents of Ag NPs, preventing their aggregation. LDHs doped with rare-earth elements behave as highly basic structures or precursors of mixed oxides with tunable redox properties.
The CD/Ag@MgAlCe-LDH nanocomposite was obtained by dispersion of a previously coprecipitated Ce-doped MgAl-LDH (MgAlCe-LDH) into an aqueous solution of AgNO3 followed by the addition of CD. The obtained mixed solution was exposed to UV light (254 nm) to induce a photoreaction (Table 2, entry 26).
The introduction of Ce3+ into the brucite-like layers in CD/Ag@MgAlCe-LDH was confirmed. Additionally, a cerium phase and Ag NPs were identified. Photoluminescence experiments revealed electron transfer from CD to Ag NPs, and transient photocurrent measurements showed that the introduction of CD enhanced the photocurrent value. Moreover, CD/Ag@MgAlCe-LDH exhibited a photocurrent intensity two-fold higher than Ag@MgAlCe-LDH which evidenced a synergistic effect of CD and Ag NPs and a more efficient electron–hole pair separation.
The complete reduction of 4-NP into 4-AP in the presence of NaBH4 (4-NP/NaBH4 = 3:200) occurred at approximately 120 s upon addition of the CD/Ag@MgAlCe-LDH catalyst. On the contrary, the Ag@MgAlCe-LDH, CD@MgAlCe-LDH, and MgAlCe-LDH catalysts were practically inactive after reaction times above 840 s. CD/Ag@MgAlCe-LDH was also very active for the reduction of various organic dyes, with complete degradation achieved for rhodamine 6G (R6G) (90 s), MB, MO, and RhB (120 s), and Congo red (CR) (440 s). Based on the values of the apparent kinetic rate constants (kapp) (pseudo-first-order reaction) for the degradation of the different substrates, the activity of the catalysts ranged as follows: CD/Ag@MgAlCe-LDH > Ag@MgAlCe-LDH > CD@MgAlCe-LDH > MgAlCe-LDH. The kapp for the reduction of 4-NP (38 × 10−3 s−1) is comparable to that obtained with the best-performing CuMgAl-LDH/rGO and CuNiAl-LDH/rGO catalysts, leading to kapp values in the range from 25.11 × 10−3 to 55.35 × 10−3 s−1 (Table 5) [39,41]. The CD/Ag@MgAlCe-LDH catalyst can be recycled and reused eight times without loss of activity.
In this catalyst, CD acted as a reducing agent of Ag+ and coordinatively bonded to the Ag NPs, preventing their aggregation. Moreover, the small size of the CD and Ag NPs increased the number of accessible active sites and provided an effective interface, improving the electron transfer. Moreover, the MgAlCe-LDH improved the catalytic activity through its high specific surface area and behaved as a basic co-catalyst. The LDH’s hydrophilic surface also facilitated the adsorption of BH4− on Ag NPs and the adsorption of the 4-NP or organic dye substrates. Ce ions were involved in the redox reaction through the reversible Ce3+/Ce4+ oxidation state. The CD NPs on the Ag NPs and the Ce ions on the MgAlCe-LDH support both promoted the electron transfer and migration.
Yang et al. achieved the combination of Pd NPs as an electron transfer system and CD as a reducing and stabilizing agent in a nanocomposite, where the originality is the use of a polymer (poly(N-isopropyl acrylamide) surface-modified LDH as a support (Figure 1; Table 2, entry 27) [22]. This multicomponent nanocatalyst displayed good water dispersibility.
The content and average diameter of the Pd NPs were 5.86 wt% and 3.7 nm, respectively, and the average diameter of CD was 4.25 nm in LDH@PDA@PNIPAM@Pd/CD. Pd NPs exhibited a more uniform size and distribution on the surface than on a reference LDH@PDA@PNIPAM@Pd composite reduced with NaBH4, due to the stabilizing and reducing effects of CD. The sequential increase in intensity of the N 1s XPS peak after PDA modification, PNIPAM grafting, and CD loading in the LDH@PDA@PNIPAM@Pd/CD material confirmed that these components were supported on the LDH surface. Moreover, the characteristic Pd 3d5/2 and Pd 3d3/2 XPS peaks of Pd NPs were attributed to three different Pd2+ species: unreduced Pd2+; PdO in contact with air, and ultra-small Pd clusters.
Reduction of MB into leucomethylene blue in the presence of an excess of NaBH4 was completed after 32 s for LDH@PDA@PNIPAM@Pd/CDs and 64, 32, 80, and 80 s for LDH@PDA@PNIPAM@Pd, LDH@Pd/CD, LDH@Pd, and Pd/CD control catalysts, respectively. Meanwhile, LDH, LDH@PDA@PNIPAM, and CD were almost inactive. LDH@PDA@PNIPAM@Pd/CD was also active for the reduction of CR, MO, R6G, and RhB and the reduction of 4-NP, o-NP, and m-NP.
These results showed that both the grafting of PNIPAM and the presence of CD improve the catalytic performance. The polymer grafting increases the dispersibility in water and the surface area of the LDH@PDA. This favors the dispersion of Pd NPs of small size immobilized on the polymer brushes. Meanwhile, the high water dispersibility improves the contacting surface area between the reactants and the catalytically active sites. The reactants and BH4− are strongly adsorbed on the LDH support around the Pd NPs. These latter are stabilized and poorly aggregated due to the presence of CD, which improves the electronic transfer between the species. All these synergistic effects serve to enhance the catalytic activity and reusability.
Interestingly, LDH@PDA@PNIPAM@Pd/CD was also investigated as a bifunctional catalyst for the one-pot Knoevenagel condensation–reduction tandem reaction of 4-nitrobenzaldehyde and malonitrile. The Knoevenagel reaction was first performed (60 °C for 6 h) and the resulting mixture subjected to reduction with NaBH4. Conversion reached 78% in the first condensation step and 64% after the second step with reduction of the alkenyl and nitro groups to alkane and amine groups. These results need to be optimized but represent a promising approach for the development of other types of tandem reactions.
NiO has been scarcely investigated for the reduction of 4-NP, although CuO, Co3O4, Fe2O3, and NiO exhibit almost similar activity [116]. Akbarzadeh and Gholami prepared a Pt-modified NiO-Al2O3 nanocomposite derived from GO-supported NiAl-LDH as a catalyst for 4-NP reduction (Table 2, entry 28) [64].
The characteristic XRD reflections of NiAl-LDH in the NiAl-LDH/G precursor moved to those of NiO after calcination at 600 °C in Pt-NiO/G. The highly dispersed Pt0 NPs in low amounts (2.3 wt%) were not detected. TEM images showed that they were highly dispersed on the mixed oxide.
The performance of the Pt-NiO/G catalyst was compared to that of NiO, NiO/G, and Pt-NiO catalysts obtained by calcination of NiAl-LDH, NiAl-LDH/G, and Pt-NiAl-LDH precursors, respectively. The latter was obtained by impregnation of H2PtCl6 on NiAl-LDH. The reduction of 4-NP was achieved with NaBH4:4-NP = 1:1000.
Both NiO (kapp = 1.37 × 10−3 s−1) and NiO/G (kapp = 2.07 × 10−3 s−1) were poorly active (pseudo-first-order kinetics). The activity was greatly improved upon introduction of Pt in Pt-NiO (kapp = 14.87 × 10−3 s−1) due to the synergistic effect between NiO and well-dispersed Pt0 NPs. Supporting Pt-NiO on the G support in the Pt-NiO/G composite led then to a two-fold increase in the kapp value, reaching 33.9 × 10−3 s−1. However, the Pt-NiO/G was poorly stable because the reaction time for reduction increased by approximately 50% after five consecutive cycles.
The mechanism of the reduction was explored by varying the BH4− and 4-NP concentrations. The non-linear dependence of the kapp values with the concentration of substrate and 4-NP suggested a Langmuir–Hinshelwood mechanism. The hydrogen transfer from the adsorbed BH4− to 4-phenolate anions was the rate-limiting step of the reaction. The higher catalytic efficiency of Pt-NiO/G is likely due to the electron transfer from Pt to NiO, leading to an electron-rich area. The active hydrogen species and electrons formed from BH4− reacting with the Pt-NiO surface are transferred to the adsorbed 4-NP, thus generating 4-AP. The authors did not consider the promoting effect of rGO, which likely also improved the electron transfer as Pt-NiO was less active than Pt-NiO/G.
4.3.2. Hydrogenation of Nitro Compounds and α-β-Unsaturated Aldehydes
Previous works have shown that Ni-supported or Ni alloys efficiently replace noble-metal-containing catalysts in the selective hydrogenation of o-chloronitrobenzene (o-CNB) to o-chloroaniline (o-CAN) [117,118]. Wang et al. prepared dispersion-enhanced Ni-supported catalysts derived from nanocomposites based on NiAl-LDH and PAA (polyacrylic acid) and L-cysteine-functionalized CNT components (Ni-L/P-CNT) (Table 2, entry 29) [47]. In these catalysts, Ni NPs with a narrow size distribution of ca. 6.0 nm highly dispersed on the CNT support exhibited thin and faceted aspects accounting for SMSI. Ni0 NPs were larger and highly aggregated in Ni/P-CNT. Dispersion of Ni0 NPs was ~11.8% and ~20.5% in Ni/P-CNT and Ni-L/P-CNT, respectively. It was improved in Ni-L/P-CNT due to the optimized dispersion of the precursor NiAl-LDH on CNT.
The small LDH crystallite size in LDH-L/P-CNT made the reduction of Ni2+ in Ni(Al)O mixed oxide easier if compared to the LDH/P-CNT catalyst (without L-cysteine), where a mixture of NiO and Ni0 was found after reduction.
Ni-L/P-CNT exhibited superior catalytic performance in the liquid-phase hydrogenation of o-CNB (140 °C, P H2 2 MPa, 150 min), compared with the LDH/P-CNT catalyst and that prepared by conventional impregnation. For similar Ni content (~21 wt%), Ni-L/P-CNT presented o-CNB conversion of 99.3% and selectivity to o-CAN of 98.8%. In contrast, Ni/P-CNT presented conversion of 2.4% and selectivity of 98.5% due to the higher specific surface area (206 vs. 109 m2 g−1), reduction degree (95 vs. 46%), and dispersion (20.5 vs. 11.8%) of Ni0 species in the former catalyst, as well as the existence of an electronic interaction between metal and support. This latter modified the mode of the metal–reactant interaction and thus enhanced the selectivity towards the desired products. The SMSI increased the high electronic density for π-electrons of the CNT support, inducing the enhanced oriented adsorption of o-CNB with the nitro group and repulsion of the chlorine group, whose hydrogenolysis was inhibited. This accounted for the higher o-CAN selectivity of the Ni/P-CNT and Ni-L/P-CNT catalysts compared to single Ni-impregnated CNT. Ni-L/P-CNT could be recycled up to five times, maintaining an o-CAN yield of 95% without Ni leaching.
Xie et al. used a NiAl-LDH/G composite as a precursor for catalysts to perform the liquid-phase hydrogenation of cinnamaldehyde (CALD) into hydrocinnamaldehyde [65]. NiAl-LDH/G was synthesized via the classical coprecipitation method of LDH in the presence of GO (Table 2, entry 30). Several comparative samples, i.e., NiAl-LDH, NiAl-LDH/C composite (C: active carbon), coprecipitated Ni(OH)2/G, and impregnated Ni(NO3)2/G, were also prepared. The catalysts (Ni-L/G, Ni-L/C, Ni-Co/G, and Ni-Im/G) were obtained by the heating of the precursors at 600 °C under N2 flow.
In the NiAl-LDH/G composite, GO was reduced into graphene, supporting highly dispersed LDH nanosheets (average size 22 nm) homogeneously dispersed and anchored on both sides of the exfoliated graphene sheets. Subsequently, small, highly homogeneous supported Ni NPs were obtained upon thermal treatment.
The activity of the Ni-L/G catalyst was evaluated in the hydrogenation reaction of CALD (120 °C, 1 MPa H2) and compared to that of Ni-Co/G, Ni-Im/G, and Ni-L/C catalysts.
The products of the reaction are hydrocinnamaldehyde (HCAL), cinnamyl alcohol (COL), and hydrocinnamyl alcohol (HCOL). Ni-L/G exhibited the lowest particle size (12.6 nm), highest dispersion (36.5%), and largest specific surface area (182.6 m2 g−1) and it was the most active (100% conversion) and selective catalyst toward HCAL (94.8%). The strong interaction between NiAl-LDH and graphene, which inhibited both the aggregation of the Ni2+-containing LDH sheets and the restacking of the graphene sheets, accounted for the high dispersion of the Ni0 NPs upon in situ reduction and the improvement in the catalytic activity. On the contrary, weak interaction between the metal precursor species and the support in Ni(OH)2/G and Ni(NO3)2/G samples led to poorly dispersed and large Ni NPs in Ni-Co/G (dispersion 19.8%; particle size 38.6 nm) and Ni-Im/G (dispersion 10.7%; particle size 68.4 nm), giving rise to lower conversion of 62% and 37.9%, respectively. Despite the similar specific surface areas (175.2 vs. 182.6 m2 g−1), Ni particle sizes (13.4 vs. 12.6 nm), and dispersion (34.6 vs. 36.5%) of Ni-L/C and Ni-L/G, the former catalyst led to lower conversion (56.5%). This shows that the 2D structure and the electronic properties of graphene, favoring the accessibility of the reactant to the active sites and electronic transfer toward the Ni NPs, improve the catalytic efficiency. Ni-L/G could be recycled up to five times while maintaining its catalytic performance.
4.3.3. Hydrogenolysis of Glycerol
The rising production of biodiesel gives rise to huge amounts of glycerol. Several means of revalorizing glycerol have been implemented using LDH-based materials, such as base-catalyzed transesterification with diethyl carbonate, dimethyl carbonate, or methyl stearate; carboxylation with urea; selective etherification to short-chain polyglycerols; acetalization with acetone; oxidation into commodity chemicals, and steam reforming into H2 [59,60,119,120,121,122,123,124,125,126]. It has been already reported that LDH/CNF and LDH/CF composites can act as efficient catalysts for the transesterification of glycerol with diethylcarbonate [59,60].
