
Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides


GIR-QUESCAT, Departamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad de Salamanca, 37008 Salamanca, Spain
*
Author to whom correspondence should be addressed.
ChemEngineering 2022, 6(4), 53; https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040053
Submission received: 3 June 2022
/
Revised: 30 June 2022
/
Accepted: 4 July 2022
/
Published: 6 July 2022
(This article belongs to the Special Issue A Themed Issue in Honor of Prof. Dr. Vicente Rives)
Abstract
:Amines with two carbon atoms in the organic chain [ethylamine (EA), diethylamine (DEA), triethylamine (TEA)] have been used as precipitant agents to obtain a hydrotalcite-like compound with Zn (II) and Al (III) as layered cations and with nitrate anions in the interlayered region to balance the charge. This Layered Double Hydroxide was prepared following the coprecipitation method, and the effect on the crystal and particle sizes was studied. Also, the effect of submitting the obtained solids to hydrothermal post-synthesis treatment by conventional heating and microwave assisted heating were studied. The obtained solids were exhaustively characterized using several instrumental techniques, such as X-ray diffraction, Thermal Analysis (DTA and TG), Chemical Analysis, Infrared Spectroscopy (FT-IR), determination of Particle Size Distribution and BET-Surface area. Well crystallized solids were obtained showing two possible LDH phases, depending on the orientation of the interlayer anion with respect to the brucite-like layers. The results indicated that there is a certain influence of the amine, when used as a precipitating agent, and as a consequence of the degree of substitution, on the crystallinity and particle size of the final solid obtained. The LDHs obtained using TEA exhibited higher crystallinity, which was improved after a long hydrothermal treatment by conventional heating. Regarding the shape of the particles, the formation of aggregates in the former solid was detected, which could be easily disintegrated using ultrasound treatments, producing solid powder with high crystallinity and small particle size, with homogeneous size distribution.
1. Introduction
The term Layered Double Hydroxides (LDHs) is used to name synthetic or natural hydroxides with a layered structure and with at least two types of metal cations in the main layers, which are positively charged, and contain anionic species in the interlayer space. This large family of compounds is also called anionic clays, in comparison with cationic clays, which, in their interlayer region, contain cations to balance the negative charge of the layers [1]. They are also known as hydrotalcite-like compounds, because hydrotalcite is the most abundant mineral with this layered structure in Nature. These materials are not as abundant in Nature as the analogous cationic clays, but they are very easy to synthesize with a tuned composition and are generally not very expensive.
The layered structure of the LDH consists of brucite-like layers with divalent cations occupying octahedral spaces formed by OH− ions [M(OH)6], in which an isomorphic, and partial, substitution of divalent cations by trivalent cations has taken place, leaving the layers positively charged. The electroneutrality of the compound is achieved by the incorporation of anions in the interlayer space [2]. The chemical composition of LDHs is described by the chemical formula: , where MII and MIII are the divalent and trivalent metal cations, respectively, and A− is the interlayer anion, with defined as the ratio [1,3]. Both organic and inorganic anions can be incorporated into the interlayer space, in a wide range of sizes and charges [1,4]. In addition, in the interlayer space there are randomly arranged water molecules, and this interlayer region has quasi-liquid behavior. The wide range of anions and divalent and trivalent cations that can be used to prepare LDH, provide them with a diversity of compositions.
In recent decades, LDHs have established themselves as promising materials, due to their properties and applications in a number of fields, such as water remediation [5,6,7], catalysis [8,9,10], drug delivery [11,12], electroactivity [13], biomedicine [14,15], and others. Their applications as anion exchanging solids depend on the interlayer anion and its affinity for the LDH layers. Thus, Miyata et al. [16] have reported an order of anionic selectivity for MgAl hydrotalcite-like materials, which could be applicable to other combinations of elements [17]. According to this, for applications involving anion exchanging, LDHs containing nitrate have a higher ion exchange facility than those containing carbonate in the interlayer space.
As above mentioned, the synthesis of LDHs in the laboratory is relatively simple and cheap, and they could be synthesized by many different methods. The method selected has an impact on the properties of the final solid and, therefore, on its subsequent application. Among the different methods described, the most widely used is the coprecipitation method, due to its great ease of use and reproducibility [3]. This method is based on precipitation by the slow dropwise addition of a solution containing the mixture of MII and MIII salts solution in a fixed ratio and the anion to an alkaline solution, working at constant pH. The addition of a second alkaline solution allows the pH of the precipitation medium to be maintained during the precipitation of the cations [3]. Many parameters are relevant to monitoring the process, such as the type and concentration of cations and anions, the precipitation medium, the pH and the temperature [18,19]. The optimal pH depends on the nature of the cations to be incorporated into the structure; thus, Kloprogge et al. [20] reported that the best crystallinity for samples of Zn/Al LDH were exhibit in the pH range 11–12. Moreover, when the coprecipitation method is carried out, the incorporation of the carbonate anion into the interlayer space is very difficult to avoid, due to the fact that it is the anion with the highest affinity for the LDH layers. However, its incorporation can be prevented by using decarbonated water and bubbling N2.
LDH synthetized by the coprecipitation method generally exhibits low crystallinity with a high degree of aggregation and a wide particle size distribution. The most common way to obtain more uniform particle properties, with an improved crystallinity is by an aging process. For this reason, the coprecipitation method is generally followed by a long aging period, from 10 to 80 h, and often longer [19]. Also, post-synthesis treatment heating at moderate temperatures is used; for instance, by gentle-to-gentle reflux or by hydrothermal treatment. The aging presumably occurs through the Ostwald ripening process, in which larger and more perfect crystallites grow at the expense of smaller particles in solution by dissolution/precipitation processes [21]. Therefore, microwave-assisted hydrothermal treatment, or hydrothermal treatment by conventional heating, have been widely used with the aim of improving structural and textural properties [22,23,24,25,26].
In our previous work [27], Zn/Al-LDH in molar ratio 2:1 was synthesized using amines with one carbon atom in the organic chain [methylamine (MMA), dimethylamine (DMA) and trimethylamine (TMA)], such as precipitant agents. Furthermore, the evolution on the properties caused by hydrothermal treatment of samples synthetized with amines using the following two ways of heating was studied: conventional heating and microwave assisted heating. Highly crystalline LDH with nitrate anion in the interlayer was obtained using DMA as the precipitant agent and, after submitting the solid to conventional hydrothermal treatment. In the present work, we reported the use of amines with two carbon atoms in the organic chain [ethylamine (EA), diethylamine (DEA), triethylamine (TEA)] as precipitant agents in the synthesis of Zn,Al-LDH in molar ratio 2:1. Furthermore, the solids were treated hydrothermally by conventional heating and microwave-assisted heating to improve the crystallinity and properties of the solids. The effect of the treatment methods was analyzed for each amine-assisted synthesis condition.
2. Materials and Methods
2.1. Materials
Zn(NO3)2·6H2O (98–102%), Al(NO3)3·9H2O (98–102%) and NaOH (98%) were purchased from Panreac and used as received. An aqueous solution of ethylamine (70% in H2O) was purchased from Alfa Aesar (Ward Hill, MA, USA). Diethylamine (99.5%) was purchased from Panreac (Barcelona, Spain). Triethylamine (≥99%) was purchased from Sigma Aldrich (Burlington, MA, USA).
2.2. Synthesis
The coprecipitation method was used to prepare the desired solids [3]. In order to avoid the intercalation of carbonate anions, decarbonated water solutions and nitrogen atmosphere were used during the synthesis. In order to prepare the solution of the metal cations, 0.3 L in 2.5 M concentration of their nitrate salts in a MII/MIII molar ratio 2:1 was prepared. For the precipitation medium, an aqueous solution of 4.5 M concentration of the desired amine was prepared. The metal cation solution was added dropwise to the amine solution, which was maintained under vigorous magnetic stirring. The pH of the precipitation media was kept at a preselected pH value of 10 by adding the required amount of a 2 M NaOH solution using a 240 CRISON pH-burette. After complete addition, the aqueous suspension of the precipitated solid was stirred for 1 h at room temperature and, then, the slurry obtained was subjected to different ageing treatments: (i) a portion of the sample without any hydrothermal treatment was kept as a reference; (ii) a portion was subjected to a hydrothermal treatment by microwave-assisted heating (MW) for 60 or 300 min at 90 °C; (iii) a portion was subjected to a hydrothermal treatment by conventional heating (HT) for 1 or 7 days at 90 °C using a home-made stainless-steel bomb lined with Teflon. Hydrothermal treatment by microwave-assisted heating was carried out in a MILESTONE ETHOS PLUS microwave oven, where the aqueous suspension was placed in Teflon digestion vessels, sealed and mounted on a turntable inside the oven. The programmed temperature was controlled by a thermocouple immersed in a reference vessel and software provided by the manufacturer. The different portions of the solid suspensions were separated and washed by centrifugation with distilled water until reaching a pH close to 7, in order to eliminate the equilibrium ions of the starting salts. Finally, the solids were dried at 40 °C in an oven under air atmosphere.
The samples were labelled according to the preparation procedure: reaction medium, hydrothermal treatment method and time period to which they had been subjected. So, a sample labeled as ZA2XYt, ZA2 represents the cations (Zn–Al, molar ratio 2/1); X represents the precipitation media (depending on the amine used EA, DEA or TEA); Y represents the aging treatment (STH for the reference sample, HT or MW); and t the treatment duration (in minutes for MW, in days for HT).
2.3. Characterization
A Yobin Ivon Ultima II apparatus at NUCLEUS (University of Salamanca, Salamanca, Spain) was used for elemental chemical analysis of Zn and Al by ICP-OES.
A Siemens D-5000 instrument was used to record Powder X-ray diffraction (PXRD) patterns using Cu-Kα radiation (λ = 1.54050 Å) with a scanning rate of 2°/min from 5° to 70° (2θ). The Scherrer equation was used to calculate the crystallite sizes from the FWHM (Full Width at Half Maximum) of the diffraction maximum (00l). The Warren correction for instrumental line broadening was taken into account, but the possible contribution of disorder effects and/or lattice strains to the peak broadening was ignored.