Xia et al. have used a nanocomposite obtained from CuMgAl-LDH and MWCNT components to valorize glycerol through hydrogenolysis to propanediols (PDOs) [66].
A series of MWCNT-CuMgAl-LDH composites were prepared by coprecipitation of CuMgAl-LDH into an aqueous suspension of pretreated MWCNT. The MWCNT’s weight varied between 1.5 and 6 wt%. The precursors were heated at 400 °C and subsequently reduced at 300 °C to obtain the MWCNT-Cu/MgAlO catalysts (Table 2, entry 31). The MWCNT enabled the ordered accumulation of lamellae, bringing narrow-sized, doublet pore channels and high surface area of the catalysts. Indeed, the specific surface areas of the MWCNT-Cu/MgAlO catalysts, in the range of 130–218 m2 g−1, were higher than that of the LDH Cu-supported catalyst Cu/MgAlO (128 m2 g−1). The MWCNT-Cu/MgAlO catalysts showed a first group of mesopores whose average size decreased from 12.8 to 4.7 nm when the MWCNT content increased from 1.5 to 6 wt%. Other mesopores of 40–50 nm were attributed to the gap of MWCNT-pillared Cu/MgAlO lamellae. The close contact of Cu/MgAlO with MWCNT also enhanced the reducibility of CuO due to H2 dissociation on MWCNT and spillover to Cu2+, and improved Cu0 dispersion.
The conversion of glycerol (26%) and the selectivity to 1,2-PDO (97.8%) both increased with the incorporation of MWCNT in the catalysts, having a maximum for (3%) MWCNT-Cu/MgAlO, with conversion and selectivity reaching 64.8 and 99.3%, respectively. At the same time, the activity per exposed Cu atom increased from 8.6 to 17.3 h−1. Remarkably, the conversion and selectivity to 1,2-PDO were higher than those obtained with Pd0.04Cu/MgAlO (conversion 35.5% and selectivity 97.2%) and Rh0.02Cu/MgAlO (conversion 32.1% and selectivity 96.3%) catalysts, which is of practical interest due to the lower price of MWCNT than noble metals.
The 1,2-PDO yield decreased from 64.3% to 41.3% after five catalytic cycles performed with (3%)MWCNT-Cu/MgAlO,, but this was mainly due to the loss of catalyst upon separation, because the activity of surface Cu atoms decreased only slightly (17.2 to 16.6 h−1) and no leaching of Cu occurred.
The results obtained in hydrogenation reactions deserve several general comments regarding the properties of the LDH/nanocarbon catalysts. Assemblies of Ni- or Cu-containing LDH and carbon supports succeeded to obtain active and selective catalysts, mainly resulting from the ability to concurrently obtain highly dispersed metal NPs at high loading, SMSI, and tailored acid–base and textural properties, leading to synergistic effects. The large majority of the LDH/carbon-derived supported catalysts combine NiAl-LDH and amorphous carbon, CNT, or rGO. A comparison of the mean size and dispersion of Ni NPs of the most efficient catalysts at high Ni loading (22–42 wt% normalized to the Al content) and almost similar activation conditions allows us to highlight the influence of the nature of the carbon support. For an Ni particle size of ca. 13 nm, GO led to higher dispersion. This suggests higher metal–support interaction with the former support, which is consistent with the uniform size of the particles observed by TEM [65]. The high dispersion (20.5%) observed at Ni loading of ~40 wt% (based on the same Al content in the catalyst of ~6.5 wt%) on the PAA and L-cysteine-functionalized CNT support confirms that the surface groups of L-cysteine interacting with the Ni2+ and Al3+ cations allowed a highly dispersed NiAl-LDH phase to be obtained. It led subsequently to highly dispersed Ni0 NPs of small size after reduction. It is noteworthy that the reduction of NiO depended on the nature of the carbon support. Treatment under nitrogen of composites composed of LDH graphene induced the complete reduction of NiO, where the supports acted as reducing agents. Meanwhile, although reduction was performed under hydrogen, Ni2+ was not totally reduced in the case of LDH/CNT composites, indicating that CNT is a less efficient reducing agent.
The works dealing with the series of hierarchical nanosheet array-like CuMgAl-LDH/rGO composites highlight the influence of their composition and activation mode [35,39,41]. With Cu and Ni (CuNiAl-LDH), the active species in the 4-NP hydrogenation is Cu2O, even though Ni is the major species. There is electron transfer from Cu to the more electronegative Ni, and the more electrophilic surface of Cu2O is more stabilized by the Ni-OH groups of the LDH. There is a strong synergistic effect among the in situ reduced Cu2O species, Ni-OH (of LDH layers), and rGO, with an enhanced electron transfer ability [35].
It can be pointed out that in the CD/Ag@MgAlCe-LDH nanocomposite, the carbon compound did not act as a support but as NPs, decorating the active Ag NPs, thus improving electron transfer. It also produced the reduction of Ag+. The Ce-doped MgAl-LDH was not only a support of high specific surface area but also a co-catalyst, providing basic sites and the redox ability of the cerium ions. The CD/Ag@MgAlCe-LDH nanocomposite represents a remarkable example of a synergistic effect and electronic interactions between the components.
4.4. HAS and Direct Synthesis of Isobutanol from Syngas
Catalysts based on Co, Fe, or Ni (e.g., CuFe- and CuCo- catalysts) have attracted particular interest for syngas (CO + H2) conversion to higher alcohols, albeit suffering from several drawbacks such as poor stability, low alcohol productivity, and low selectivity. LDHs are very promising to obtain efficient catalysts for HAS. CO conversion from 17 to 57% and alcohol selectivity from 45 to 60% have been reported using catalysts prepared from CuFeMg-LDH, CuFeMgAl-LDH, CoZnGaAl-LDH, CoMn-LDH, and CuCoAl-LDH precursors [127,128,129,130,131,132].
Dispersion of the LDH on a carbonaceous support greatly improved the performance of the obtained composite catalysts by avoiding hot spots during the reaction and the agglomeration of LDH particles, which decrease selectivity to higher alcohols.
Several works deal with catalysts combining CuCoAl-LDH with CF or CNT and CuFeMg-LDH with CF highly selective to C2+OH alcohols, and combining CuZnAl-LDH with CF for the direct synthesis of isobutanol from syngas [43,48,67,68,69]. The different nanocomposites were prepared by coprecipitation of LDH on acid-treated CF or CNT. The LDH crystallite size decreased with the CF or CNT content, in agreement with the increasing dispersion of the nucleation centers, generating more sheets and preventing their stacking. All nanocomposites presented a mesoporous structure, where the CF and CNT supports were completely wrapped by the highly dispersed LDH nanosheets. Metal NPs or alloys were formed upon reduction. The reduced composites exhibited the same lamellar morphology as the precursors without agglomeration of the particles and metal NPs with a uniform spherical-like shape.
The preparation of the LDH/carbon nanocatalysts and the conditions of activation are summarized in Table 2, entries 32–35.
CO conversions in the range of 32.5%–38.5% for CuCo/Al2O3/CFs (220 °C) and of 39.1–44.6% for CuCo-Red/CNT catalysts (230 °C), with selectivities to C+2OH in the range from 40.6% to 61.2% in the HAS from syngas, were higher than that found for reduced CuCoAl-LDH (CO conversion 30.1 and 34.7% at 220 and 230°C, respectively, and selectivity ~24.5%). Increasing the CF or CNT content in the nanocomposite favored the CO conversion and C+2OH selectivity at the expense of hydrocarbon, CO2, and methanol. The rise in CO conversion can be related to the smaller size of the CuCo-alloy NPs, their higher dispersion, and the H2 adsorption/activation on the supports. Indeed, CF or CNT was found to promote the reduction of Cu2+ and Co2+ due to the activation and spillover of H2 and the lowering of the crystallite size of the LDH. The Cu and Co interaction in the alloy is highly favorable to C+2OH selectivity. Cu is the active species responsible for CO molecular activation and insertion, whereas Co is responsible for CO dissociative activation and chain propagation.
The improved selectivity to C+2OH also accounted for the good thermal conductivity of CF and CNT supports, which suppress hot spots responsible for the hydrocarbons’ formation. Simultaneously, water produced during HAS can react with CO via the water–gas shift reaction (WGSR) to produce CO2. This reaction, faster at higher temperatures, is lowered by the suppression of hot spots and by the higher diffusion of water in the porous structure of the supported catalysts. The lower hydrocarbon selectivity observed with CNT-containing nanocomposites suggests that thermal conductivity is probably better with CNT than CF.
In the CuFeMg-LDH/CF composite developed by Cao et al., Cu acts as the active site for CO molecular activation and insertion, and Fe for CO dissociation and chain propagation, with a cooperative effect between these sites [67]. Moreover, dispersion of the CuFe dual sites on the CF support improved the catalytic performance in comparison to an unsupported catalyst.
The CuFeMg-LDH/CF (Cu/Fe/Mg molar ratio 1:1:1) composite was prepared as CuCo-LDH/CF composites, leading to LDH nanosheets (200–230 nm) uniformly attached to the CF support and not agglomerated (Table 2, entry 32) [48]. The CuFe-Red/CF catalyst was obtained by direct reduction at 450 °C of the CuFeMg-LDH/CF precursor without intermediate calcination to avoid the reduction of Cu2+ to Cu0 by CF. It contained Cu0 and Fe0 particles supported on MgO and CF (30 wt%), with Cu0 particles size (5.2 nm) smaller than in the unsupported catalyst (6.1 nm).
Both CuFe-Red/CF and CuFe-Red presented higher CO conversion and C+2OH selectivity than Cu-Fe catalysts previously reported in the literature for HAS, according to the strong synergistic effect between the Cu and Fe species obtained from the CuFe-LDH precursor [133,134]. CuFe-Red/CF performed better than CuFe-Red, with a significant decrease in hydrocarbon and CO2 selectivity due to the improvement in the thermal conductivity upon introduction of CF. Only a slight deactivation in CuFe-Red/CF was observed during 500 h of reaction, with CO conversion decreasing from 35.4% to 30.1% and selectivity to alcohols from 41.1% to 33.9%. The spent CuFe-Red/CF catalyst contained Cu0, MgO, and CF, as in the fresh catalyst, but Fe0 NPs converted into iron carbide (Fe2C) species. Cu and Fe2C were active species for HAS. After 500 h of reaction, there was no sintering of the particles, but separation of the Cu and Fe species was the key reason for the deactivation and the enhancement of hydrocarbon selectivity.
With CO conversion of 35.4% at 280 °C, CuFe-Red/CF showed lower activity than CuCo/Al2O3/30%CF, whose conversion reached 38.5% at 230 °C, both catalysts having the same CF loading of 30%. Selectivity to CO2 was enhanced with CuFe-Red/CF compared to CuCo/Al2O3/30%CF due to the higher reaction temperature, causing the WGSR to occur more rapidly, generating CO2.
Isobutanol is a valuable platform chemical obtained from the HAS from syngas. Cu/ZnO/Al2O3-based catalysts are the most interesting for the industrial synthesis of isobutanol from syngas [135]. Alkali-doped Cu/Zn catalysts have been largely investigated for HAS in order to increase the yield of higher alcohols [136,137,138,139,140].
Huang et al. have prepared CuZnAl-LDH/CF composites by coprecipitation for the direct synthesis of isobutanol from syngas, focusing on the CF content and on the addition of K (Table 2, entry 35) [68,69]. Well-crystallized CuO and weakly crystallized ZnO were the only phases detected in the composites. Reduction at 320 °C led to Cu0 NPs and to a CuO/ZnO solid solution whose formation was promoted upon the addition of ACF or K. The content of CuO/ZnO solid solution increased from 0.7 to 3.7 and 5.1 wt% when moving from CuO/ZnO/Al2O3 to CuO/ZnO/Al2O3/30%ACF and CuO/ZnO/Al2O3/30%ACF/K. The reducibility of Cu2+ into the CuO/ZnO/Al2O3/ACF catalysts was improved with the ACF content (0 to 30 wt%). Interestingly, EDS analysis revealed similar amounts of Cu and Zn on CuO/ZnO/Al2O3/30%ACF and CuO/ZnO/Al2O3/30%ACF/K samples, while a Cu enrichment was produced on the Cu/ZnO/Al2O3 surface (without ACF), showing that K and/or ACF promoted the formation of the CuO/ZnO solid solution.
Moderate CO adsorption is beneficial to alcohol synthesis and particularly for isobutanol selectivity resulting from CO insertion by reversal aldol condensation at the β-carbon of n-propanol [141]. CO-TPD analysis showed that the peak area of the moderately CO adsorbed species increased as CuO/ZnO/Al2O3/30%ACF/K > CuO/ZnO/Al2O3/30%ACF > CuO/ZnO/Al2O3. Then, both ACF and K promoted the adsorption of CO species with moderate strength.
The CuO/ZnO/Al2O3/ACF composites with CO conversion in the range from 24.94% to 47.27% were more active than the non-supported CuO/ZnO/Al2O3 mixed oxide with conversion of 24.52%. This agreed with an increase in the specific surface area and dispersion of the LDH nanoflakes in the composites, leading to more accessible Cu2+ active sites. Conversion decreased at 40% ACF content (42.61%) due to the low amount of CuO/ZnO/Al2O3 active phase. Selectivity to alcohols decreased upon addition of ACF (>10%) at the expense of alkanes. However, methanol and isobutanol were the two main products. Addition of ACF increased significantly the isobutanol selectivity, reaching a maximum of 19.88% for 30% ACF. CO conversion and isobutanol selectivity further increased upon addition of K, although to a lower extent than upon addition of ACF. The best-performing catalyst was CuO/ZnO/Al2O3/30%ACF/K, with CO conversion of 49.77% and isobutanol selectivity of 22.13% owing to the synergistic effect between ACF and K. Both promoted electron transfer to Cu and adsorption of CO with moderate strength, favorable to CO insertion and carbon chain propagation. Otherwise, they enhanced the content of active CuO/ZnO solid solution.