A Perkin-Elmer Spectrum One instrument was used to record the FT-IR spectra by transmission with a nominal resolution of 2 cm−1 from 4000 cm−1 to 450 cm−1, using KBr pressed pellets.
SDT Q600 equipment from TA Instruments was used to carried out the thermogravimetric (TG) and differential thermal analyses (DTA). The thermal analyses were carried out by heating from room temperature to 900 °C at a rate of 10 °C/min under continuous oxygen (L’Air Liquide, 99.995%) flow (50 mL/min).
A Micromeritics Gemini VII 2390t apparatus was used to record the nitrogen (L’Air Liquide, 99.999%) adsorption–desorption isotherms at −196 °C, and to calculate the specific surface area and porosity data. The apparatus was calibrated with He (L’Air Liquide, 99.999%). Before measurements, the samples were pretreated at 110 °C for 2 h under a stream of N2 in a Micromeritics FlowPrep 060 Sample Degass System.
A Diffraction Mastersizer 2000 equipment from Malvern Instruments was used to determine the particle size distribution (PSD) by Laser Diffraction. Using the dispersion unit Hydro 2000 from Malvern Instruments, the solid was dispersed in water at 25 °C (approx. 0.05 vol.%), and, after measuring the PSD for the dispersed samples, ultrasounds were applied in situ to disaggregate the particles.
3. Results and Discussion
3.1. Element Chemical Analysis
Table 1 gives the Zn/Al molar ratio values and chemical formulae of the samples synthesized in the presence of the different amines used as precipitant agents. The chemical formulae were deduced from the results of elemental chemical analysis and thermogravimetric analysis (see below).
In all cases, the Zn/Al molar ratio approached the value of 2, suggesting a complete precipitation of the existing cations in the synthesis medium. In some cases, a small deviation could be observed, but never more than 5%.
For the determination of the chemical formula of each of the samples, the amount of nitrate anion was calculated from the Al/(Al + Zn) molar ratio, assuming that it was the only interlayered anion, as observed by FT-IR spectroscopy, neutralizing the positive charge excess of the layers.
Unlike samples synthesized without amines or in the presence of methylamine, for dimethylamine or trimethylamine samples synthesized in the presence of EA, DEA or TEA have water content per chemical formula which is generally lower, as reported in our previous work [27] The more regular stacking of the octahedral layers, as observed by PXRD, leads to a decrease in the number of water molecules per unit formula, and this fact is more evident in the samples having HT hydrothermal treatment.
On the other hand, samples ZA2EAHT1 and ZA2EAHT7 were mostly or completely formed by ZnO, as deduced from the PXRD analysis. However, after chemical analysis, the presence of Al in the solid sample could be determined, indicating the formation of an amorphous phase containing aluminum [28], not observed by X-ray diffraction. Therefore, as there was a mixture of phases in the solid sample, it was not possible to determine the amount of Zn and Al forming the LDH structure (if its collapse was not complete); thus, making it difficult to determine its chemical formula.
3.2. Powder X-ray Diffraction (PXRD)
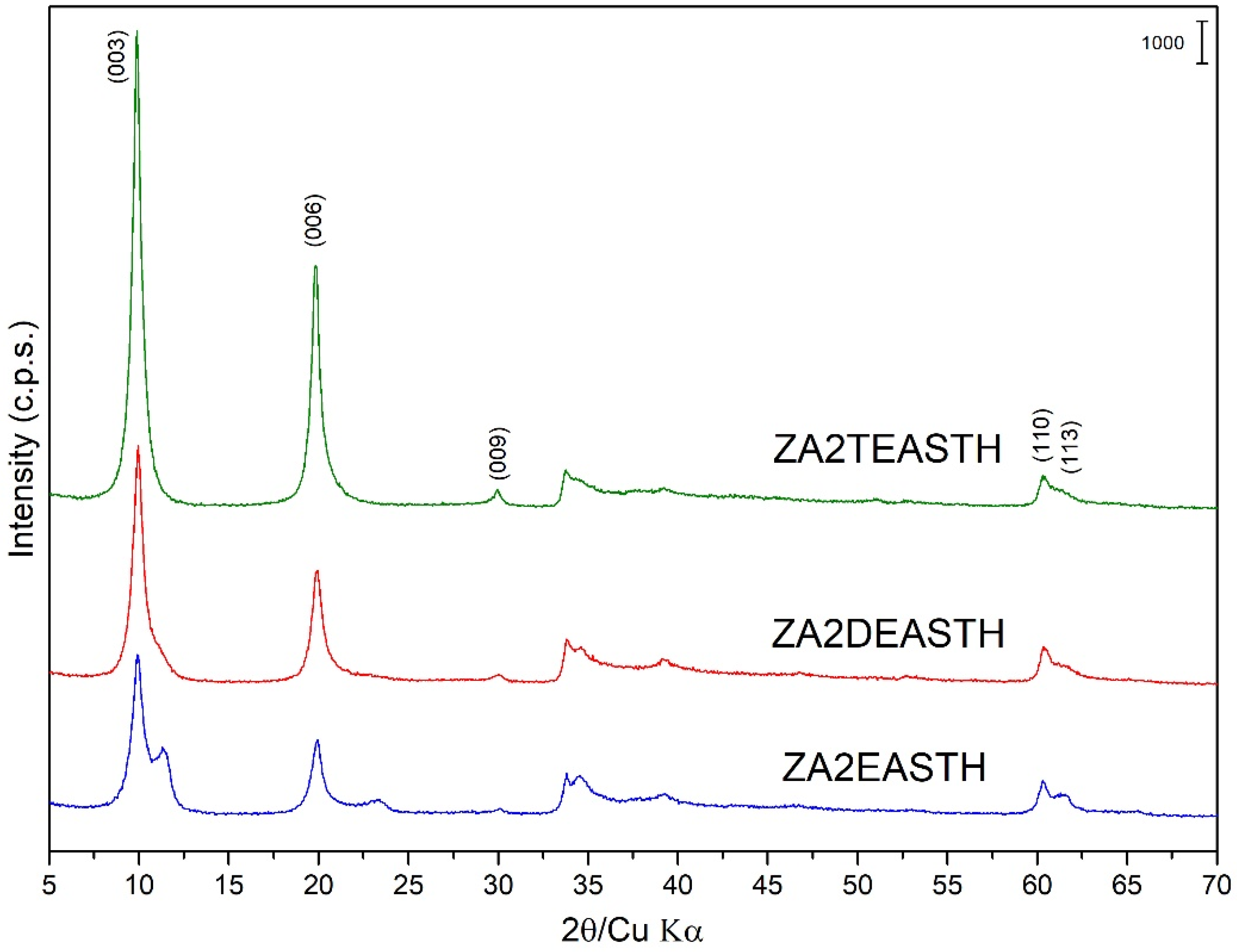
The samples synthesized using EA, DEA and TEA as modifiers of the precipitation medium were also obtained in the form of microcrystalline powder. Figure 1 shows the PXRD diagrams of these samples without hydrothermal treatment. The positions and relative intensities of the recorded diffraction peaks revealed a layered structure of the solids, characteristic of an ordered 3R1 polytype of solids with the LDH structure (JCPDS: 22-0700) [29,30,31]. In all cases, the most intense diffraction peak, attributed to the diffraction plane (003) of the crystal structure, was recorded at a position 10.0° (2θ), with a spacing of 8.93 Å. This spacing is in agreement with the values reported by Miyata et al. [16] for LDH with nitrate as the interlayer anion arranged in a perpendicular orientation to the brucite-like layers and with a M2+/M3+ molar ratio close to 2. Confirming the layered structure, diffraction peaks corresponding to crystallographic planes (006) and (009) were recorded at values close to 19.9° (2θ) and 30.0° (2θ), respectively, and with spacings of 4.46 Å and 2.98 Å. Reflections corresponding to diffraction planes (110) and (113) were recorded at 60.3° (2θ) and 61.3° (2θ), with spacings of 1.53 Å and 1.51 Å, respectively.
As can be seen in Figure 1, the solids prepared using DEA or TEA as precipitant agents showed a single crystallographic phase. However, in the case of the sample prepared in the presence of EA, diffraction peaks corresponding to a second LDH phase could be distinguished in the PXRD diagram. Thus, it is possible to distinguish the most characteristic peak of this secondary phase, which was recorded close to 11.33° (2θ) and ascribed to the diffraction peak (003), with a spacing of 7.81 Å. In this case, the diffraction angle for this diffraction peak was slightly higher than that found for the sample synthesized in the absence of amines [27], where, unlike the shoulder observed for the sample prepared in the absence of amines, a well-defined maximum could be clearly distinguished. In this second phase with a spacing of 7.81 Å for the peak, due to the (003) planes, the interlaminar nitrate anions were arranged with their molecular plane parallel to the plane of the brucite-like layers [32,33,34].
Observing the width and profile of the peaks ascribed to the diffraction plane (003) close to 10° (2θ), the crystallinity of these samples decreased in the order: ZA2TEASTH > ZA2DEASTH > ZA2EASTH. Again, it could be observed how the use of amines (and the nature of these) in the precipitation medium modifies the crystallinity of the solids, to the point of two phases coexisting, with nitrate anion in the interlayer space depending on the amine used.