A comparison of the results obtained from the CuCoAl-LDH/CF, CuCoAl-LDH/CNT, and CuFeMg-LDH/CF composites, on one hand, and from the CuZnAl-LDH/CF composite, on the other hand, can hardly be achieved due to the different reaction temperatures in the range of 220–280 °C in the former case and of 320 °C in the latter. However, the results deserve several comments. A different product distribution was obtained at almost similar CO conversion when CuCo alloy and Cu with Fe2C NPs or CuO/ZnO solid solution were the main active species. The former species gave rise to higher hydrocarbon selectivities despite the lower reaction temperature, which suggests better thermal conductivity in the presence of ZnO. Ethanol and C4+OH were the main alcohols formed. The CuO/ZnO solid solution as the active species led to higher alcohol selectivity, with methanol and isobutanol as the main products. The higher alcohol yield was accompanied by a higher CO2 yield according to the formation of a large amount of water and then WGSR with CO.
5. Concluding Remarks and Perspectives
This review evidences that the catalysts derived from LDH/nanocarbon composites deserve significant attention due to their huge potentialities apart from electro- and photo-catalysis. They are yet employed with remarkable efficiency in a large range of reactions, including C–C cross-coupling (e.g., aldolization, Michael, Knoevenagel, Ullman, Sonogashira, Heck, synthesis of chalcone, etc.), oxidation, hydrogenation, HAS, and cascade reactions (e.g., Knoevenagel–Michael and oxidation–Knoevenagel). These hierarchical catalysts will be undeniably developed in the near future due to their unique tailoring for targeted reactions. A dramatic variety of assemblies can be obtained, as illustrated through several remarkable examples. Assembling hollow flower-like LDH and N, S-doped graphene acting as a support of Pd NPs led to composites with hollow inner and mesoporous hierarchically flower-like outer structures. Sandwich-like superstructures resulted from the intercalation of LDH and rGO and of MMO and MWCNT. Magnetic properties can be obtained combining Fe3O4 NP, GO, and LDH. Colloidal suspensions of LDH/GO composites resulted from the self-assembly of previously exfoliated LDH and GO. A uniform nanoarray-like framework was obtained by the coprecipitation of CuMgAl-LDH on exfoliated and citrate-functionalized GO.
Several specific properties account for the various morphologies and the structural and textural features. LDH nucleates from smaller sp2 graphitic domains and some unrepaired defect sites on the nanocarbons. The resulting growth of highly dispersed and poorly aggregated LDH nanosheets in turn impedes the restacking and agglomeration of the graphene sheets. Both features give rise to highly accessible and dispersed active sites. Dispersion of the LDH nanosheets can be also greatly improved through pre-functionalization of the carbon support, as achieved with PAA-CNT and subsequent immobilization bridging linkers, e.g., L-cysteine. Moreover, the supersaturation rate of the LDH during coprecipitation allows control of the orientation of the nanosheets on the nanocarbon.
Other specific properties of the LDH/nanocarbon composites, such as the hydrophobic/hydrophilic balance and the amphiphilic property exhibited by LDH/CNT nanocomposites used as highly thermodynamically stable Pickering emulsifiers, are emerging. Adsorption of aromatic reactants via π-π stacking with graphene-like and CNT supports is highly favorable to catalytic reactivity.
One of the main specificities of the LDH/carbon composites is the interfacial electron transfer. It is facilitated by the considerable inherent electronic mobility in the carbon components, leading to charge redistribution upon interaction with the less conductive LDH phases. Differences have been previously reported among the variety of nanocarbons with, for instance, higher charge transfer ability of the planar π-electrons of the highly graphitized structure of graphene, compared to CNT with a more distorted sp2 structure [142,143,144,145].
Evidence of the in situ self-reduction of cations such as Ni2+ induced by different carbon supports, i.e., rGO or CNT with SMSI, has been given. It occurs upon heating in the absence of H2 in LDH/rGO composites or under hydrogen in LDH/CNT composites. Reduction of the cations can also occur during the reaction processes using chemical agents (NaBH4) thus avoiding the pre-reduction step.
The textures that improve the mass transport and accessibility of the reactants to the active sites, the electron transfer leading to SMSI and the improved adsorption of the reactants, and the dispersion of the active sites are the main features responsible for the synergistic effects observed in the nanocomposites in comparison to physical mixtures of the components. However, a specificity of the LDH/nanocarbon catalysts is the multi-phase synergistic effect illustrated through outstanding examples.
It must be underlined for future developments that basic sites can be introduced through several routes in LDH/nanocarbon composites. LDH or LDO components can indeed provide Lewis- or Brønsted-type basic sites. Another approach is the doping of the LDH/nanocarbon with an alkaline cation (e.g., K+) or the introduction of N atoms in GO (N,S-GO), amorphous carbon, or CD (NCD).
Some strong tendencies can be drawn from the reported results. The large majority of the composites are prepared by coprecipitation of the LDH phase in the presence of the nanocarbon. Otherwise, well-known methods for controlling the size and avoiding aggregation of the LDH particles have been employed. We can underline the separated nucleation and aging step process, the exfoliation of the nanosheets, and the use of urea as an alkaline controlling agent. Regarding the composition of the LDH, MgAl-LDH is the most widely used for base-catalyzed reactions, and the Cu- or Ni-containing LDH for redox reactions.
GO is undeniably the most significant nanocarbon in all the reported works. It is generally exfoliated by sonication, leading to almost individual nanosheets. Its reduction in the alkaline media creates defect sites very useful as nucleation sites to improve the dispersion of the coprecipitated LDH phase.
It is important to highlight the good durability and cyclability of the LDH/nanocarbon catalysts, which is a critical property for practical applications. It is consistent with the high mechanical strength of the nanocarbons and the robustness of the assemblies with LDH, the SMSI leading to very weak leaching. Moreover, regeneration of the active sites is facilitated by the high dispersion and accessibility.
The above overview suggests several perspectives for the larger development of LDH/nanocarbon catalysts:
- Those based on N-doped nanocarbons (N-GO, N-CD, N-graphene quantum dots) would lead to hierarchical structures exhibiting original basic properties. They will efficiently perform a larger range of base-catalyzed reactions, particularly those requiring strong basic strength (e.g., isomerization of olefines).
- The ability to introduce basic sites by different routes (LDH and/or carbon compound) and metal NPs through transition metal cations in the LDH or by impregnation offers unlimited possibilities to design multifunctional catalysts. This will allow large implementation of one-pot and cascade reactions. It is perhaps the most promising application.
- LDHs intercalated with catalytically active anionic species by direct exchange or reconstruction have not been yet exploited in LDH/carbon compounds. This would be a new, original route to synthesize multifunctional catalysts.
- There is a great need to optimize the interface between the nanocarbons and the LDHs, determining to a large extent the charge transfer and cyclability of the obtained catalysts. This will result from control of the synthesis conditions. Because direct growth of LDH on the nanocarbon by coprecipitation appears the most convenient and largely used method, great effort must be devoted to control the supersaturation level during synthesis. Moreover, coprecipitation of LDH through separated nucleation and aging steps will be more extensively exploited and, when large LDH particle size is needed, the urea synthesis method will be preferred. Furthermore, exfoliation of LDH could be more largely developed, particularly using organic solvents, leading to exceptional results.
- CD-based LDH/nanocarbons offer possibilities to improve the electron transfer and to decorate the metal NPs. These composites have been scarcely studied, although they are potential multifunctional catalysts for cascade reactions.
Therefore, the first concern in the new directions of research involving LDH/nanocarbon catalysts will be the tailored combination of components chosen to provide the specific sites involved in the targeted reactions. Original LDH compositions including rare earth and noble metal cations must be considered. Besides the most adapted nanocarbon, other oxides and/or dopants (e.g., Na+, K+, F−) able to induce, for instance, magnetic properties or improve basicity could be added. A preparation method exploiting several intrinsic properties of the LDHs (exfoliation, reconstruction, anionic exchange) will largely contribute to inducing synergistic effects and porous hierarchical structures. At present, one of the most promising catalytic applications of these materials concerns the valorization of biomass. The LDH/nanocarbon catalysts can be adapted to one-pot and cascade reactions in liquid media, including aqueous media, which has gradually led to their use in this application field. The noticeable advantage of the LDH/nanocarbon composites to obtain good-performing supported metal catalysts at low noble metal loading or avoiding the use of highly dispersed transition metals (Ni, Co, Cu, etc.) without leaching and good recyclability will be largely exploited to develop industrial processes of biomass valorization at low cost. Another promising direction is the synthesis of fine chemicals for pharmaceutical applications, where cascade reactions are particularly suitable to reduce costs and improve the atom economy of the processes.
Author Contributions
Conceptualization, D.T. and M.G.Á.; resources, D.T. and M.G.Á.; writing—original draft preparation, D.T. and M.G.Á.; writing—review and editing D.T. and M.G.Á.; supervision, D.T. and M.G.Á. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Varadwaj, G.B.B.; Nyamori, V.O. Layered double hydroxide- and graphene-based hierarchical nanocomposites: Synthetic strategies and promising applications in energy conversion and conservation. Nano Res. 2016, 9, 3598–3621. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, Q.; Li, B.; Xue, H.; Pang, H.; Chen, C. Recent progress in layered double hydroxide based materials for electrochemical capacitors: Design, synthesis and performance. Nanoscale 2017, 9, 15206–15225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.Q.; Zhang, Q.; Huang, J.Q.; Wei, F. Hierarchical Nanocomposites Derived from Nanocarbons and Layered Double Hydroxides–Properties, Synthesis, and Applications. Adv. Func. Mater. 2012, 22, 675. [Google Scholar] [CrossRef]
- Kulandaivalu, S.; Azman, N.H.N.; Sulaiman, Y. Advances in Layered Double Hydroxide/Carbon Nanocomposites Containing Ni2+ and Co2+/3+ for Supercapacitors. Front. Mater. 2020, 7, 147. [Google Scholar] [CrossRef]
- Tang, C.; Titirici, M.M.; Zhang, Q. A review of nanocarbons in energy electrocatalysis: Multifunctional substrates and highly active sites. J. Energy Chem. 2017, 26, 1077–1093. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Wang, H.F.; Zhu, X.L.; Li, B.Q.; Zhang, Q. Advances in Hybrid Electrocatalysts for Oxygen Evolution Reactions: Rational Integration of NiFe Layered Double Hydroxides and Nanocarbon. Part. Part. Syst. Charact. 2016, 33, 473–486. [Google Scholar] [CrossRef]