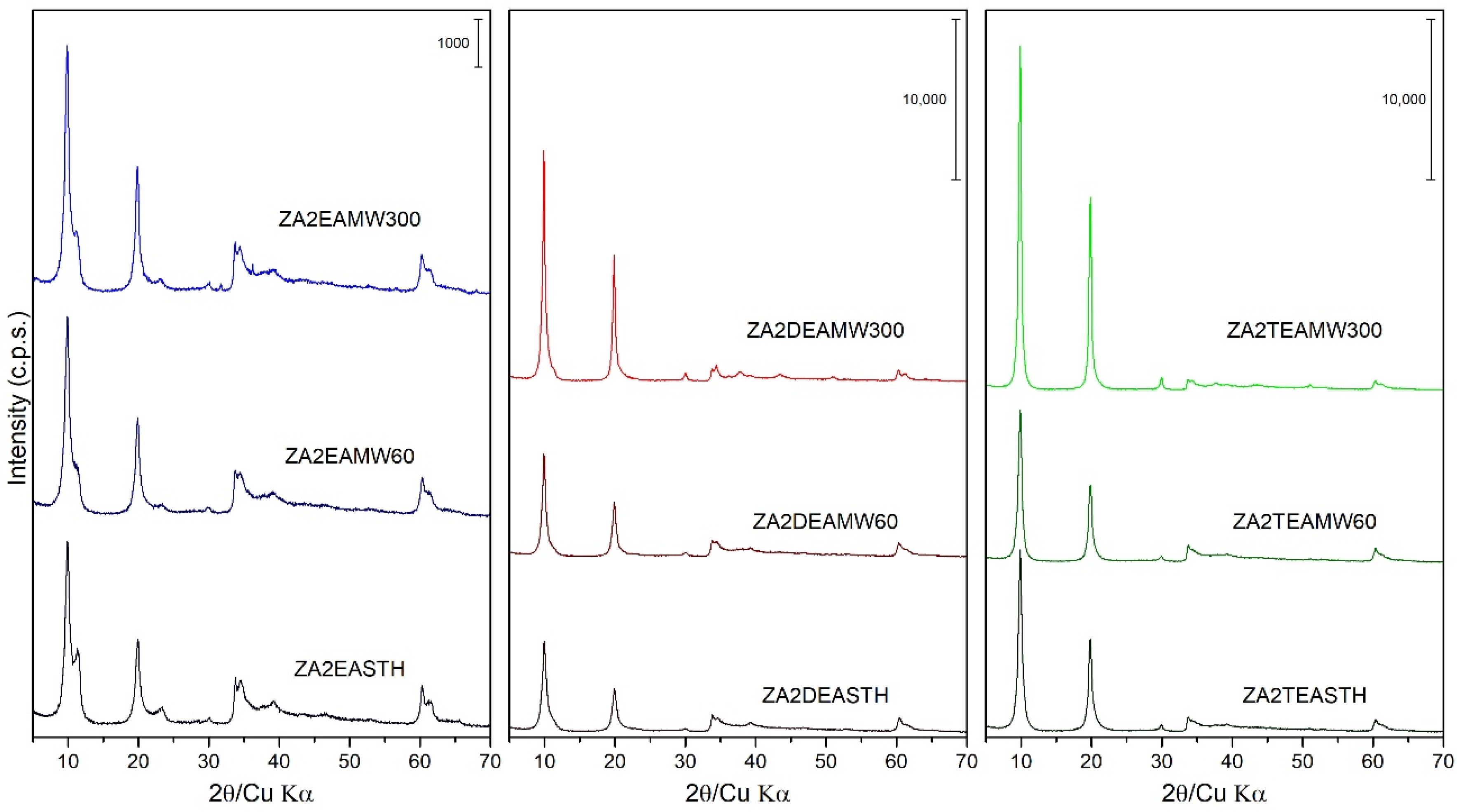
The samples synthesized in the presence of EA, DEA and TEA were also subjected to hydrothermal treatments, using two heating routes: microwave-assisted heating (MW) and conventional oven heating (HT). Figure 2 includes the PXRD plots of these samples with different MW hydrothermal treatment periods. Observing the profile of the diffraction peaks and their relative intensities as the treatment time increased, it can be seen how, unlike the samples prepared in our previous work [27], the application of MW hydrothermal treatment increased the crystallinity of the solids as the MW treatment time increased. Comparing the width of the diffraction peaks (003) of the samples subjected to a longer treatment time, that is, the samples with 300 min of treatment, the following decreasing order of crystallinity could be established as a function of the amine used: ZA2TEAMW300 > ZA2DEAMW300 > ZA2EAMW300. It is noteworthy that, as the MW hydrothermal treatment time was prolonged on the samples synthesized with EA, the relative intensity of the diffraction peak (003) of the phase with the nitrate anion parallel to the brucite-like layers did not increase as the treatment time increased, going from being a well-defined peak in sample ZA2EASTH to being a shoulder of the peak (003) in sample ZA2EAMW300.
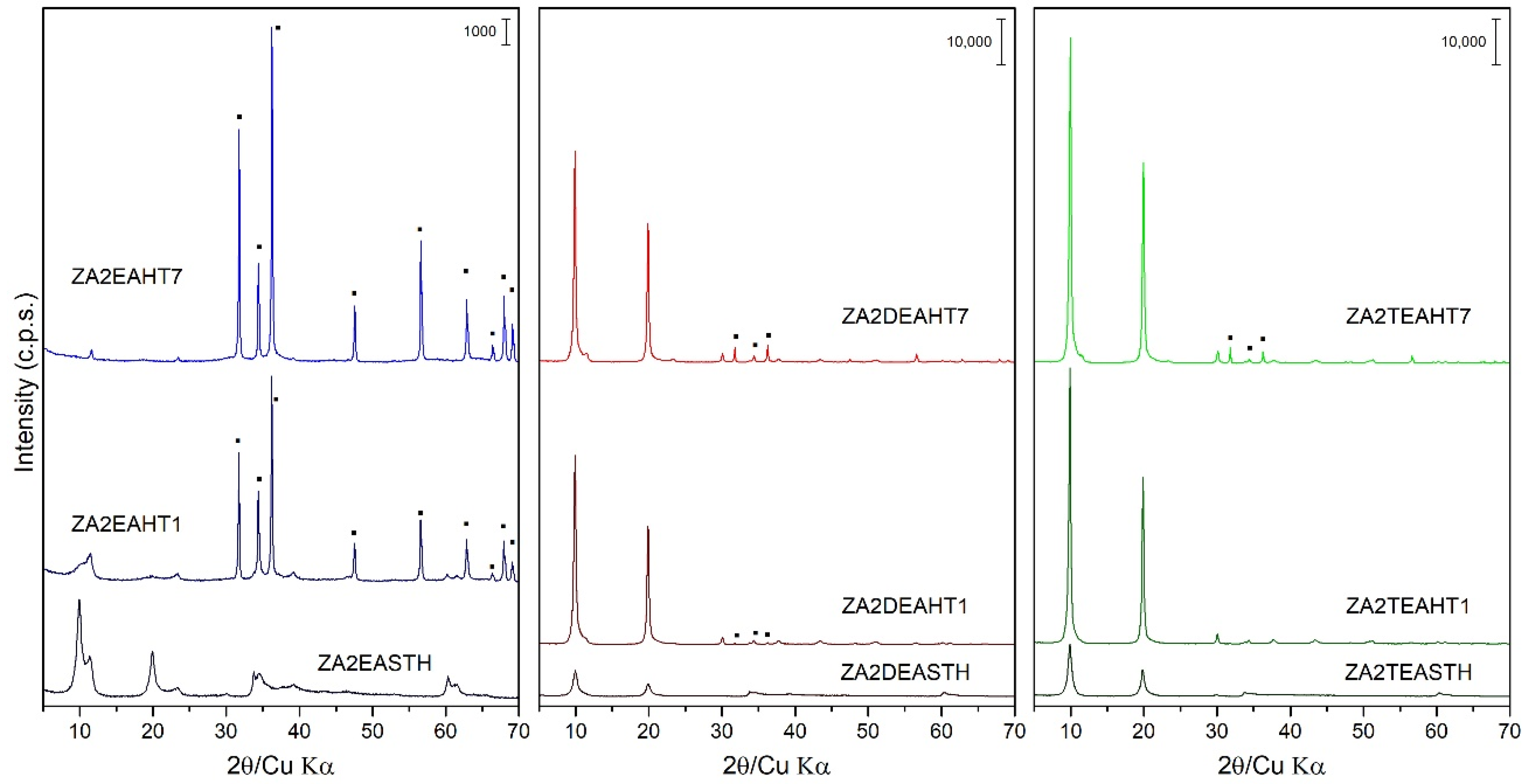
The application of a conventional hydrothermal treatment (HT) resulted in a greater increase in the crystallinity of the solids synthesized in the presence of DEA and TEA. However, the same effect is not observed for the samples synthesized in the presence of EA, where the application of HT treatment resulted in the collapse of the layered structure, as can be observed in Figure 3. Thus, the PXRD recorded for samples synthetized using EA showed the formation of a zinc oxide (ZnO) like zincite phase (JCPDS: 00-036-1451 [29]). This ZnO phase is identified mainly by the reflections recorded in the range of 28°–38° (2θ). While with 1 day of HT treatment the collapse of the LDH phase was not complete, as can be seen from its corresponding PXRD diagram, after 7 days of treatment the collapse was complete. So, the main diffraction peaks of sample ZA2EAHT7 corresponded to the ZnO phase and, with very little intensity, the diffraction peaks at 11.6° and 23.4° (2θ) corresponded to the crystallographic planes (003) and (006), respectively, of the LDH phase with the interlayer nitrate arranged parallel to the plane of the brucite-like layers.
In the case of the samples synthetized using DEA or TEA, segregation of a small amount of the zincite phase (ZnO) could also be observed as the HT treatment time increased. Thus, from the profile of the diffraction peaks (003) and their relative intensities for these two series of samples, it can be observed how with 1 day of treatment, solids of high crystallinity were obtained without segregation of the ZnO phase when TEA was used as the precipitant agent, and with a very small amount of this phase when DEA was used. Therefore, the segregation of ZnO decreased as the degree of substitution of the amino group in the compound used as precipitating agent increased. Comparing the width of the diffraction peaks (003) of the samples subjected to a longer treatment time (samples with 7 days of treatment) the following decreasing order of crystallinity can be established as a function of the amine used: ZA2TEAHT7 > ZA2DEAHT7. Table 2 shows the amount of zincite phase present in the sample calculated from the calibration line with the ratio of the areas of the characteristic peaks of the zincite phase and the LDH phase [35]. Thus, it is shown how the amount of zincite phase increased as the HT treatment time increased, finding an amount of approximately 2% of ZnO in the sample ZA2DEAHT7. By extrapolation of the calibration line to samples synthetized using EA, for the ZA2EAHT1 sample, 67% was ZnO and for longer treatments it was close to 100% (Table 2).