- Song, B.; Zeng, Z.; Zeng, G.; Gong, J.; Xiao, R.; Ye, S.; Chen, M.; Lai, C.; Xu, P.; Tang, X. Powerful combination of g-C3N4 and LDHs for enhanced photocatalytic performance: A review of strategy, synthesis, and applications. Adv. Colloid Interface Sci. 2019, 272, 101999. [Google Scholar] [CrossRef]
- Daud, M.; Kamal, M.S.; Shehzad, F.; Al Harthi, M. Graphene/Layered Double Hydroxides Nanocomposites: A Review of Recent Progress in Synthesis and Applications. Carbon 2016, 104, 241–252. [Google Scholar] [CrossRef]
- Gu, P.; Zhang, S.; Li, X.; Wang, X.; Wen, T.; Jehan, R.; Alsaedi, A.; Hayat, T.; Wang, X. Recent advances in layered double hydroxide-based nanomaterials for the removal of radionuclides from aqueous solution. Environ. Pollut. 2018, 240, 493–505. [Google Scholar] [CrossRef]
- Pang, H.; Wu, Y.; Wang, X.; Hu, B.; Wang, X. Recent Advances in Composites of Graphene and Layered Double Hydroxides for Water Remediation: A Review. Chem. Asian J. 2019, 14, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, G.; Li, X. Graphene/layered double hydroxide nanocomposite: Properties, synthesis, and applications. Chem. Eng. J. 2016, 292, 207–223. [Google Scholar] [CrossRef]
- Fan, G.; Li, F.; Evans, D.G.; Duan, X. Catalytic applications of layered double hydroxides: Recent advances and perspectives. Chem. Soc. Rev. 2014, 43, 7040–7066. [Google Scholar] [CrossRef] [PubMed]
- Winter, F.; Van Dillen, A.J.; de Jong, K.P. Supported hydrotalcites as highly active solid base catalysts. Chem. Commun. 2005, 31, 3977–3979. [Google Scholar] [CrossRef] [PubMed]
- Winter, F.; Koot, V.; van Dillen, A.J.; Geus, J.W.; de Jong, K.P. Hydrotalcites supported on carbon nanofibers as solid base catalysts for the synthesis of MIBK. J. Catal. 2005, 236, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.N.; Oh, W.D.; Li, Y.; Hu, X. Nanocarbons as platforms for developing novel catalytic composites: Overview and prospects. Appl. Catal. A Gen. 2018, 562, 94–105. [Google Scholar] [CrossRef]
- Bhuyan, M.S.A.; Uddin, M.N.M.; Islam, M.; Bipasha, F.A.; Hossain, S.S. Synthesis of graphene. Int. Nano Lett. 2016, 6, 65–83. [Google Scholar] [CrossRef] [Green Version]
- Mallakpour, S.; Khadem, E. Carbon nanotube–metal oxide nanocomposites: Fabrication, properties and applications. Chem. Eng. J. 2016, 302, 344–367. [Google Scholar] [CrossRef]
- Sousa, H.B.A.; Martins, C.S.M.; Prior, J.A.V. You Don’t Learn That in School: An Updated Practical Guide to Carbon Quantum Dots. Nanomaterials 2021, 11, 611. [Google Scholar] [CrossRef]
- Feng, L.; Xie, N.; Zhong, J. Carbon Nanofibers and Their Composites: A Review of Synthesizing, Properties and Applications. Materials 2014, 7, 3919–3945. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Tichit, D.; Medina, F.; Llorca, J. Role of the synthesis route on the properties of hybrid LDH-graphene as basic catalysts. Appl. Surf. Sci. 2017, 396, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.S.; Menzel, R.; Wang, Y.; Garcia-Gallastegui, A.; Bawaked, S.M.; Obaid, A.Y.; Basahel, S.N.; Mokhtar, M. Graphene-oxide-supported CuAl and CoAl layered double hydroxides as enhanced catalysts for carbon-carbon coupling via Ullmann reaction. J. Solid State Chem. 2017, 246, 130–137. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, W.; Cui, D.; Lü, C. Mussel-inspired preparation of temperature-responsive polymer brushes modified layered double hydroxides@Pd/carbon dots hybrid for catalytic applications. Appl. Clay Sci. 2021, 200, 105958. [Google Scholar] [CrossRef]
- Tichit, D.; Coq, B. Catalysis by Hydrotalcites and Related Materials. Cattech 2003, 7, 206–217. [Google Scholar] [CrossRef]
- Takehira, K. Recent development of layered double hydroxide-derived catalysts − Rehydration, reconstitution, and supporting, aiming at commercial application. Appl. Clay Sci. 2017, 136, 112–141. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, Tuning, and Exploiting the Basicity of Hydrotalcites for Applications in Heterogeneous Catalysis. Chem. Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef]
- Li, C.; Wei, M.; Evans, D.G.; Duan, X. Layered Double Hydroxide-based Nanomaterials as Highly Efficient Catalysts and Adsorbents. Small 2014, 10, 4469. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Lim, S.Y.; Shen, W.; Gao, Z. Carbon quantum dots and their applications. Chem. Soc. Rev. 2015, 44, 362–391. [Google Scholar] [CrossRef]
- Inagaki, M.; Tsumura, T.; Kinumoto, T.; Toyoda, M. Graphitic carbon nitrides (g-C3N4) with comparative discussion to carbon materials. Carbon 2019, 141, 580–607. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Marcu, I.C.; Tichit, D. Progress in Layered Double Hydroxides—From Synthesis to New Applications; Nocchetti, M., Costantino, U., Eds.; World Scientific Publishing Ltd.: Singapore, 2022; pp. 189–362. [Google Scholar]
- Zheng, Y.; Liu, J.; Liang, J.; Jaroniec, M.; Qiao, S.Z. Graphitic Carbon Nitride Materials: Controllable Synthesis and Applications in Fuel Cells and Photocatalysis. Energy Environ. Sci. 2012, 5, 6717–6731. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, L.; Guan, S.; Zhang, X.; Wang, B.; Cao, X.; Yu, Z.; He, Y.; Evans, D.G.; Feng, J.; et al. Ultrathin and Vacancy-Rich CoAl-Layered Double Hydroxide/Graphite Oxide Catalysts: Promotional Effect of Cobalt Vacancies and Oxygen Vacancies in Alcohol Oxidation. ACS Catal. 2018, 8, 3104–3115. [Google Scholar] [CrossRef]
- Zeynizadeh, B.; Gilanizadeh, M. Synthesis and characterization of a magnetic graphene oxide/Zn–Ni–Fe layered double hydroxide nanocomposite: An efficient mesoporous catalyst for the green preparation of biscoumarins. New J. Chem. 2019, 43, 18794–18804. [Google Scholar] [CrossRef]
- Iqbal, K.; Iqbal, A.; Kirillov, A.M.; Shan, C.; Liu, W.; Tang, Y. A new multicomponent CDs/Ag@Mg–Al–Ce-LDH nanocatalyst for highly efficient degradation of organic water pollutants. J. Mater Chem. A 2018, 6, 4515–4524. [Google Scholar] [CrossRef]
- Wei, Z.; Li, Y.; Dou, L.; Ahmad, M.; Zhang, H. Cu3–xNixAl-Layered Double Hydroxide-Reduced Graphene Oxide Nanosheet Array for the Reduction of 4-Nitrophenol. ACS Appl. Nano Mater. 2019, 2, 2383. [Google Scholar] [CrossRef]
- Rohani, S.; Ziarani, G.M.; Ziarati, A.; Badiei, A. Designer 3D CoAl-layered double hydroxide@N, S doped graphene hollow architecture decorated with Pd nanoparticles for Sonogashira couplings. Appl. Surf. Sci. 2019, 496, 143599. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Crivoi, D.G.; Medina, F.; Tichit, D. Synthesis of Chalcone Using LDH/Graphene Nanocatalysts of Different Compositions. ChemEngineering 2019, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Fan, G.; Yang, L.; Li, F. Highly Efficient Hybrid Cobalt–Copper–Aluminum Layered Double Hydroxide/Graphene Nanocomposites as Catalysts for the Oxidation of Alkylaromatics. ChemCatChem 2016, 8, 363–371. [Google Scholar] [CrossRef]
- Dou, L.; Zhang, H. Facile assembly of nanosheet array-like CuMgAl-layered double hydroxide/rGO nanohybrids for highly efficient reduction of 4-nitrophenol. J. Mater. Chem. A 2016, 4, 18990–19002. [Google Scholar] [CrossRef]
- Miao, M.Y.; Feng, J.T.; Jin, Q.; He, Y.F.; Liu, Y.N.; Du, Y.Y.; Zhang, N.; Li, D.Q. Hybrid Ni–Al layered double hydroxide/graphene composite supported gold nanoparticles for aerobic selective oxidation of benzyl alcohol. RSC Adv. 2015, 5, 36066–36074. [Google Scholar] [CrossRef]
- Dou, L.; Wang, Y.; Li, Y.; Zhang, H. Novel core–shell-like nanocomposites xCu@Cu2O/MgAlO-rGO through an in situ self-reduction strategy for highly efficient reduction of 4-nitrophenol. Dalton Trans. 2017, 46, 15836–15847. [Google Scholar] [CrossRef]
- Shan, Y.; Yu, C.; Yang, J.; Dong, Q.; Fan, X.; Qiu, J. Thermodynamically Stable Pickering Emulsion Configured with Carbon-Nanotube-Bridged Nanosheet-Shaped Layered Double Hydroxide for Selective Oxidation of Benzyl Alcohol. ACS Appl. Mater. Interfaces 2015, 7, 12203–12209. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Liu, G.; Wang, L.; Liu, J.; Yue, Y.; Zhang, L.; Liu, Y. Growing layered double hydroxides on CNTs and their catalytic performance for higher alcohol synthesis from syngas. J. Mater. Sci. 2016, 51, 5216–5231. [Google Scholar] [CrossRef]
- Celaya-Sanfiz, A.; Morales-Vega, N.; De Marco, M.; Iruretagoyena, D.; Mokhtar, M.; Bawaked, S.M.; Basahel, S.N.; Al Thabaiti, S.A.; Alyoubi, A.O.; Shaffer, M.S.P. Self-condensation of acetone over Mg–Al layered double hydroxide supported on multi-walled carbon nanotube catalysts. J. Mol. Catal. A Chem. 2015, 398, 50–57. [Google Scholar] [CrossRef]
- Lan, M.; Fan, G.; Sun, W.; Li, F. Synthesis of hybrid Zn–Al–In mixed metal oxides/carbon nanotubes composite and enhanced visible-light-induced photocatalytic performance. Appl. Surf. Sci. 2013, 282, 937–946. [Google Scholar] [CrossRef]
- Wang, H.; Xiang, X.; Li, F. Hybrid ZnAl-LDH/CNTs nanocomposites: Noncovalent assembly and enhanced photodegradation performance. AIChE J. 2010, 56, 768–778. [Google Scholar] [CrossRef]
- Wang, J.; Fan, G.; Li, F. A hybrid nanocomposite precursor route to synthesize dispersion-enhanced Ni catalysts for the selective hydrogenation of o-chloronitrobenzene. Catal. Sci. Technol. 2013, 3, 982–991. [Google Scholar] [CrossRef]
- Wang, L.; Cao, A.; Liu, G.; Zhang, L.; Liu, Y. Bimetallic CuCo nanoparticles derived from hydrotalcite supported on carbon fibers for higher alcohols synthesis from syngas. Appl. Surf. Sci. 2016, 360, 77–85. [Google Scholar] [CrossRef]
- Guo, Y.; Fan, L.; Liu, M.; Yang, L.; Fan, G.; Li, F. Nitrogen-Doped Carbon Quantum Dots-Decorated Mg-Al Layered Double Hydroxide-Supported Gold Nanocatalysts for Efficient Base-Free Oxidation of Benzyl Alcohol. Ind. Eng. Chem. Res. 2020, 59, 636–646. [Google Scholar] [CrossRef]
- Roelofs, J.C.A.A.; Lensveld, D.J.; van Dillen, A.J.; de Jong, K.P. On the Structure of Activated Hydrotalcites as Solid Base Catalysts for Liquid-Phase Aldol Condensation. J. Catal. 2001, 203, 184–191. [Google Scholar] [CrossRef]
- Roelofs, J.C.A.A.; van Dillen, A.J.; de Jong, K.P. Base-catalyzed condensation of citral and acetone at low temperature using modified hydrotalcite catalysts. Catal. Today 2000, 60, 297–303. [Google Scholar] [CrossRef]
- Roelofs, J.C.A.A.; van Dillen, A.J.; de Jong, K.P. Condensation of citral and ketones using activated hydrotalcite catalysts. Catal. Lett. 2001, 74, 91–94. [Google Scholar] [CrossRef]
- Wang, Y.; Dou, L.; Zhang, H. Nanosheet Array-Like Palladium-Catalysts Pdx/rGO@CoAl-LDH via Lattice Atomic-Confined in Situ Reduction for Highly Efficient Heck Coupling Reaction. ACS Appl. Mater. Interfaces 2017, 9, 38784–38795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, Z.; Zhao, Y.; Miras, H.N.; Song, Y.F. Precise Control of the Oriented Layered Double Hydroxide Nanosheets Growth on Graphene Oxides Leading to Efficient Catalysts for Cascade Reactions. ChemCatChem 2019, 11, 5466–5474. [Google Scholar] [CrossRef]
- Huang, P.; Liu, J.; Wei, F.; Zhu, Y.; Wang, X.; Cao, C.; Song, W. Size-selective adsorption of anionic dyes induced by the layer space in layered double hydroxide hollow microspheres. Mater. Chem. Front. 2017, 1, 1550–1555. [Google Scholar] [CrossRef]
- Costantino, U.; Marmottini, F.; Nocchetti, M.; Vivani, R. New Synthetic Routes to Hydrotalcite-Like Compounds—Characterisation and Properties of the Obtained Materials. Eur. J. Inorg. Chem. 1998, 10, 1439–1446. [Google Scholar] [CrossRef]
- Adachi-Pagano, M.; Forano, C.; Besse, J.P. Synthesis of Al-rich hydrotalcite-like compounds by using the urea hydrolysis reaction—control of size and morphology. J. Mater. Chem. 2003, 13, 1988–1993. [Google Scholar] [CrossRef]