The lattice parameters of the prepared solids were calculated from the positions of the diffraction peaks due to the (003) and (110) planes [30]; being, c = 3 · d(003) ≈ 26.6–26.8 Å, and a = 2 · d(110) ≈ 3.069–3.077 Å. In addition, from the value of the Full Width at Half Maximum (FWHM) of reflection 003, the crystallite size (D) in the c direction was calculated using the Scherrer equation, D = kλ/βcosθ [36,37], where k is a constant, taken in this case as 0.9; λ is the wavelength of the radiation used; β the FWHM and θ the diffraction angle; correction due to instrumental broadening was not applied. The values calculated for the samples obtained are included in Table 3, together with the calculated values for the number of stacked layers.
The similarity of the lattice parameter a value, with differences of less than 1%, are coherent with the homogeneity of metals composition in the samples.
The crystallite size (D) values highlight how the conventional hydrothermal treatment led to a greater increase in the crystallinity of the solids. When the MW treatment was applied, it was observed that, as the treatment time increased the crystal size increased, resulting in the obtaining of, in both the DEA and TEA series, crystal sizes around 200 Å. However, when HT treatment was applied, it could be observed that the samples synthesized in the presence of TEA with only one day of treatment reached a crystal size close to 320 Å, which was practically maintained, even if the HT treatment time increased. In the case of samples prepared in the presence of DEA, with one day of HT treatment a crystal size higher than 280 Å was obtained; reaching a crystal size of 334 Å when the treatment was extended to 7 days.
3.3. FT-IR Spectroscopy
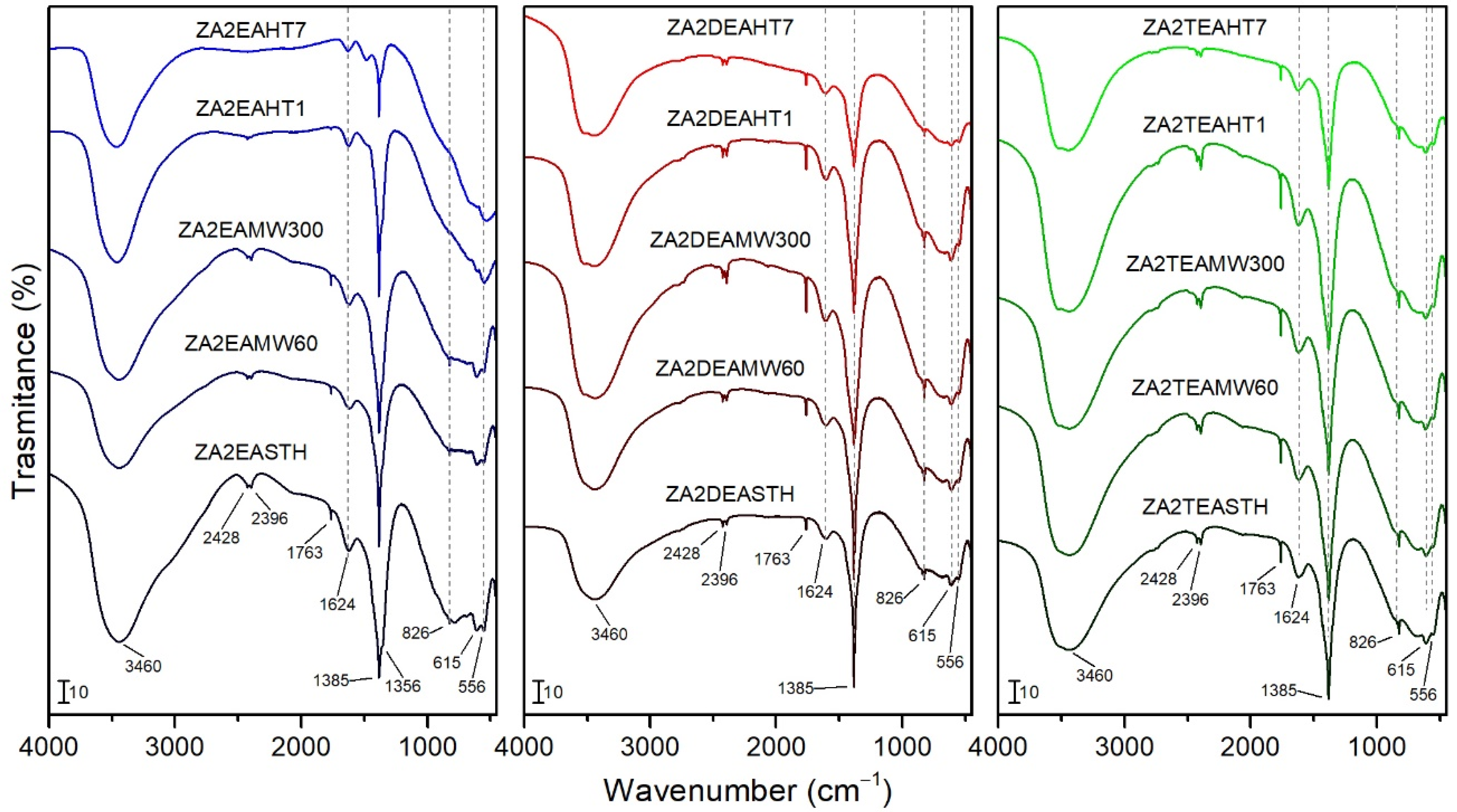
FT-IR spectra of the synthesized solids are plotted in Figure 4. The broad band at 3460 cm−1 is ascribed to the stretching vibration modes of the hydroxyl groups and the water molecules of the interlayer space. At lower wavenumbers it was possible to observe a bands at 615 and 556 cm−1 caused by M-OH vibration modes. The vibration ascribed to the bending mode of the interlayer water molecules was recorded at 1624 cm−1 [18,38,39]. The bands at 2396 and 2428 cm−1 could be attributed to atmospheric CO2 weakly bonded on the LDH surface.
In all cases, the characteristic bands of the vibrational modes of the nitrate anion molecules were recorded, confirming its presence as an interlayer anion. Thus, bands at 1385 cm−1 and 826 cm−1 can be observed, assigned to the ν3(E’) and ν2 (A2’’) vibrational modes, respectively, of with a D3h symmetry [19,38]. At 1763 cm−1 a narrow band can be observed, corresponding to the combination of the vibrational modes ν1(A1′) at 1068 cm−1 and ν4(E’) at 692 cm−1 of nitrate, the latter not clearly observed in the infrared spectra. For sample ZA2EASTH, a shoulder can be seen at 1356 cm−1 (Figure 4), which could be due to nitrate anions in parallel orientation in the second LDH phase observed by PXRD. As observed by PXRD for the samples synthesized in the presence of EA after MW hydrothermal treatment, a crystallinity increase of the phase with the anions in perpendicular orientation took place in detriment of the phase with the anions in parallel orientation to the layers. This effect was reflected in the FT-IR spectra, where the shoulder at 1356 cm−1 became less evident (Figure 4). On the other hand, when DEA or TEA were used as precipitaton agents, such a shoulder was also not observed at 1356 cm−1.
When the sample synthesized in the presence of EA was subjected to a HT hydrothermal treatment process, as observed in the PXRD studies, the structure collapsed segregating the zinc oxide in its zincite phase, being almost complete with long treatment times; this caused the FT-IR spectra of these samples to change slightly. In Figure 4 the bands attributed to the vibrational modes of the hydroxyl groups and water molecules (band positions already mentioned above) can be identified. In addition, the vibration band at 1385 cm−1, attributed to the presence of the nitrate anion of the LDH phase recorded with low intensity in the PXRD diagrams of the sample, can be clearly observed. As a result of the formation of ZnO in the HT-treated samples, a band at the limit of the spectrum, around 470 cm−1, attributed to the stretching vibrational mode of ZnO, was observed in the FT-IR spectra [40].
In the spectra of the samples synthesized in the presence of DEA and TEA, no different bands were observed for the hydrothermally treated samples, both MW and HT, with respect to that of the samples that received no treatment. It should be noted that the higher crystallinity of the samples resulted in a better ordering of both the interlaminar anions and the layered structure, giving rise to a regularity that was reflected in a subtle increase in intensity and narrowing of the vibration bands.
Neither in the FT-IR spectra recorded for the samples without hydrothermal treatment, nor for the samples subjected to MW or HT hydrothermal treatments, were bands corresponding to the vibrational modes of the amines used during the synthesis observed.
3.4. Thermal Analysis
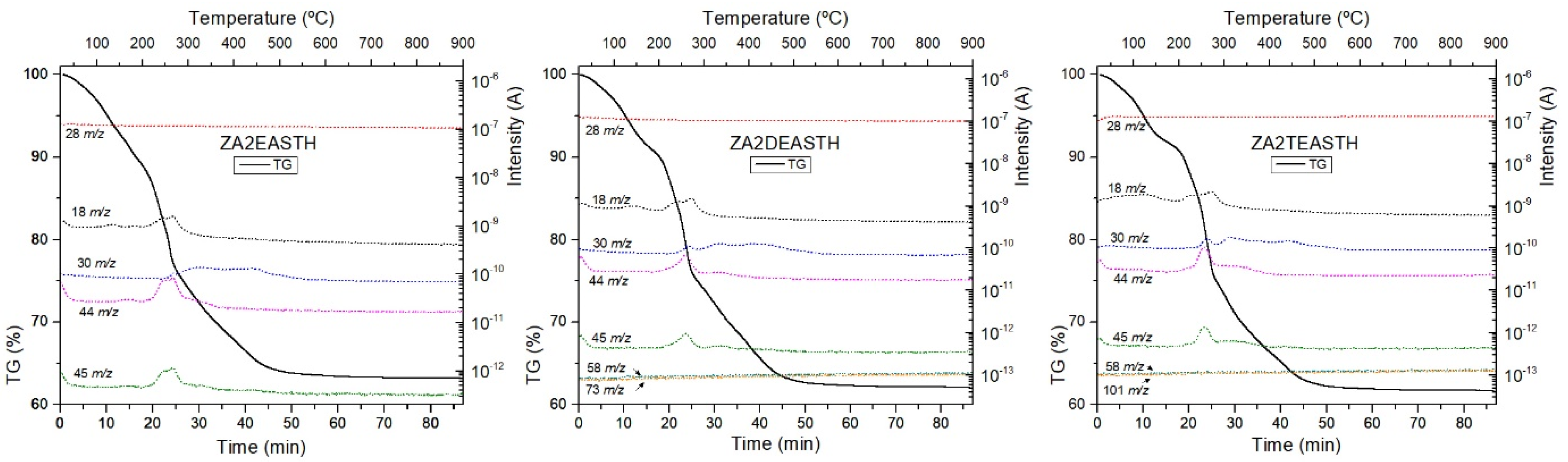
The thermal analysis of the samples prepared using EA, DEA or TEA as precipitan agents were carried out to determine their stability and evolution to mixed oxides. During the thermal analysis, mass spectrometry (MS) of the gases and vapors formed during the process (EGA, evolved gas analysis) was carried out. To identify the masses of the generated species, a complete mass spectrum was initially recorded and, in a second step, the MS analysis was performed by fixing these masses and following the change in their intensities throughout the analysis. The reference MS of the expected evolved gases was also taken into account. The signals monitorized corresponded to H2O (m/z = 18), N2 and CO (m/z = 28), NO (m/z = 30), N2O (m/z = 44), NO2 (m/z = 45), EA (fragment at m/z = 30, 44 and 45), DEA (fragments at m/z = 58 and 73) and TEA (fragments at m/z = 58 and 101).