- Ogawa, M.; Kaiho, H. Homogeneous Precipitation of Uniform Hydrotalcite Particles. Langmuir 2002, 18, 4240–4242. [Google Scholar] [CrossRef]
- Modesto-Lopez, L.B.; Chimentao, R.J.; Álvarez, M.G.; Rosell-Llompart, J.; Medina, F.; Llorca, J. Direct growth of hydrotalcite nanolayers on carbon fibers by electrospinning. Appl. Clay Sci. 2014, 101, 461–467. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Frey, A.M.; Bitter, J.H.; Segarra, A.M.; de Jong, K.P.; Medina, F. On the role of the activation procedure of supported hydrotalcites for base catalyzed reactions: Glycerol to glycerol carbonate and self-condensation of acetone. Appl. Catal. B Environ. 2013, 134, 231–237. [Google Scholar] [CrossRef]
- Stamate, A.E.; Pavel, O.D.; Zavoianu, R.; Brezestean, I.; Ciorita, A.; Birjega, R.; Neubauer, K.; Koeckritz, A.; Marcu, I.C. Ce-Containing MgAl-Layered Double Hydroxide-Graphene Oxide Hybrid Materials as Multifunctional Catalysts for Organic Transformations. Materials 2021, 14, 7457. [Google Scholar] [CrossRef]
- Zhao, Y.; Xie, R.; Lin, Y.; Fan, G.; Li, F. Highly efficient solvent-free aerobic oxidation of ethylbenzene over hybrid Zn–Cr layered double hydroxide/carbon nanotubes nanocomposite. Catal. Commun. 2018, 114, 65–69. [Google Scholar] [CrossRef]
- Shen, J.; Ye, S.; Xu, X.; Liang, J.; He, G.; Chen, H. Reduced graphene oxide based NiCo layered double hydroxide nanocomposites: An efficient catalyst for epoxidation of styrene. Inorg. Chem. Commun. 2019, 104, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, E.; Gholami, M.R. Pt–NiO–Al2O3/G derived from graphene-supported layered double hydroxide as efficient catalyst for p-nitrophenol reduction. Res. Chem. Intermed. 2017, 43, 5829–5839. [Google Scholar] [CrossRef]
- Xie, R.; Fan, G.; Ma, Q.; Yang, L.; Li, F. Facile synthesis and enhanced catalytic performance of graphene-supported Ni nanocatalyst from a layered double hydroxide-based composite precursor. J. Mater. Chem. A 2014, 2, 7880–7889. [Google Scholar] [CrossRef]
- Xia, S.; Zheng, L.; Ning, W.; Wang, L.; Chen, P.; Hou, Z. Multiwall carbon nanotube-pillared layered Cu0.4/Mg5.6Al2O8.6: An efficient catalyst for hydrogenolysis of glycerol. J. Mater. Chem. A 2013, 1, 11548–11552. [Google Scholar] [CrossRef]
- Cao, A.; Yang, Q.; Wei, Y.; Zhang, L.; Liu, Y. Synthesis of higher alcohols from syngas over CuFeMg-LDHs/CFs composites. Int. J. Hydrogen Energy 2017, 42, 17425–17434. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Gao, Z.H.; Kou, W.; Liu, Y.; Huang, W. Direct Synthesis of Isobutanol from Syngas over Nanosized Cu/ZnO/Al2O3 Catalysts Derived from Hydrotalcite-like Materials Supported on Carbon Fibers. Energy Fuels 2017, 31, 8572–8579. [Google Scholar] [CrossRef]
- Kou, J.W.; Cheng, S.Y.; Gao, Z.H.; Cheng, F.Q.; Huang, H. Synergistic effects of potassium promoter and carbon fibers on direct synthesis of isobutanol from syngas over Cu/ZnO/Al2O3 catalysts obtained from hydrotalcite-like compounds. Solid State Sci. 2019, 87, 138–145. [Google Scholar] [CrossRef]
- Tichit, D.; Naciri Bennani, M.; Figueras, F.; Tessier, R.; Kervennal, J. Aldol condensation of acetone over layered double hydroxides of the meixnerite type. Appl. Clay Sci. 1998, 13, 401–415. [Google Scholar] [CrossRef]
- Abello, S.; Medina, F.; Tichit, D.; Perez Ramirez, J.; Cesteros, Y.; Salagre, P.; Sueiras, J.E. Nanoplatelet-based reconstructed hydrotalcites: Towards more efficient solid base catalysts in aldol condensations. Chem. Commun. 2005, 11, 1453–1455. [Google Scholar] [CrossRef]
- Abello, S.; Medina, F.; Tichit, D.; Perez Ramirez, J.; Groen, J.C.; Sueiras, S.E.; Salagre, P.; Cesteros, Y. Aldol Condensations over Reconstructed Mg–Al Hydrotalcites: Structure–Activity Relationships Related to the Rehydration Method. Chem. Eur. J.— Eur. J. 2005, 11, 728–739. [Google Scholar] [CrossRef]
- Evranos Aksoz, B.; Ertan, R. Chemical and Structural Properties of Chalcones. IFABAD J. Pharm. Sci. 2011, 36, 223–242. [Google Scholar]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yáñez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A Valid Scaffold for Monoamine Oxidases Inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Hargrove-Leak, S.C.; Amiridis, M.D. Substitution effects in the heterogeneous catalytic synthesis of flavanones over MgO. Catal. Commun. 2002, 3, 557–563. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S.; Velty, A. Activated hydrotalcites as catalysts for the synthesis of chalcones of pharmaceutical interest. J. Catal. 2004, 221, 474–482. [Google Scholar] [CrossRef]
- Solhy, A.; Tahir, R.; Sebti, S.; Skouta, R.; Bousmina, M.; Zahouily, M.; Lazrek, M. Efficient synthesis of chalcone derivatives catalyzed by re-usable hydroxyapatite. Appl. Catal. A Gen. 2010, 374, 189–193. [Google Scholar] [CrossRef]
- Jioui, I.; Danoun, K.; Solhy, A.; Jouiad, M.; Zahouily, M.; Essaid, B.; Len, C.; Fihri, A. Modified fluorapatite as highly efficient catalyst for the synthesis of chalcones via Claisen–Schmidt condensation reaction. J. Ind. Eng. Chem. 2016, 39, 218–225. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Segarra, A.M.; Contreras, S.; Sueiras, J.E.; Medina, F.; Figueras, F. Enhanced use of renewable resources: Transesterification of glycerol catalyzed by hydrotalcite-like compounds. Chem. Eng. J. 2010, 161, 340–345. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Chimentão, R.J.; Figueras, F.; Medina, F. Tunable basic and textural properties of hydrotalcite derived materials for transesterification of glycerol. Appl. Clay Sci. 2012, 58, 16–24. [Google Scholar] [CrossRef]
- Albadi, J.; Mansournezhad, A.; Salehnasab, S. Green synthesis of biscoumarin derivatives catalyzed by recyclable CuO–CeO2 nanocomposite catalyst in water. Res. Chem. Intermed. 2015, 41, 5713–5721. [Google Scholar] [CrossRef]
- Sadeghi, B.; Tayebe, Z. A Fast, Highly Efficient, and Green Protocol for Synthesis of Biscoumarins Catalyzed by Silica Sulfuric Acid Nanoparticles as a Reusable Catalyst. J. Chem. 2013, 2013, 179013. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, P.; Katyal, A.; Kalra, R.; Dass, S.K.; Prakash, S.; Chandra, R. Phosphotungstic Acid: An Efficient Catalyst for the Aqueous Phase Synthesis of Bis-(4-hydroxycoumarin-3-yl)methanes. Catal. Lett. 2010, 134, 303–308. [Google Scholar] [CrossRef]
- Jo, J.W.K.; Kumar, S.; Tonda, S. N-doped C dot/CoAl-layered double hydroxide/g-C3N4 hybrid composites for efficient and selective solar-driven conversion of CO2 into CH4. Compos. Part B 2019, 176, 107212. [Google Scholar] [CrossRef]
- Nava Andrade, K.; Knauth, P.; López, Z.; Hirata, G.A.; Guevara Martinez, S.J.; Carbajal Arízaga, G.G. Assembly of folate-carbon dots in GdDy-doped layered double hydroxides for targeted delivery of doxorubicin. Appl. Clay Sci. 2020, 192, 105661. [Google Scholar] [CrossRef]
- Liu, W.; Liang, R.; Lin, Y. Confined synthesis of carbon dots with tunable long-wavelength emission in a 2-dimensional layered double hydroxide matrix. Nanoscale 2020, 12, 7888–7894. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Antonietti, M. Polymeric Graphitic Carbon Nitride as a Heterogeneous Organocatalyst: From Photochemistry to Multipurpose Catalysis to Sustainable Chemistry. Angew. Chem. Int. Ed. 2012, 51, 68–89. [Google Scholar] [CrossRef]
- Watanabe, H.; Asano, S.; Fujita, S.I.; Yoshida, H.; Arai, M. Nitrogen-Doped, Metal-Free Activated Carbon Catalysts for Aerobic Oxidation of Alcohols. ACS Catal. 2015, 5, 2886–2894. [Google Scholar] [CrossRef]
- Xie, R.; Fan, G.; Yang, L.; Li, F. Solvent-free oxidation of ethylbenzene over hierarchical flower-like core–shell structured Co-based mixed metal oxides with significantly enhanced catalytic performance. Catal. Sci. Technol. 2015, 5, 540–548. [Google Scholar] [CrossRef]
- Xie, R.; Fan, G.; Yang, L.; Li, F. Hierarchical flower-like Co–Cu mixed metal oxide microspheres as highly efficient catalysts for selective oxidation of ethylbenzene. Chem. Eng. J. 2016, 288, 169–178. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, S.J.; Gao, S.Y.; Liu, X.F.; Long, S.F.; Kong, Y. In situ embedding of ultra-fine nickel oxide nanoparticles in HMS with enhanced catalytic activities of styrene epoxidation. Microporous Mesoporous Mater. 2017, 238, 69–77. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chen, T.T.; Jian, P.M.; Wang, L.X. Hierarchical 0D/2D Co3O4 hybrids rich in oxygen vacancies as catalysts towards styrene epoxidation reaction. Chin. J. Catal. 2018, 39, 1942–1950. [Google Scholar] [CrossRef]
- Huang, C.L.; Zhang, H.Y.; Sun, Z.Y.; Zhao, Y.F.; Chen, S.; Tao, R.T.; Liu, Z.M. Porous Fe3O4 nanoparticles: Synthesis and application in catalyzing epoxidation of styrene. J. Colloid Interface Sci. 2011, 364, 298–303. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Z.; Fan, G.; Li, F. The promotional effect of ZnO addition to supported Ni nanocatalysts from layered double hydroxide precursors on selective hydrogenation of citral. Catal. Sci. Technol. 2014, 4, 1123–1131. [Google Scholar] [CrossRef]
- Ma, N.; Song, Y.; Han, F.; Waterhouse, G.I.N.; Li, Y.; Ai, S. Highly selective hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran at low temperature over a Co–N–C/NiAl-MMO catalyst. Catal. Sci. Technol. 2020, 10, 4010–4018. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Fan, G.; Li, F. Synthesis of a novel Ni/C catalyst derived from a composite precursor for hydrodechlorination. Catal. Commun. 2012, 19, 56–60. [Google Scholar] [CrossRef]
- Wang, J.; Fan, G.; Li, F. Carbon-supported Ni catalysts with enhanced metal dispersion and catalytic performance for hydrodechlorination of chlorobenzene. RSC Adv. 2012, 2, 9976–9985. [Google Scholar] [CrossRef]
- Jiang, J.; Lim, Y.S.; Park, S.; Kim, S.H.; Yoon, S.; Piao, L. Hollow porous Cu particles from silica-encapsulated Cu2O nanoparticle aggregates effectively catalyze 4-nitrophenol reduction. Nanoscale 2017, 9, 3873–3880. [Google Scholar] [CrossRef]
- Bai, S.; Shen, X.P.; Zhu, G.X.; Li, M.Z.; Xi, H.T.; Chen, K.M. In situ Growth of NixCo100–x Nanoparticles on Reduced Graphene Oxide Nanosheets and Their Magnetic and Catalytic Properties. ACS Appl. Mater. Interf. 2012, 4, 2378–2386. [Google Scholar] [CrossRef]
- Bordbar, M.; Negahdar, N.; Nasrollahzadeh, M.M. Melissa Officinalis L. leaf extract assisted green synthesis of CuO/ZnO nanocomposite for the reduction of 4-nitrophenol and Rhodamine B. Sep. Purif. Technol. 2018, 191, 295–300. [Google Scholar] [CrossRef]
- Pang, J.J.; Li, W.T.; Cao, Z.H.; Xu, J.J.; Li, X.; Zhang, X.K. Mesoporous Cu2O–CeO2 composite nanospheres with enhanced catalytic activity for 4-nitrophenol reduction. Appl. Surf. Sci. 2018, 439, 420–429. [Google Scholar] [CrossRef]
- Ye, W.C.; Yu, J.; Zhou, X.X.; Gao, D.Q.; Wang, D.A.; Wang, C.M.; Xue, D.S. Green synthesis of Pt–Au dendrimer-like nanoparticles supported on polydopamine-functionalized graphene and their high performance toward 4-nitrophenol reduction. Appl. Catal. B 2016, 181, 371–378. [Google Scholar] [CrossRef]
- Konar, S.; Kalita, H.; Puvvada, N.; Tantubay, S.; Mahto, M.K.; Biswas, S.; Pathak, A. Shape-dependent catalytic activity of CuO nanostructures. J. Catal. 2016, 336, 11–22. [Google Scholar] [CrossRef]
- Sasmal, A.K.; Dutta, S.; Pal, T. A ternary Cu2O–Cu–CuO nanocomposite: A catalyst with intriguing activity. Dalton Trans. 2016, 45, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Chen, R.F.; Liu, W.K.; Wu, J.M.; Gao, D. Catalytic reduction of 4-nitrophenol over Ni-Pd nanodimers supported on nitrogen-doped reduced graphene oxide. J. Hazard. Mater. 2016, 320, 96–104. [Google Scholar] [CrossRef]
- Wang, X.; Liu, D.P.; Song, S.Y.; Zhang, H.J. Pt@CeO2 Multicore@Shell Self-Assembled Nanospheres: Clean Synthesis, Structure Optimization, and Catalytic Applications. J. Am. Chem. Soc. 2013, 135, 15864–15872. [Google Scholar] [CrossRef]