TG curves of the samples without hydrothermal treatment (STH) are included in Figure 5, together with the tracked masses of the gases generated during the process. In all curves, the typical decomposition stages of LDH compounds can be identified. Three decomposition stages could be identified in all TG curves. First, below 180 °C, water removal was observed, as shown by the MS peak at m/z = 18. The second stage of decomposition was observed up to around 300 °C, and corresponded to the release of water from the condensation of hydroxyl groups of the brucite-like layers. Finally, a steady mass loss was observed between 300 and 700 °C, which corresponded to the removal of interlayer nitrate species. The MS signals recorded in this temperature range corresponded to formation of species such as NO, NO2, and N2O, from nitrate decomposition. In all the curves, the process of elimination of the hydroxyl groups practically overlapped with the last stage of decomposition/removal of the interlaminar anion. However, there was better differentiation of the first stage from the second decomposition stage as the degree of substitution of the amino group in the compound used as precipitating agent increased. Thus, for the ZA2TEASTH sample a small plateau could be observed around 200 °C.
From the tracking of the m/z signals of each amine, the absence of amine residues in the final solids could be concluded, as could also be observed by FT-IR spectroscopy. Only in the case of the sample prepared in the presence of EA, could the mass at 45 m/z be attributed to that amine. However, some of the masses associated with EA overlap with the signals of the decomposition products of the nitrate anion (N2O, NO and NO2). The absence of EA was confirmed because the mass tracking curves had the same profile as those of NOx gas formation in the solids synthesized in the presence of DEA and TEA.
For the hydrothermally treated samples, both MW and HT, had similar TG curves recorded, in which the aforementioned plateau was more evident as a result of the increase in the crystallinity of the samples.
On the other hand, it can also be observed that, as the crystalline regularity of the solids increases, the total mass loss decreased (Table 4), due to the lower amount of water retained in the solids. Table 4 shows that as the hydrothermal treatment time increased, the total mass loss was lower, being in all cases between 30 and 40%. Only in the case of the samples synthesized in the presence of EA and with HT hydrothermal treatment was a total mass loss less than 30% observed. This was due to the formation of ZnO in these samples, where after 7 days of treatment practically the whole structure had collapsed and the mass loss observed in the TG curve was due to the small amount of the LDH phase, the retained water and the decomposition of the nitrate anion.
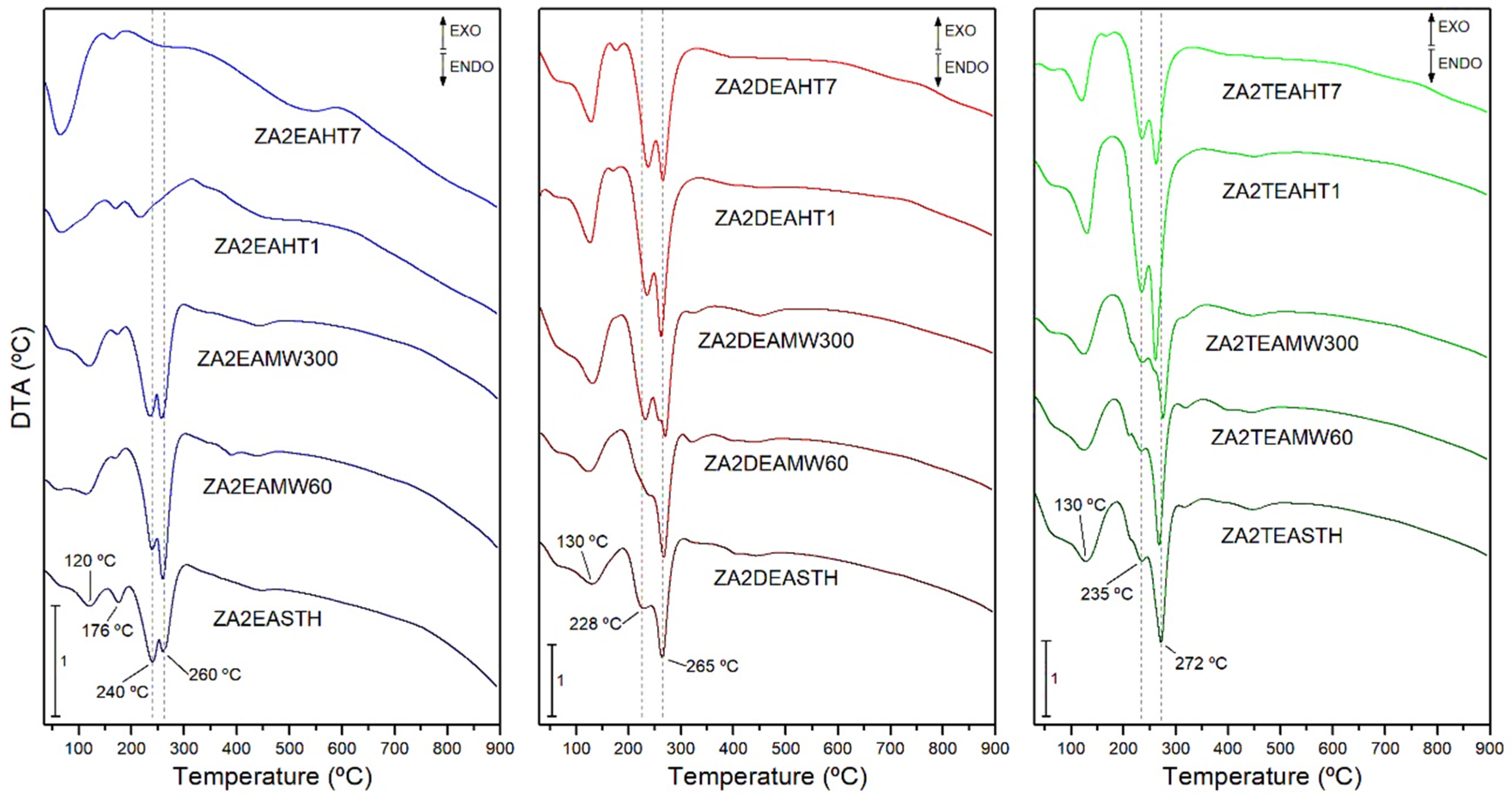
The DTA curves of the samples both without hydrothermal treatment and with hydrothermal treatments, MW or HT, are included in Figure 6. In all cases, endothermic minima associated with the different decomposition processes of the samples can be observed.
The minimum recorded at 120–130 °C in the DTA curves was associated with the process of release of the water retained in the interlayer space. However, in the case of the samples synthesized in the presence of EA, another minimum could be observed at 176 °C, which could correspond to the elimination of water molecules retained more strongly in the structure of the layered solid. On the other hand, at temperatures above 200 °C a minimum associated with the process of elimination of hydroxyl groups in the form of water vapor and the decomposition of the nitrate anion was found. In many cases this minimum presented a shoulder at lower temperature, or was even dissociated into two clearly distinguishable minima. This dissociation and, therefore, differentiation in the decomposition processes, became more evident as the crystalline regularity in the solids increased after the application of a hydrothermal treatment. On the other hand, it is worth mentioning that in all cases the main minimum was found at higher temperatures than in the case of the solids synthesized in our previous work using methylamine, dimethylamine or trimethylamine as precipitant agents, and even in the case of the solids synthesized in the absence of amines [27]. Thus, for the sample synthesized in the presence of DEA, this minimum was found at 260 °C, while for the sample synthesized in the presence of TEA, the minimum shifted to 272 °C.
3.5. Specific Surface Area and Porosity
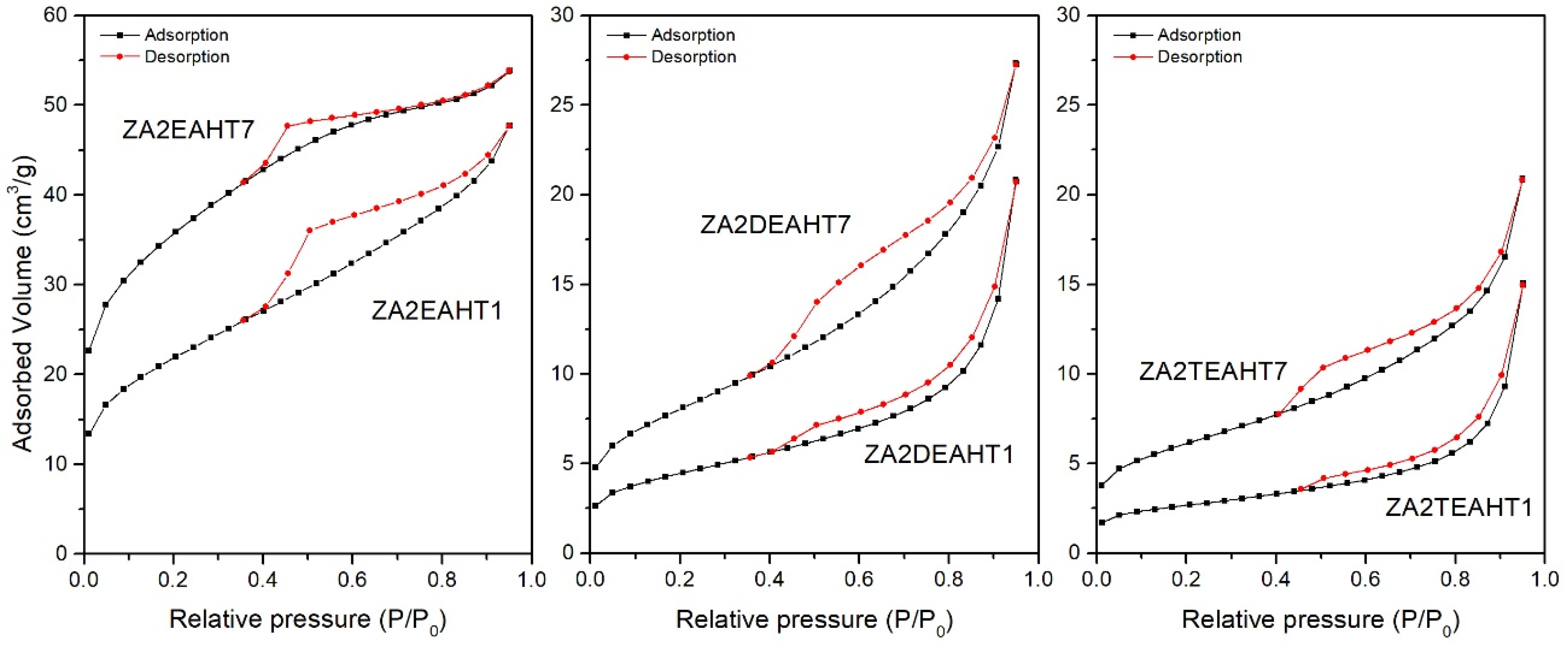
The textural properties of the synthesized solids were studied from the N2 adsorption–desorption isotherms at −196 °C. Table 5 includes the values of the specific surface areas calculated by the BET (SBET) method [41,42], the pore volume (Vpore) and the average pore diameter calculated by the BJH method [42,43] for the synthesized samples. For the samples prepared in the presence of DEA and TEA, both without hydrothermal treatment and with MW treatment, the adsorption capacity was below the confidence limit of the equipment used. Only for the samples with HT hydrothermal treatment (and sample ZA2DEAMW300) did the adsorption measurements present confidence values. This behavior was not observed for samples prepared in the presence of EA, although the samples presented low SBET values, close to the detection limit. Figure 7 and Figure 8 include the corresponding adsorption–desorption isotherms, which correspond to type II according to the IUPAC classification [44,45], corresponding to adsorption on non-porous or mesoporous adsorbents, where adsorption can occur without monolayer-multilayer restrictions. Moreover, it can be observed how all of them presented a hysteresis cycle, corresponding to the H3 type according to the IUPAC classification [42], indicating that adsorption took place in slit-shaped pores formed by layer-like particles.
In all cases SBET values were higher than those obtained for the samples obtained with the amines used in our previous work and, also, the values were higher than those found for the samples synthesized in the absence of amines [27]. On the contrary, smaller pore diameter sizes were recorded.
In view of the results included in Table 5, the segregation of ZnO in the samples synthesized in the presence of EA with HT treatment led to a substantial increase in the specific surface area. Solids with higher porosity were obtained, where the pore volume increased, with smaller pore diameter sizes with respect to the sample without hydrothermal treatment.
In the case of the solids synthesized in the presence of DEA and TEA, after the application of a HT hydrothermal treatment, according to the PXRD results, an increase in the crystallinity took place, as a consequence of better ordering of the brucite-like layers. Together with the increase in the crystallinity of the solids, an increase in the SBET could be observed and adsorption–desorption curves could be recorded in both cases. Moreover, the prolongation of the HT treatment resulted in higher crystallinity linked to an increase of the SBET value, where, in both series of samples, the SBET value for sample with 7 days of treatment was twice that for the sample with one day of treatment. However, it is important to remember the presence of approximately 2% of ZnO in sample ZA2DEAHT7, which could justify the difference in specific surface area with respect to that of sample ZA2TEAHT7.
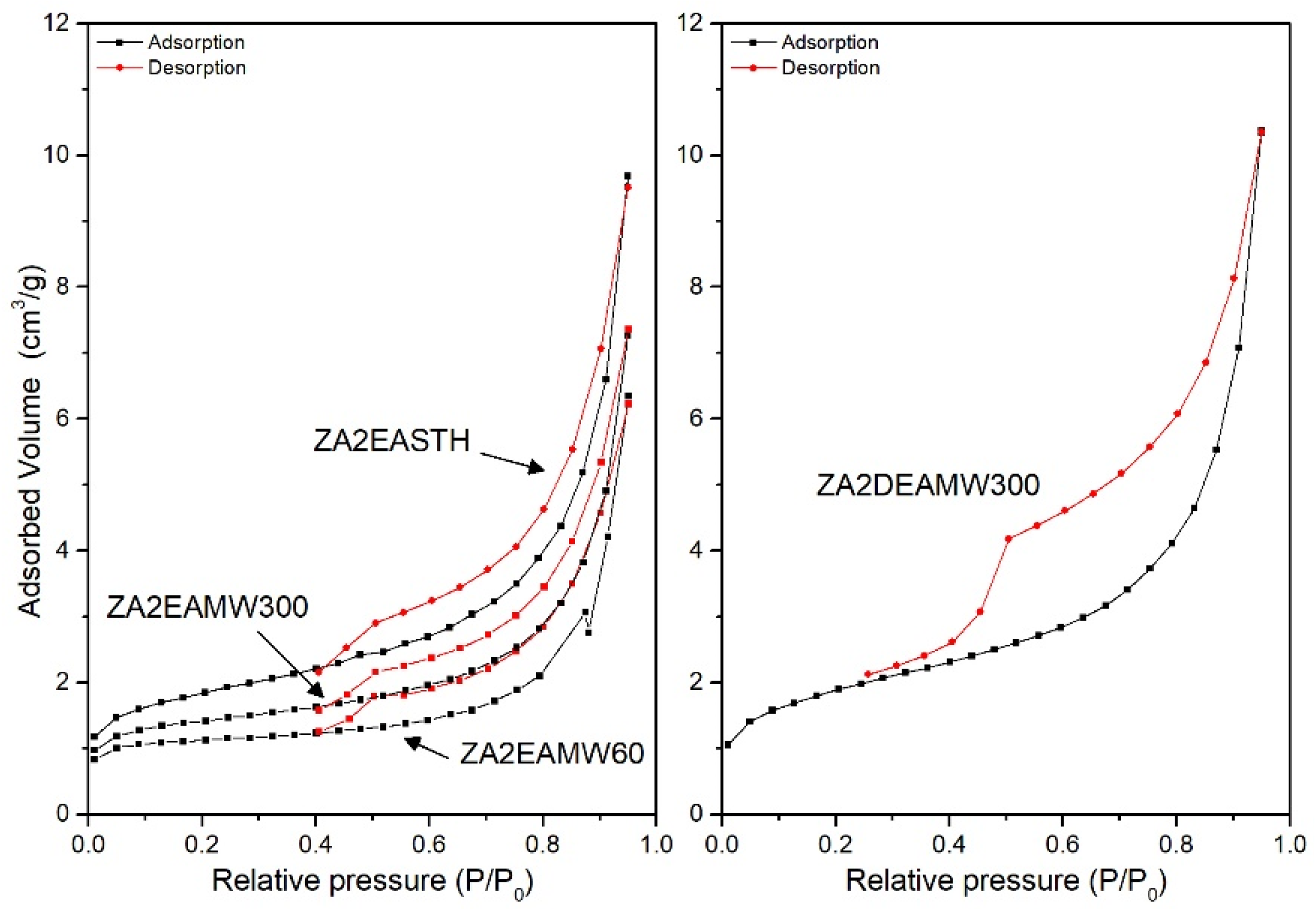
Figure 8 shows the adsorption–desorption curves for the samples without hydrotermal treatment and with MW treatment. While the sample without hydrothermal treatment had an SBET value close to 6 m2/g, for samples synthetized using EA as precipitant agent, when a MW hydrothermal treatment was applied the surface area decreased. In the three curves a similar behavior against the desorption process can be observed, with slightly larger pore sizes when MW treatment was applied. On the other hand, when DEA or TEA were used as precipitant agents, only the adsorption–desorption curve for sample ZA2DEAMW300 was recorded. The SBET value for this sample was 6.4 m2/g, higher than that found for the analogous sample synthesized in the presence of EA.