- Jiang, Y.F.; Yuan, C.Z.; Xie, X.; Zhou, X.; Jiang, N.; Wang, X.; Imran, M.; Xu, A.W. A Novel Magnetically Recoverable Ni-CeO2–x/Pd Nanocatalyst with Superior Catalytic Performance for Hydrogenation of Styrene and 4-Nitrophenol. ACS Appl. Mater. Interfaces 2017, 9, 9756–9762. [Google Scholar] [CrossRef]
- Wu, G.Q.; Liang, X.Y.; Zhang, L.J.; Tang, Z.Y.; Al-Mamun, M.; Zhao, H.J.; Su, X.T. Fabrication of Highly Stable Metal Oxide Hollow Nanospheres and Their Catalytic Activity toward 4-Nitrophenol Reduction. ACS Appl. Mater. Interfaces 2017, 9, 18207–18214. [Google Scholar] [CrossRef]
- Yu, C.; Fu, J.J.; Muzzio, M.; Shen, T.; Su, D.; Zhu, J.J.; Sun, S.H. CuNi Nanoparticles Assembled on Graphene for Catalytic Methanolysis of Ammonia Borane and Hydrogenation of Nitro/Nitrile Compounds. Chem. Mater. 2017, 29, 1413–1418. [Google Scholar] [CrossRef]
- Fang, H.; Wen, M.; Chen, H.X.; Wu, Q.S.; Li, W.Y. Graphene stabilized ultra-small CuNi nanocomposite with high activity and recyclability toward catalysing the reduction of aromatic nitro-compounds. Nanoscale 2016, 8, 536–542. [Google Scholar] [CrossRef]
- Kohantorabi, M.; Gholami, M.R. Kinetic Analysis of the Reduction of 4-Nitrophenol Catalyzed by CeO2 Nanorods-Supported CuNi Nanoparticles. Ind. Eng. Chem. Res. 2017, 56, 1159–1167. [Google Scholar] [CrossRef]
- Krishna, R.; Fernandes, D.M.; Ventura, J.; Freire, C.; Titus, E. Novel synthesis of highly catalytic active Cu@Ni/RGO nanocomposite for efficient hydrogenation of 4-nitrophenol organic pollutant. Int. J. Hydrogen Energy 2016, 41, 11608–11615. [Google Scholar] [CrossRef]
- Qi, H.T.; Yu, P.; Wang, Y.X.; Han, G.C.; Liu, H.B.; Yi, Y.P.; Li, Y.L.; Mao, L.Q. Graphdiyne Oxides as Excellent Substrate for Electroless Deposition of Pd Clusters with High Catalytic Activity. J. Am. Chem. Soc. 2015, 137, 5260–5263. [Google Scholar] [CrossRef]
- Mallick, K.; Witcomb, M.; Scurrell, M. Silver nanoparticle catalysed redox reaction: An electron relay effect. Mater. Chem. Phys. 2006, 97, 283–287. [Google Scholar] [CrossRef]
- Mandlimath, T.R.; Gopal, B. Catalytic activity of first row transition metal oxides in the conversion of p-nitrophenol to p-aminophenol. J. Mol. Catal. A Chem. 2011, 350, 9–15. [Google Scholar] [CrossRef]
- Chen, J.; Yao, N.; Wang, R.; Zhang, J. Hydrogenation of chloronitrobenzene to chloroaniline over Ni/TiO2 catalysts prepared by sol–gel method. Chem. Eng. J. 2009, 148, 164–172. [Google Scholar] [CrossRef]
- Yan, X.; Sun, J.; Wang, Y.; Yang, J. A Fe-promoted Ni–P amorphous alloy catalyst (Ni–Fe–P) for liquid phase hydrogenation of m- and p-chloronitrobenzene. J. Mol. Catal. A Chem. 2006, 252, 17–22. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, X.; Yang, N.; Tantai, X.; Xiao, X.; Jiang, B.; Zhang, L. Morphology-Controlled Synthesis of Three-Dimensional Hierarchical Flowerlike Mg–Al Layered Double Hydroxides with Enhanced Catalytic Activity for Transesterification. Ind. Eng. Chem. Res. 2019, 58, 7937–7947. [Google Scholar] [CrossRef]
- Balsamo Mendieta, S.; Heredia, A.; Crivello, M. Nanoclays as dispersing precursors of La and Ce oxide catalysts to produce high-valued derivatives of biodiesel by-product. Mol. Catal. 2020, 481, 110290. [Google Scholar] [CrossRef]
- Álvarez, M.G.; Chimentão, R.; Barrabés, N.; Föttinger, K.; Gispert-Guirado, F.; Kleymenov, E.; Tichit, D.; Medina, F. Structure evolution of layered double hydroxides activated by ultrasound induced reconstruction. Appl. Clay Sci. 2013, 83, 1–11. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Cong, X.; Liu, S.; Zhou, D. Influence of Zr on the performance of Mg-Al catalysts via hydrotalcite-like precursors for the synthesis of glycerol carbonate from urea and glycerol. Appl. Catal. A Gen. 2018, 555, 36–46. [Google Scholar] [CrossRef]
- Sangkhum, P.; Yanamphorn, J.; Wangriya, A.; Ngamcharussrivichai, C. Ca–Mg–Al ternary mixed oxides derived from layered double hydroxide for selective etherification of glycerol to short-chain polyglycerols. Appl. Clay Sci. 2019, 173, 79–87. [Google Scholar] [CrossRef]
- Li, X.; Zheng, L.; Hou, Z. Acetalization of glycerol with acetone over Co[II](Co[III]xAl2−x)O4 derived from layered double hydroxide. Fuel 2018, 233, 565–571. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S.; Rajaei, K.; Tarighi, S. Oxidation of bio-renewable glycerol to value-added chemicals through catalytic and electro-chemical processes. Appl. Energy 2018, 230, 1347–1379. [Google Scholar] [CrossRef]
- Jing, F.; Liu, S.; Wang, R.; Li, X.; Yan, Z.; Luo, S.; Chu, W. Hydrogen production through glycerol steam reforming over the NiCexAl catalysts. Renew. Energy 2020, 158, 192–201. [Google Scholar] [CrossRef]
- Gao, W.; Zhao, Y.; Liu, J.; Huang, Q.; He, S.; Li, C.; Zhao, J.; Wei, M. Catalytic conversion of syngas to mixed alcohols over CuFe-based catalysts derived from layered double hydroxides. Catal. Sci. Technol. 2013, 3, 1324–1332. [Google Scholar] [CrossRef]
- Han, X.; Fang, K.; Sun, Y. Effects of metal promotion on CuMgFe catalysts derived from layered double hydroxides for higher alcohol synthesis via syngas. RSC Adv. 2015, 5, 51868–51874. [Google Scholar] [CrossRef]
- Han, X.; Fang, K.; Zhou, J.; Zhao, L.; Sun, Y. Synthesis of higher alcohols over highly dispersed Cu–Fe based catalysts derived from layered double hydroxides. J. Colloid Interface Sci. 2016, 470, 162–171. [Google Scholar] [CrossRef]
- Ning, X.; An, Z.; He, J. Remarkably efficient CoGa catalyst with uniformly dispersed and trapped structure for ethanol and higher alcohol synthesis from syngas. J. Catal. 2016, 340, 236–247. [Google Scholar] [CrossRef]
- Liao, P.; Zhang, C.; Zhang, L.; Yang, Y.; Zhong, L.; Wang, H.; Sun, Y. Higher alcohol synthesis via syngas over CoMn catalysts derived from hydrotalcite-like precursors. Catal. Today 2018, 311, 56–64. [Google Scholar] [CrossRef]
- Cao, A.; Liu, G.; Yue, Y.; Zhang, L.; Liu, Y. Nanoparticles of Cu–Co alloy derived from layered double hydroxides and their catalytic performance for higher alcohol synthesis from syngas. RSC Adv. 2015, 5, 58804–58812. [Google Scholar] [CrossRef]
- Xiao, K.; Bao, Z.; Qi, X.; Wang, X.; Zhong, L.; Lin, M. Unsupported CuFe bimetallic nanoparticles for higher alcohol synthesis via syngas. Catal. Commun. 2013, 40, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Qiu, M.; Liu, J.; Li, Y.; Wang, T.; Ma, L.; Wu, C. Influence of manganese promoter on co-precipitated Fe–Cu based catalysts for higher alcohols synthesis. Fuel 2013, 109, 21–27. [Google Scholar] [CrossRef]
- Kattel, S.; Ramirez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.J.; Anderson, R.B. A chain growth scheme for the higher alcohols synthesis. J. Catal. 1984, 85, 428–436. [Google Scholar] [CrossRef]
- Nunan, J.G.; Bogdan, C.E.; Klier, K.; Smith, K.J.; Young, C.W.; Herman, R.G. Methanol and C2 oxygenate synthesis over cesium doped CuZnO and Cu/ZnO/Al2O3 catalysts: A study of selectivity and 13C incorporation patterns. J. Catal. 1988, 113, 410–433. [Google Scholar] [CrossRef]
- Nunan, J.G.; Herman, R.G.; Klier, K. Higher alcohol and oxygenate synthesis over Cs/Cu/ZnO/M2O3 (M = Al, Cr) catalysts. J. Catal. 1989, 116, 222–229. [Google Scholar] [CrossRef]
- Spivey, J.J.; Egbebi, A. Heterogeneous catalytic synthesis of ethanol from biomass-derived syngas. Chem. Soc. Rev. 2007, 36, 1514–1528. [Google Scholar] [CrossRef]
- Subramani, V.; Gangwal, S.K. A Review of Recent Literature to Search for an Efficient Catalytic Process for the Conversion of Syngas to Ethanol. Energy Fuels 2008, 22, 814–839. [Google Scholar] [CrossRef]
- Herman, R.G. Advances in catalytic synthesis and utilization of higher alcohols. Catal. Today 2000, 55, 233–245. [Google Scholar] [CrossRef]
- Li, L.; Zhu, Z.H.; Yan, Z.F.; Lu, G.Q.; Rintoul, L. Catalytic ammonia decomposition over Ru/carbon catalysts: The importance of the structure of carbon support. Appl. Catal. A Gen. 2007, 320, 166–172. [Google Scholar] [CrossRef]
- Nie, R.; Wang, J.; Wang, L.; Qin, Y.; Chen, P.; Hou, Z. Platinum supported on reduced graphene oxide as a catalyst for hydrogenation of nitroarenes. Carbon 2012, 50, 586–596. [Google Scholar] [CrossRef]
- Ye, A.; Fan, W.; Zhang, Q.; Deng, W.; Wang, Y. CdS–graphene and CdS–CNT nanocomposites as visible-light photocatalysts for hydrogen evolution and organic dye degradation. Catal. Sci. Technol. 2012, 2, 969–978. [Google Scholar] [CrossRef]
- Li, Y.; Gao, W.; Ci, L.; Wang, C.; Ajayan, P.M. Catalytic performance of Pt nanoparticles on reduced graphene oxide for methanol electro-oxidation. Carbon 2010, 48, 1124–1130. [Google Scholar] [CrossRef]
- Luo, X.; Yuan, S.; Pan, X.; Zhang, C.; Du, S.; Liu, Y. Synthesis and Enhanced Corrosion Protection Performance of Reduced Graphene Oxide Nanosheet/ZnAl Layered Double Hydroxide Composite Films by Hydrothermal Continuous Flow Method. ACS Appl. Mater. Interfaces 2017, 9, 18263–18275. [Google Scholar] [CrossRef]
- Tichit, D.; Layrac, G.; Gérardin, C. Synthesis of layered double hydroxides through continuous flow processes: A review. Chem. Eng. J. 2019, 369, 302–332. [Google Scholar] [CrossRef]
Figure 1.
(a) Schematic synthesis of LDH@PDA@PNIPAM@Pd/CDs and LDH@PDA@PNIPAM@Pd catalysts. Reproduced with permission [22]. Copyright 2021, Elsevier. (b) Schematic synthesis strategy for nanoarray-like xCu-LDH/rGO nanohybrids. Reproduced with permission [35]. Copyright 2019, ACS. (c) Formation process of hollow flower-like and SEM images of CoAl-LDH@N,S-G@Pd. Reproduced with permission [36]. Copyright 2019, Elsevier.
Figure 1.
(a) Schematic synthesis of LDH@PDA@PNIPAM@Pd/CDs and LDH@PDA@PNIPAM@Pd catalysts. Reproduced with permission [22]. Copyright 2021, Elsevier. (b) Schematic synthesis strategy for nanoarray-like xCu-LDH/rGO nanohybrids. Reproduced with permission [35]. Copyright 2019, ACS. (c) Formation process of hollow flower-like and SEM images of CoAl-LDH@N,S-G@Pd. Reproduced with permission [36]. Copyright 2019, Elsevier.

Figure 2.
(a) −ln(1−C) against time (t) and Arrhenius plots for the Heck reaction of iodobenzene and styrene on Pdx/rGO@CoAl-LDH at varied temperatures; (b) plausible mechanism for the Heck coupling reaction of aryl halides with styrene on Pdx/rGO@CoAl-LDH catalysts. Reproduced with permission [53]. Copyright 2017, ACS. (c) The possible reaction pathways for the oxidation of BA over the CoAl-ELDH/GO catalyst. Reproduced with permission [32]. Copyright 2018, ACS.
Figure 2.
(a) −ln(1−C) against time (t) and Arrhenius plots for the Heck reaction of iodobenzene and styrene on Pdx/rGO@CoAl-LDH at varied temperatures; (b) plausible mechanism for the Heck coupling reaction of aryl halides with styrene on Pdx/rGO@CoAl-LDH catalysts. Reproduced with permission [53]. Copyright 2017, ACS. (c) The possible reaction pathways for the oxidation of BA over the CoAl-ELDH/GO catalyst. Reproduced with permission [32]. Copyright 2018, ACS.

Figure 3.
(a) HRTEM image of ZnCr-LDH/CNT composite; (b) ZnCr-LDH/CNT performance with single molecular oxygen, single TBHP (0.5 mL, 70% aqueous solution), and mixed oxidants of O2 and TBHP, respectively (reaction conditions: EB, 10 mL; PO2, 1.0 MPa; 130 °C; catalyst, 0.1 g). Reproduced with permission [62]. Copyright 2018, Elsevier. (c,d) SEM and TEM images of NiCo-LDH/RGO5 and (e) proposed reaction pathway for the epoxidation for styrene. Reproduced with permission [63]. Copyright 2019, Elsevier.
Figure 3.
(a) HRTEM image of ZnCr-LDH/CNT composite; (b) ZnCr-LDH/CNT performance with single molecular oxygen, single TBHP (0.5 mL, 70% aqueous solution), and mixed oxidants of O2 and TBHP, respectively (reaction conditions: EB, 10 mL; PO2, 1.0 MPa; 130 °C; catalyst, 0.1 g). Reproduced with permission [62]. Copyright 2018, Elsevier. (c,d) SEM and TEM images of NiCo-LDH/RGO5 and (e) proposed reaction pathway for the epoxidation for styrene. Reproduced with permission [63]. Copyright 2019, Elsevier.