3.6. Particle Size Distribution
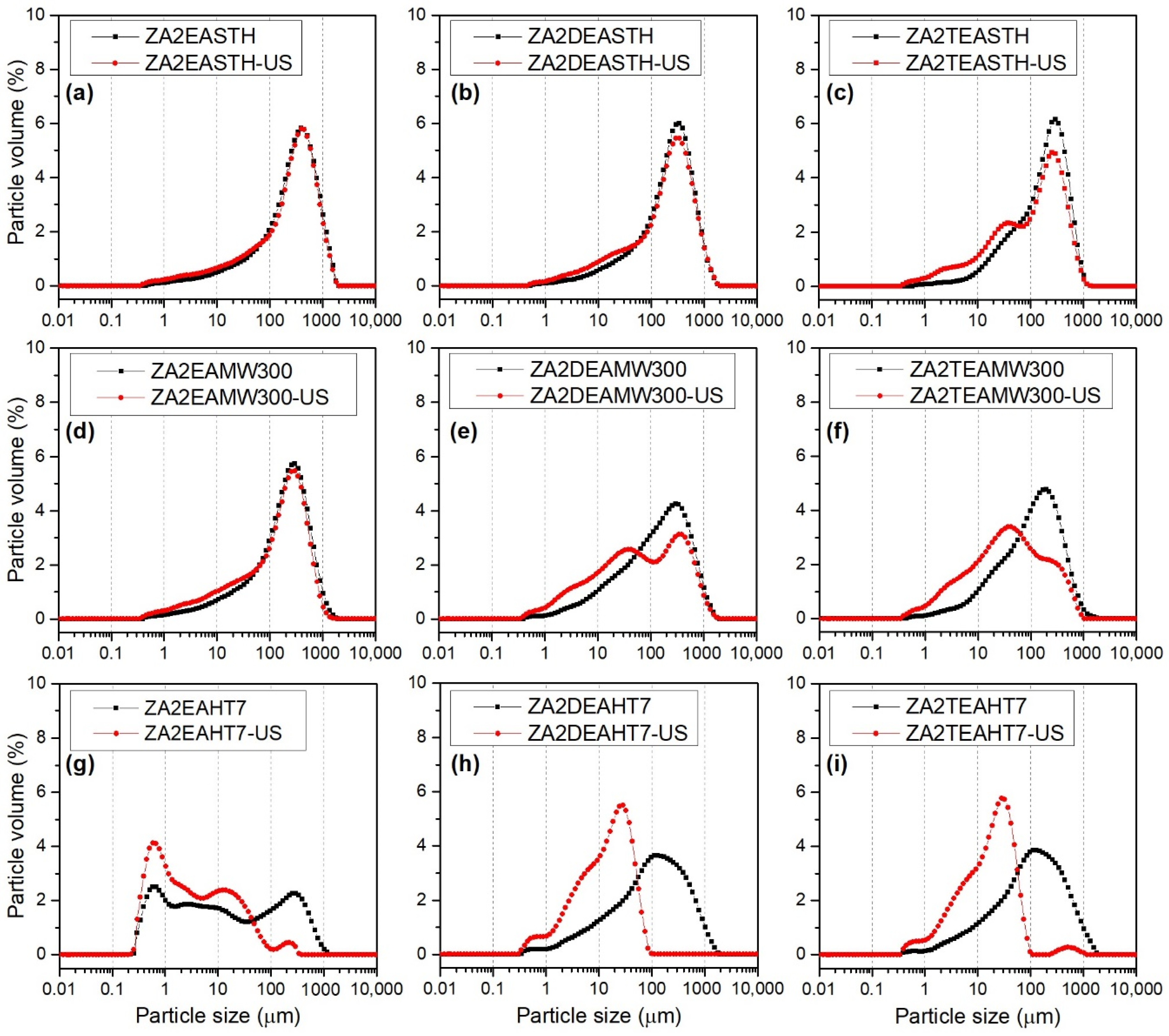
Figure 9 shows the particle size distribution curves of the samples synthesized in the presence of EA, DEA and TEA without hydrothermal treatment and after the application of the longest periods of both hydrothermal treatments: MW and HT. For each of the samples two distribution curves are represented: (i) the distribution curve of the sample in aqueous suspension, black curve, and (ii) the distribution curve of the sample in aqueous suspension subjected to sonication treatment for 15 min, directly in the particle size analyzer, red curve. Sonication treatment is often used to disaggregate primary particles, changing the distribution curves.
The samples without hydrothermal treatment presented a monomodal size distribution centered between 300 and 400 µm. Although, as the degree of amino group substitution increased, a small shoulder could be observed at lower size values, approximately at 20 µm, as observed in Figure 9a–c. The application of ultrasound for 15 min did not have a great impact on the distribution curves; only in the case of sample ZA2TEASTH was the aforementioned shoulder accentuated at 20 µm. This fact indicates that samples prepared in the presence of EA and DEA give rise to robust particles that are more difficult to disaggregate than particles obtained using TEA as the precipitant agent.
The particle size distribution curves for the samples subjected to MW hydrothermal treatment for 300 min are included in Figure 9d–f. While the sample synthesized in the presence of EA presented the same profile as its version without hydrothermal treatment, broader distribution curves were observed for samples ZA2DEAMW300 and ZA2TEAMW300. After the application of ultrasound, practically no changes in the distribution were observed for sample ZA2EAMW300, indicating the difficulty in desaggregating its particles, showing the same robustness as the STH sample. In the case of samples prepared in the presence of DEA and TEA, after the application of ultrasound, disaggregation to smaller particle size took place, resulting in bimodal distribution curves with maxima at 40 µm. Moreover, after the application of ultrasound, it can be observed how these distributions became wider.
The distribution curves for the samples subjected to HT hydrothermal treatment for 7 days are included in Figure 9g–i, where it is observed that the samples synthesized in the presence of DEA and TEA presented the same behavior as the samples with MW hydrotermal treatment over 300 min, obtaining monomodal particle size distributions that covered a wide range of sizes. However, when an ultrasound treatment was applied, the particles disaggregated, obtaining monomodal distributions at smaller particle sizes with a maxima between 20 and 30 µm, which presented, in both cases, a shoulder at even smaller particle sizes, around 5 µm. Furthermore, in both samples it was observed how the size distributions became narrower. For example, the particle size distribution of sample ZA2DEAHT7 decreased from a maximum at 130 µm to a maximum at 30 µm, with a shoulder at 6 µm. In contrast, the curve of sample ZA2EAHT7 presented slightly different behavior, where a multimodal distribution was obtained over a wide range of particle sizes, with maxima at 0.6, 2.7, 10 and 300 µm. After the application of ultrasound, the disintegration of the larger particles occurred, also resulting in a multimodal distribution with maxima at 0.6, 2.5, 12 and 225 µm, with a higher percentage of smaller particles. The different behavior of sample ZA2EAHT7 was due to fact that in this sample the hydrotalcite-type structure had completely collapsed and the only phase detected by PXRD was ZnO (Figure 3).
Table 6 includes the volume-weighted mean particle diameter, D[4,3], values for each of the samples, both before and after ultrasound treatment [46,47]. In general, the lowest particle size values were obtained for the samples synthesized in the presence of TEA, obtaining values even lower than those of the samples obtained without amines in the reaction medium (reported in our previous work [27]). For all series of samples, similar D[4,3] values were found, regardless of the MW treatment time, whereas, when HT hydrothermal treatment was applied, as the treatment time increased, the D[4,3] value slightly decreased. The smallest particle size, 114 µm, was found for sample ZA2EAHT7, where, as previously commented, the structure had collapsed to zinc oxide.
The application of a sonication treatment had a great impact on the D[4,3] value of the samples subjected to HT hydrothermal treatment, as can be deduced from the data collected in Table 6. Thus, the lowest value was obtained for sample ZA2DEAHT7, with a mean particle diameter of 36 µm.
It can be concluded that the samples subjected to MW treatment were formed by tightly bound particle agglomerates, deduced by the low disintegration after the sonication treatment. Only for samples with long periods of MW treatment was a slight disintegration of the agglomerates observed, obtaining the lowest D[4,3] value for the sample ZA2TEAMW300, with 112 µm.
4. Conclusions
The effect on the properties of Zn-Al-NO3 LDHs, prepared using amines with two carbon atoms in the organic chain, together with the application of hydrothermal treatments using different energy sources, conventional or microwave heating, was studied. Well crystallized compounds were obtained, the crystallinity of which improved a lot after prolonged conventional hydrothermal treatment when diethylamine or triethylamine were used as precipitant agents. When ethylamine was used in the synthesis media, two LDH phases were obtained, in which the nitrate anion had two different orientations in the interlayer space. Furthermore, the LDH structure of solids prepared using ethylamine collapsed easily with a conventional hydrothermal treatment to form zinc oxide. When the samples were subjected to conventional hydrothermal treatment, the formation of ZnO was also observed when DEA or TEA were used, although to a smaller amount. So, the ZnO content is lower as the degree of substitution of the amino group of the compound used as precipitating agent increases. The results also showed the formation of aggregates which could be disaggregated by sonication. So, it was possible to obtain solids with high crystal sizes and low particle size distributions when a conventional treatment was used.