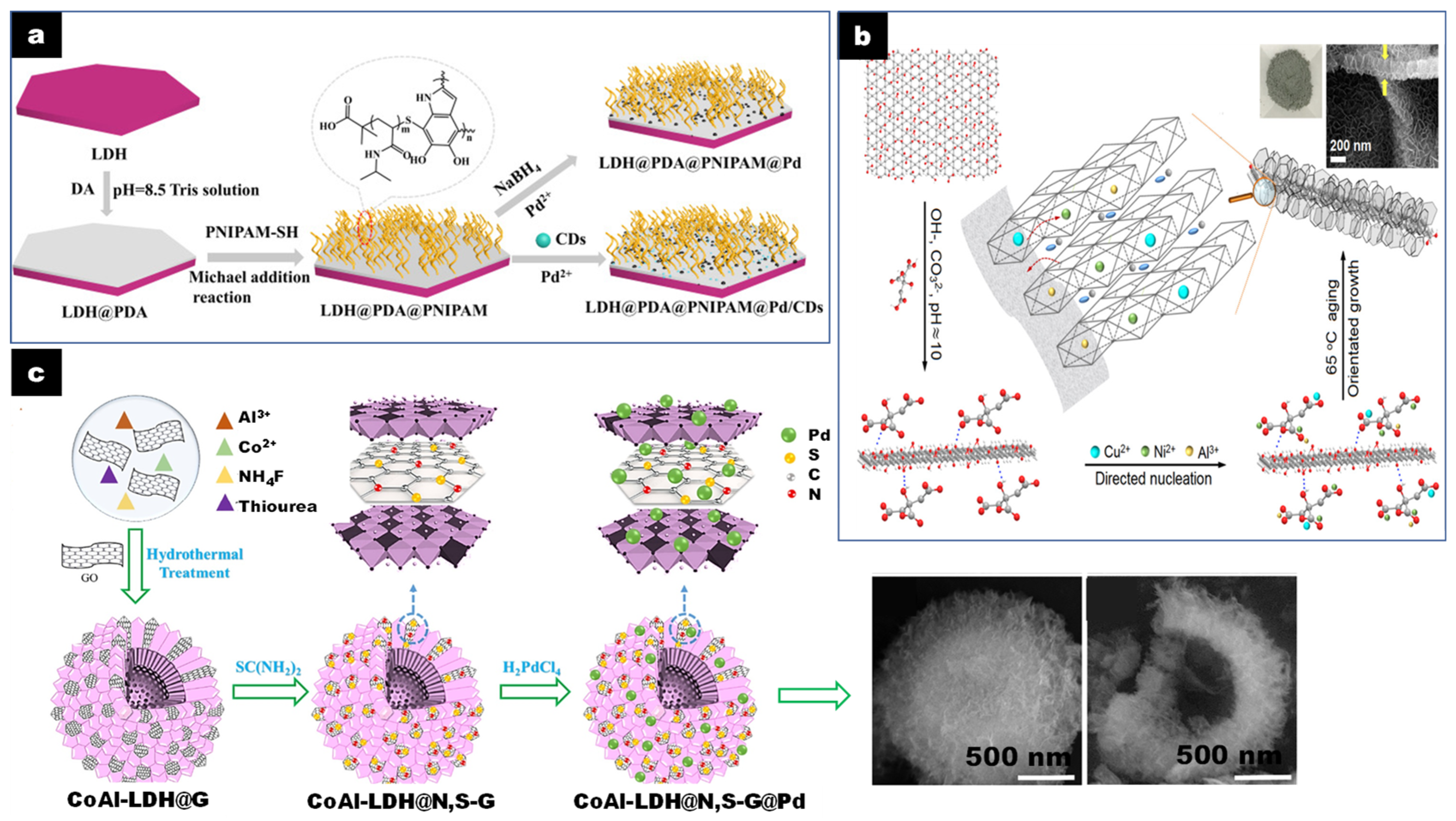{kind=link}
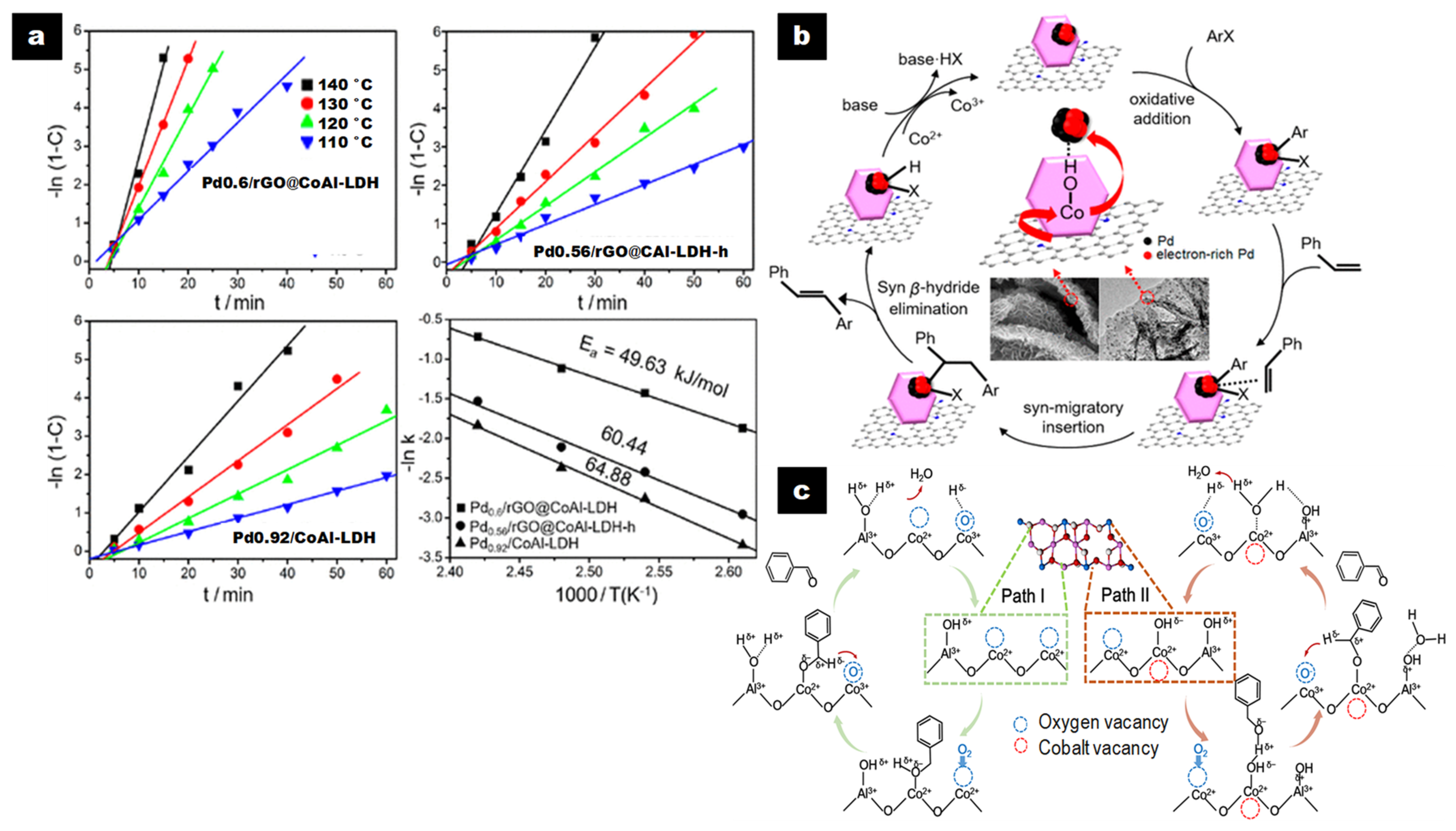{kind=link}
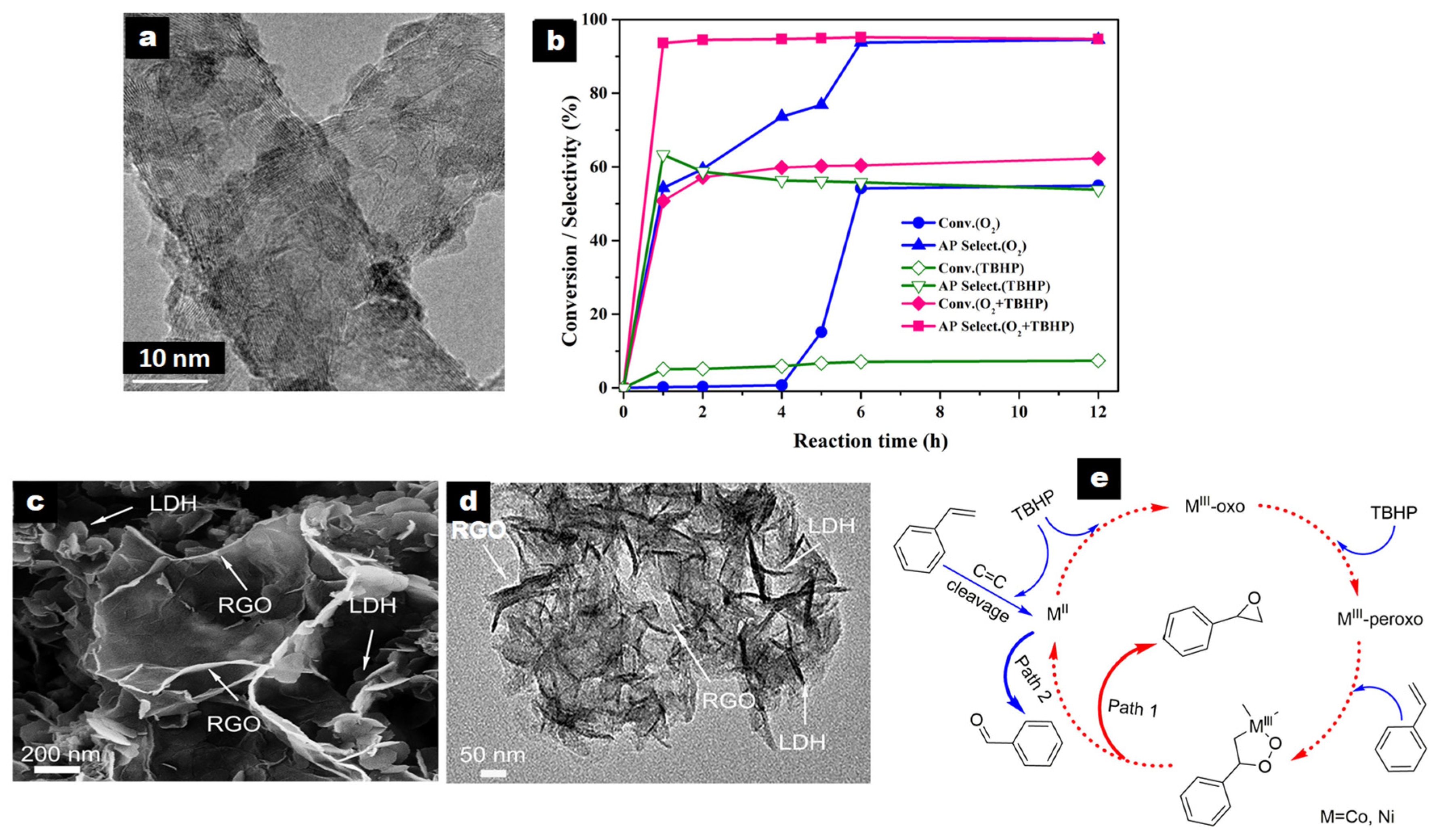{kind=link}
Table 1.
Properties and synthesis methods of different types of nanocarbons.
| Nanocarbon | Properties | Synthesis Methods |
|---|---|---|
| Graphene-like materials | sp2-sp3 electronic configuration with free π-electrons Semiconductor, fast electron transfer Highly functionalizable Strong hybridization with electronic state of catalyst species, strong interfacial coupling π-π conjugation interaction of reactants Extremely high theoretical surface area | Confined self-assembly Chemical vapor deposition Arc discharge Epitaxial growth on SiC layer Unzipping of carbon nanotubes Mechanical exfoliation of graphite Sonication of graphite Electrochemical exfoliation/functionalization of graphene Chemical synthesis/exfoliation |
| CNT | sp2 electronic configuration with free π-electrons Highly graphitic Enhanced charge transport Tailorable acid/basicity and easy functionalization High surface area Good thermal stability | Arc discharge Laser ablation Chemical vapor deposition |
| CNF | Facile and eco-friendly preparation Semiconductive, electronic structure similar to graphite Chemically active edges, easy functionalization Excellent thermal resistance High surface area | Chemical vapor deposition Floating catalyst method Electrospinning/carbonization |
| CD | sp2 hybridization Water solubility, easy functionalization Low cost and toxicity Quantum confinement properties, semiconductor | Microwave-assisted Combustion/hydrothermal Supporting synthesis method Arc discharge Laser ablation Electrochemical synthesis Chemical oxidation |
Table 2.
Composition and preparation method of the LDH/nanocarbon composites, and catalytic reactions performed.
Table 2.
Composition and preparation method of the LDH/nanocarbon composites, and catalytic reactions performed.
| Comp. | Preparation Method | Reaction (Substrate) | Reaction Conditions | Ref. | |
|---|---|---|---|---|---|
| 1 | CNF/MgAl-LDH | Pore precipion by incipient impion | Condension (Acetone) | 1.8 mol acetone; mcat: 1 g; activated 450 °C; rehyd. gas phase; T = 0 °C | [13] |
| 2 | CNF/MgAl-LDH | Pore precipion by incipient impion | MIBK synthesis (Acetone) | 1.8 mol acetone; mcat: 1 g; calcined 450 °C; red. 250 °C (H2); rehyd. gas phase. T = 60 °C, P H2 = 1.2 bar | [14] |
| 3 | CNF/MgAl-LDH | Pyrolysis/hydration of electrospun PVA/PEO/MgAl–nitrate | Transestion (DEC, glycerol) | DEC: glycerol: 16 mol/mol; mcat: 0.4 g; calcined 450 °C; rehyd. gas phase; T = 130 °C | [59] |
| 4 | MWCNT/MgAl-LDH | Coprecipion | Condension (Acetone) | 0.8 mol acetone; mcat adjusted to contain 50 mg LDH; calcined 450 °C; rehyd. liq. phase; T = 0 °C | [44] |
| 5 | CNF/MgAl-LDH | Pore precipion by incipient impion | Transestion (DEC, glycerol) | DEC: glycerol: 17 mol/mol; mcat: 0.3 g; calcined 500 °C; rehyd. liq. or gas phase; T = 130 °C | [60] |
| 6 | CNF/MgAl-LDH | Pore precipion by incipient impion | Condension (Acetone) | 100 g acetone; 1 g supported catalyst (0.3 g bulk catalyst); calcined 500 °C; rehyd. gas phase | [60] |
| 7 | rGO/MgAl-LDH | Copion or self-assembly; Freeze drying or drying in air (80 °C) | Condension (Acetone) | 27.5 mmol acetone; 1.5 wt% LDH/rGO respect to acetone; activated 450 °C; T = 0 °C | [20] |
| 8 | rGO/MgAl-LDH | Copion LDH on GO | Claisen-Schmidt condension (BALD, acetophenone) | BALD (a)/acetophenone = 1.05; mcat: 25 wt%; calcined 450 °C; T = 40 °C | [37] |
| 9 | rGO/CeMgAl-LDH | Copion LDH on GO | Knoev.el (BALD, diethylmalonate) | BALD: diethylmalonate 2:3; mcat: 1 wt%; non-activated; T = 160 °C. | [61] |
| 10 | rGO/CeMgAl-LDH | Copion LDH on GO | Cascade oxidation- Knoev.el (BA/benzoyl acetonitrile) | BA (b)/benzoyl acetonitrile 0.83; mcat: 1 g; non-calcined; P O2 =1 atm: T = 80 °C | [61] |
| 11 | GO/CuAl-LDH GO/CoAl-LDH | Copion LDH from metal nitrates with NaOH + Na2CO3 on GO) Copion LDH from metal chlorides by urea on GO (97 °C; 48 h) | Ullmann (Iodobenzene) | Iodobenzene (2 mmol), DMSO (4 mmol), mcat: 0.25 g; non-activated; T = 110 °C | [21] |
| 12 | N,S-G/CoAl-LDH/Pd | Mixture Al3+, Co2+, NH4F, and thiourea hydrothermally treated. Then + H2PdCl4 | Sonogashira (Aryl halides/Phenylacetylene) | Aryhalide (1 mmol), phenylacetylene (1.2 mmol), 0.06 mol% Pd; cat. non-activated; T = 100 °C. | [36] |
| 13 | rGO/CoAl-LDH/Pd | Copion Co and Al nitrates on the CA-modified GO (pH 10). Aging 65 °C; 4 h | Heck (Iodobenz./Styrene) | DMF (12 mL), H2O (4 mL), K2CO3 (3 mmol), aryl halide (1 mmol), styrene (1.2 mmol); cata non-activated | [53] |
| 14 | GO/ZnNiFe-LDH/Fe3O4 | Fe3O4 on GO. Then LDH immobilized on Fe3O4@GO | One-pot Knoev.el-Michael (BALD/4-hydroxycoumarin) | 4-hydroxycoumarin/BALD = 1: 0.5; mcat: 30 mg; non-activated; reflux H2O (2 mL) | [33] |
| 15 | rGO/RuCoAl-LDH | Ru, Co, and Al chlorides added into GO susp.ion. NaOH + Na2CO3 then added until pH 10. Aging 90 °C; 6 h Ru, Co, and Al chlorides and NaOH + Na2CO3 sol.ion simultaneously added to GO susp.ion until pH 10. Aging 90 °C; 6 h | One-pot oxidation + Knoev.el condion (Cinnamyl alcohol/ethyl cyanoacetate (substituted BA/ethyl cyanoacetate or malonitrile)) | BA (1 mmol) and toluene (7 mL) in 50 mL flask kept 60 °C (O2, 2 h, 0.10 MPa). After ethyl cyanoacetate (1.5 mmol) added at 60 °C: cata non-activated | [54] |
| 16 | rGO/NiAl-LDH/Au | LDH/rGO: Copion metal nitrates and exfol. GO dispersed into NaOH + Na2CO3 Au/NiAl-LDH/rGO: (1) PVA aq. sol. (PVA/Au = 1.2) + aq. HAuCl4 +NaBH4 (NaBH4/Au = 5); (2) LDH/rGO added in the Au-containing colloid | Oxidation (BA) | mcata = 0.4 g; non-activated; 40 mL BA, no solvent; T = 140 °C; P O2 = 2 bar | [40] |
| 17 | GO/CoAl-LDH | Assembly of GO and LDH exfoliated in formamide | Oxidation (BA) | mcata = 0.1 g; non-activated; 1 mmol BA, 5 mL DMF; T = 120 °C; P O2 = 1 bar | [32] |
| 18 | NCD/MgAl-LDH/Au | Copion LDH in presence NCD (pH 10.5). Then addition of HAuCl4 into aq. suspion NCD/LDH and poly(N-vinyl-2-pyrrolidone) + NaBH4 | Oxidation (BA) | mcata = 0.1 g; non-activated; 10 mL BA, no solvent; T = 120 °C; P O2 = 0.4 MPa | [49] |
| 19 | CNT/MgAl-LDH/Ru | LDH-CNT: Acid-treated CNT dispersed in NaOH + Na2CO3 then addition of metal nitrates (pH 10.5). Treatment 100 °C; 16 h. Wetness impr. LDH-CNT with RuCl3.3H2O (Ru = 1 wt%) | Oxidation (BA) | mcata = 0.2 g (0.02 mmol Ru); reduced 400 °C (H2); 2 mmol BA, 105 mL toluene, 6 mL H2O; T = 85 °C; P O2 = 1 bar; O2 flow rate 25 mL min−1 | [42] |
| 20 | GO/CoCuAl-LDH | Copion LDH from metal nitrates on GO with NaOH + Na2CO3 (pH 10). GO/LDH = 0.2–0.8 wt/wt | Oxidation (EB (c)) | EB 10 mmol; TBHP 40 mmol; mcata = 0.1 g; non-activated; T = 120 °C | [38] |
| 21 | CNT/ZnCr-LDH | Copion LDH from nitrate salts on acid-treated CNT and NaOH + Na2CO3 | Oxidation (EB) | EB: 10 mL (81.7 mmol); no solvent; PO2 =1 MPa; mcata = 0.1 g; non-activated; T = 130 °C | [62] |
| 22 | rGO/NiCo-LDH | GO colloid added in mixed sol. (VC2H5OH:VH2O = 1:1) and sonicated. Metal chloride solion (Ni/Co = 1) added dropwise to GO suspion with NH4Cl (pH 9). Treatment 120 °C; 12 h. | Oxidation (Sty (d)) | 1.1 mL Sty; mcata = 0.03 g; non activated; 10 mL acetonitrile, 1.8 mL TBHP (70%, aq. sol.); T = 80 °C | [63] |
| 23 | rGO/CuMgAl-LDH | Copion LDH into CA (e)-GO suspension | Reduction (4-NP) | 1 mM 4-NP (200 µL) + 10 mM NaBH4 (2.5 mL) + 10 µL cata. susp. (2.5 mg mL−1) non-activated | [39] |
| 24 | rGO/CuMgAl-LDH | Copion LDH into CA-GO suspension | Reduction (4-NP) | 200 µL 4-NP (200 µL, 1 mM) + NaBH4 (2.5 mL, 10 mM) + cata. (20 µL, 1 mg mL−1) calcined 600 °C | [41] |
| 25 | rGO/CuNiAl-LDH | Copion LDH into CA-GO suspension | Reduction (4-NP) | 4-NP (200 µL, 1 mM) + NaBH4 sol (2.5 mL, 10 mM) + cata. (10 µL, 2.5 mg mL−1) non-activated | [35] |
| 26 | CDs/MgAlCe-LDH/Ag | LDH Copion and dispersed into AgNO3 aq. sol. Then addition of CDs aq. sol. and UV irradiation | Reduction (4-NP, MB, MO, CR, RhB, R6G) | 4-NP (200 µL, 10 mM) + NaBH4 sol (200 µL, 0.1 M) + cata. (20 µL, 1 mg mL−1) non-activated | [34] |
| 27 | CDs/MgAl-LDH/PNIPAM/Pd | LDH functed with PNIPAM (Mussel). Then addition of Pd and CDs | Reduction; Knoev.el; One-pot Knoev. el -reduction (MB, RhB, CR, R6G, MO. 4-NP, o-NO, m-NP, BALD, malonitrile) | MB (2 mL, 0.013 mM) + NaBH4 sol (1 mL, 0.5 M) + cata. (20 µL, 0.05 mg mL−1) non-activated | [22] |
| 28 | rGO/NiAl-LDH/Pt | Copion LDH on GO. Then impregnation with H2PtCl6 | Reduction (p-nitrophenol, P-NA) | P-NA (3 mL, 0.1 M) + NaBH4 sol (0.1 mL, 0.1 M) + mcat: 1 mg activated 600 °C (N2) | [64] |
| 29 | PAA-CNT/NiAl-LDH | LDH copion in presence PAA functionalized CNT (P-CNT) and L-cysteine | Reduction o-(CNB) | T = 140 °C, P H2 = 2 MPa; cata activated 500 °C (10% v/v H2/Ar) | [47] |
| 30 | rGO/NiAl-LDH | LDH copion in presence GO | Reduction (CALD) | T = 120 °C, P H2 = 1 MPa; cata activated 600 °C (N2) | [65] |
| 31 | MWCNT/CuMgAl-LDH | LDH copion in presence pretreated MWCNT | Reduction (Glycerol) | Glycerol (5 g, 60 wt%); P H2 2 Mpa; T = 180 °C; mcat: 0.2 g activated 400 °C (air) | [66] |
| 32 | CFs/CuCoAl-LDH | Copion of Cu, Co, and Al nitrates on CF (pH 9.5–10). Aging 60 °C | HAS (H2/CO/N2) | 3 MPa syngas mixture H2/CO/N2 = 8:4:1 (GHSV: 3900 mL (gcat h)−1); T = 220 °C; cata reduced 450 °C (H2) | [48] |
| 33 | CNT/CuCoAl-LDH | Cu, Co, Al nitrates dissolved into CNT solion then + (NaOH + Na2CO3) at pH 9.5. Aging RT | HAS (H2/CO/N2) | 3 MPa syngas mixture H2/CO/N2 = 8:4:1 (GHSV: 3900 mL (gcat h)−1). T = 230 °C; cata reduced 450 °C (H2) | [43] |
| 34 | CFs/CuFeMg-LDH | Copion Cu, Fe and Mg nitrates and on CF at pH 9.5–10. Aging 65 °C | HAS (H2/CO/N2) | 3 MPa syngas mixture H2/CO/N2 = 8:4:1 (GHSV: 3900 mL (gcat h)−1). T = 280 °C; cata reduced 500 °C (H2) | [67] |
| 35 | CFs/CuZnAl-LDH/K | Copion Zn and Cu nitrates on CF and Al nitrate at pH 5.5–6.2. Aging 110 °C. K2CO3 impion | Isobutanol synthesis (H2/CO) | H2/CO = 2 (GHSV: 3900 h−1), mcat: 560 mg; T = 320 °C, P = 4 MPa; cata activated 400 °C (N2) + 320 °C (N2/H2 = 4:1) | [68,69] |
(a) Benzaldehyde; (b) Benzyl alcohol; (c) Ethlylbenzene; (d) Styrene; (e) Citric acid.
Table 3.
Basicity and catalytic activity in the self-condensation of acetone at 0 °C for different MgAl-LDH/carbon nanocomposites.
Table 3.
Basicity and catalytic activity in the self-condensation of acetone at 0 °C for different MgAl-LDH/carbon nanocomposites.
| Carbon | Activation (Rehyd.ion) | mHT [mg] | macet [mol] | mHT/macet. × 103 [g/g] | CO2 ads. [mmol gHT−1] | React. Rate [mmolDAA gHT−1 h−1] | Ref. |
|---|---|---|---|---|---|---|---|
| CNF | Calc. 450 °C (gas phase) | 110 | 1.8 | 1.05 | 0.75 (a) | 542 (b) | [14] |
| CNF | Calc. 450 °C (gas phase) | 120 | 1.72 | 1.20 | 0.37 (c) | nd | [60] |
| MWCNT | Calc. 450 °C (liq. phase) | 50 | 0.8 | 1.08 | 1.8 (c) | 196 (d) | [44] |
| rGO | Calc. 450 °C | 0.57 | 2.75 × 10−2 | 0.36 | 0.41 (c) | 0.052 (e) | [20] |
(a) CO2 adsorption at 0 °C; (b) over the first 15 min; (c) CO2 adsorption at 25 °C; (d) from the amount of DAA produced over 100 h; (e) from the amount of DAA produced over 8 h.
Table 4.
Conditions of reaction and results of aerobic oxidation of BA over different nanocomposite catalysts.
Table 4.
Conditions of reaction and results of aerobic oxidation of BA over different nanocomposite catalysts.
| Catalyst | BA [mmol] | Solv | mcat [g] | PO2 [bar] | T [°C] | t [h] | SS [m2 g−1] | Conv. [%] | BAL [%] | BB [%] | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Au/NiAl-LDH/rGO | 4.3 | no | 0.4 | 2 | 140 | 5 | 172.5 | 35 | 80 | 20 | [40] |
| CoAl-ELDH/GO | 1 | DMF | 0.1 | 1 | 120 | 4 | 92.2 | 99.3 | 0 | [42] | |
| Au/NCD/MgAl-LDH | 89 | no | 0.1 | 4 | 120 | 5 | 61 | 49 | 83.4 | 16 | [32] |
| Ru/LDH-CNT (Pickering) | 2 | H2O/Tolne | 0.2 | 1 | 85 | 5 | 89 | 92 | 99.9 | 6 | [49] |
BAL: Benzaldehyde; BB: Benzyl benzoate.
Table 5.
Physico-chemical characteristics, catalytic performance in the reduction of 4-NP, and reusability of the nanocomposites containing Cu-based LDH and rGO.
Table 5.
Physico-chemical characteristics, catalytic performance in the reduction of 4-NP, and reusability of the nanocomposites containing Cu-based LDH and rGO.
| Catalyst | SA [m2 g−1] | Active Interf. Domain | Size Cu [nm] | t [min] | kapp [×10−3 s−1] | TOF [h−1] | Rec. (a) [nb] | Ref |
|---|---|---|---|---|---|---|---|---|
| Cu1Mg2Al-LDH/rGO (1.0Cu-LDH/rGO) | 210.5 | Cu2O-Cu(LDH)-rGO | Cu2O (~6.8) | 2 | 25.11 | 161.9 | 20 | [39] |
| 1.0Cu@Cu2O/MgAlO-rGO | 160 | Cu-Cu2O-MgAlO-rGO | Cu@Cu2O (24.3/core 12.2) | 1 | 55.35 | 199.6 | 25 | [41] |
| Cu1Ni2Al-LDH/rGO | 151 | Cu2O-Ni-OH (CuNiAl-LDH)-rGO | Cu2O (~3.8) | 1.5 | 34.37 | 197.6 | 10 | [35] |
(a) Number of reuse cycles.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tichit, D.; Álvarez, M.G. Layered Double Hydroxide/Nanocarbon Composites as Heterogeneous Catalysts: A Review. ChemEngineering 2022, 6, 45. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040045
AMA Style
Tichit D, Álvarez MG. Layered Double Hydroxide/Nanocarbon Composites as Heterogeneous Catalysts: A Review. ChemEngineering. 2022; 6(4):45. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040045
Chicago/Turabian StyleTichit, Didier, and Mayra G. Álvarez. 2022. "Layered Double Hydroxide/Nanocarbon Composites as Heterogeneous Catalysts: A Review" ChemEngineering 6, no. 4: 45. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040045