Author Contributions
Conceptualization, F.M.L.; methodology, A.M., A.J. and F.M.L.; investigation, A.M.; resources, F.M.L.; writing—original draft preparation, A.M.; writing—review and editing, A.M. and F.M.L.; visualization, A.M. and A.J.; supervision, F.M.L.; funding acquisition, F.M.L. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from Universidad de Salamanca (Plan I-B3) and Proyect RTC-2014-1908-3, Ministerio de Economía y Competitividad.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
A. Misol thanks Junta de Castilla y León and ERDF for a predoctoral contract. A. Jiménez thanks Universidad de Salamanca and Banco Santander for a predoctoral contract. The authors want to express the great pleasure and privilege in collaborating with this paper for this special issue of the ChemEngineering in honor of our mentor and friend Vicente Rives, Professor of Inorganic Chemistry at University of Salamanca.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Rives, V.; Labajos, F.M.; Herrero, M. Layered Double Hydroxides as Nanofillers of Composites and Nanocomposite Materials Based on Polyethylene. In Polyethylene-Based Blends, Composites and Nanocomposites; Visakh, P.M., Morlanes, M.J.M., Eds.; Wiley: Beverly, MA, USA, 2015; pp. 163–200. [Google Scholar]
- Rives, V. Layered Double Hydroxides: Present and Future; NOVA Science Publishers, Inc.: New York, NY, USA, 2001; ISBN 978-1-61209-289-8. [Google Scholar]
- Wang, Q.; O’hare, D. Recent advances in the synthesis and application of layered double hydroxide (LDH) nanosheets. Chem. Rev. 2012, 112, 4124–4155. [Google Scholar] [CrossRef] [PubMed]
- Alonso-de-Linaje, V.; Mangayayam, M.C.; Tobler, D.J.; Dietmann, K.M.; Espinosa, R.; Rives, V.; Dalby, K.N. Sorption of chlorinated hydrocarbons from synthetic and natural groundwater by organo-hydrotalcites: Towards their applications as remediation nanoparticles. Chemosphere 2019, 236, 124369. [Google Scholar] [CrossRef]
- Dietmann, K.M.; Linke, T.; Trujillano, R.; Rives, V. Effect of chain length and functional group of organic anions on the retention ability of mgal-layered double hydroxides for chlorinated organic solvents. ChemEngineering 2019, 3, 89. [Google Scholar] [CrossRef] [Green Version]
- Alonso-de-Linaje, V.; Mangayayam, M.C.; Tobler, D.J.; Rives, V.; Espinosa, R.; Dalby, K.N. Enhanced sorption of perfluorooctane sulfonate and perfluorooctanoate by hydrotalcites. Environ. Technol. Innov. 2021, 21, 101231. [Google Scholar] [CrossRef]
- Trujillano, R.; Nájera, C.; Rives, V. Activity in the Photodegradation of 4-Nitrophenol of a Zn,Al Hydrotalcite-Like Solid and the Derived Alumina-Supported ZnO. Catalysts 2020, 10, 702. [Google Scholar] [CrossRef]
- Karásková, K.; Pacultová, K.; Jirátová, K.; Fridrichová, D.; Koštejn, M.; Obalová, L. K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx. Catalysts 2020, 10, 1134. [Google Scholar] [CrossRef]
- Li, P.; Yu, F.; Altaf, N.; Zhu, M.; Li, J.; Dai, B.; Wang, Q. Two-Dimensional Layered Double Hydroxides for Reactions of Methanation and Methane Reforming in C1 Chemistry. Materials 2018, 11, 221. [Google Scholar] [CrossRef] [Green Version]
- Rives, V.; Del Arco, M.; Martín, C. Layered double hydroxides as drug carriers and for controlled release of non-steroidal antiinflammatory drugs (NSAIDs): A review. J. Control. Release 2013, 169, 28–39. [Google Scholar] [CrossRef]
- Choi, G.; Choy, J. Recent progress in layered double hydroxides as a cancer theranostic nanoplatform. WIREs Nanomed. Nanobiotechnol. 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Patel, R.; Park, J.T.; Patel, M.; Dash, J.K.; Gowd, E.B.; Karpoormath, R.; Mishra, A.; Kwak, J.; Kim, J.H. Transition-metal-based layered double hydroxides tailored for energy conversion and storage. J. Mater. Chem. A 2018, 6, 12–29. [Google Scholar] [CrossRef]
- Saifullah, B.; Hussein, M.Z. Inorganic nanolayers: Structure, preparation, and biomedical applications. Int. J. Nanomed. 2015, 10, 5609–5633. [Google Scholar] [CrossRef] [Green Version]
- Mishra, G.; Dash, B.; Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Miyata, S. Anion-Exchange Properties of Hydrotalcite-Like Compounds. Clays Clay Miner. 1983, 31, 305–311. [Google Scholar] [CrossRef]
- Inayat, A.; Klumpp, M.; Schwieger, W. The urea method for the direct synthesis of ZnAl layered double hydroxides with nitrate as the interlayer anion. Appl. Clay Sci. 2011, 51, 452–459. [Google Scholar] [CrossRef]
- Abderrazek, K.; Frini Srasra, N.; Srasra, E. Synthesis and Characterization of [Zn-Al] Layered Double Hydroxides: Effect of the Operating Parameters. J. Chin. Chem. Soc. 2017, 64, 346–353. [Google Scholar] [CrossRef]
- Bukhtiyarova, M.V. A review on effect of synthesis conditions on the formation of layered double hydroxides. J. Solid State Chem. 2019, 269, 494–506. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Hickey, L.; Frost, R.L. The effects of synthesis pH and hydrothermal treatment on the formation of zinc aluminum hydrotalcites. J. Solid State Chem. 2004, 177, 4047–4057. [Google Scholar] [CrossRef] [Green Version]
- Galvão, T.L.P.; Neves, C.S.; Caetano, A.P.F.; Maia, F.; Mata, D.; Malheiro, E.; Ferreira, M.J.; Bastos, A.C.; Salak, A.N.; Gomes, J.R.B.; et al. Control of crystallite and particle size in the synthesis of layered double hydroxides: Macromolecular insights and a complementary modeling tool. J. Colloid Interface Sci. 2016, 468, 86–94. [Google Scholar] [CrossRef]
- Benito, P.; Herrero, M.; Barriga, C.; Labajos, F.M.; Rives, V. Microwave-assisted homogeneous precipitation of hydrotalcites by urea hydrolysis. Inorg. Chem. 2008, 47, 5453–5463. [Google Scholar] [CrossRef]
- He, J.; Wei, M.; Li, B.; Kang, Y.; Evans, D.G.; Duan, X. Preparation of Layered Double Hydroxides. In Layered Double Hydroxides. Structure and Bonding; Duan, X., Evans, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 119, pp. 89–119. [Google Scholar]
- Labajos, F.M.; Rives, V.; Ulibarri, M.A. Effect of hydrothermal and thermal treatments on the physicochemical properties of Mg-Al hydrotalcite-like materials. J. Mater. Sci. 1992, 27, 1546–1552. [Google Scholar] [CrossRef]
- Ezeh, C.I.; Tomatis, M.; Yang, X.; He, J.; Sun, C. Ultrasonic and hydrothermal mediated synthesis routes for functionalized Mg-Al LDH: Comparison study on surface morphology, basic site strength, cyclic sorption efficiency and effectiveness. Ultrason. Sonochem. 2018, 40, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zadaviciute, S.; Baltakys, K.; Bankauskaite, A. The effect of microwave and hydrothermal treatments on the properties of hydrotalcite. J. Therm. Anal. Calorim. 2017, 127, 189–196. [Google Scholar] [CrossRef]
- Misol, A.; Labajos, F.M.; Morato, A.; Rives, V. Synthesis of Zn,Al layered double hydroxides in the presence of amines. Appl. Clay Sci. 2020, 189, 105539. [Google Scholar] [CrossRef]
- Kooli, F.; Depège, C.; Ennaqadi, A.; De Roy, A.; Besse, J.P. Rehydration of Zn-Al layered double hydroxides. Clays Clay Miner. 1997, 45, 92–98. [Google Scholar] [CrossRef]
- De La Rosa-Guzmán, M.Á.; Guzmán-Vargas, A.; Cayetano-Castro, N.; Del Río, J.M.; Corea, M.; Martínez-Ortiz, M.D.J. Thermal stability evaluation of polystyrene-Mg/zn/Al LDH nanocomposites. Nanomaterials 2019, 9, 1528. [Google Scholar] [CrossRef] [Green Version]
- Drits, V.A.; Bookin, A.S. Crystal Structure and X-Ray Identification of Layered Double Hydroxides. In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; NOVA Science Publishers, Inc.: New York, NY, USA, 2001; pp. 41–100. [Google Scholar]
- Thomas, G.S.; Radha, A.V.; Kamath, P.V.; Kannan, S. Thermally induced polytype transformations among the Layered Double Hydrodides (LDHs) of Mg Zn with Al. J. Phys. Chem. B 2006, 110, 12365–12371. [Google Scholar] [CrossRef]
- Marappa, S.; Radha, S.; Kamath, P.V. Nitrate-Intercalated Layered Double Hydroxides–Structure Model, Order, and Disorder. Eur. J. Inorg. Chem. 2013, 2013, 2122–2128. [Google Scholar] [CrossRef]
- Karthikeyan, J.; Fjellvåg, H.; Bundli, S.; Sjåstad, A.O. Efficient Exfoliation of Layered Double Hydroxides; Effect of Cationic Ratio, Hydration State, Anions and Their Orientations. Materials 2021, 14, 346. [Google Scholar] [CrossRef]
- Wang, S.L.; Wang, P.C. In situ XRD and ATR-FTIR study on the molecular orientation of interlayer nitrate in Mg/Al-layered double hydroxides in water. Coll. Surf. A Physicochem. Eng. Asp. 2007, 292, 131–138. [Google Scholar] [CrossRef]
- Misol, A.; Jiménez, A.; Morato, A.; Labajos, F.M.; Rives, V. Quantification by Powder X-ray Diffraction of Metal Oxides Segregation During Formation of Layered Double Hydroxides. Eur. J. Eng. Technol. Res. 2020, 5, 1243–1248. [Google Scholar] [CrossRef]
- Brown, J.G. X-rays and Their Applications; Plenum/Ros.; Plenum Publishing Corporation: New York, NY, USA, 1966; ISBN 0-306-20021-X. [Google Scholar]
- Jenkins, R.; de Vries, J.L. Worked Examples in X-ray Analysis, 2nd ed.; Springer: New York, NY, USA, 1970; ISBN 978-1-4899-2649-4. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part A: Theory and Applications in Inorganic Chemistry, 6th ed.; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2009; ISBN 9780471743392. [Google Scholar]
- Zhang, Y.; Wang, L.; Zou, L.; Xue, D. Crystallization behaviors of hexagonal nanoplatelet MgAlCO3 layered double hydroxide. J. Cryst. Growth 2010, 312, 3367–3372. [Google Scholar] [CrossRef]
- Feng, W.; Chen, J.; Hou, C.-Y. Growth and characterization of ZnO needles. Appl. Nanosci. 2014, 4, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Brunauer, S.; Deming, L.S.; Deming, W.E.; Teller, E. On a Theory of the van der Waals Adsorption of Gases. J. Am. Chem. Soc. 1940, 62, 1723–1732. [Google Scholar] [CrossRef]
- A Basic Guide to Particle Characterization; Malvern Instruments Limited: Malvern, UK, 2015; pp. 1–24. Available online: https://www.cif.iastate.edu/sites/default/files/uploads/Other_Inst/Particle%20Size/Particle%20Characterization%20Guide.pdf (accessed on 30 June 2022).
- A Guidebook to Particle Size Analysis. Horiba Sci. 2019, 1–32. Available online: https://www.horiba.com/aut/scientific/products/particle-characterization/particle-size-essentials-guidebook/ (accessed on 30 June 2022).
Figure 1.
Powder X-ray diffraction diagrams of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment.
Figure 1.
Powder X-ray diffraction diagrams of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment.

Figure 2.
PXRD of the samples prepared in the presence of EA, DEA and TEA with 60 and 300 min of MW treatment.
Figure 2.
PXRD of the samples prepared in the presence of EA, DEA and TEA with 60 and 300 min of MW treatment.

Figure 3.
PXRD of the samples prepared in the presence of EA, DEA and TEA with 1 and 7 days of HT treatment. (▪) ZnO phase.
Figure 3.
PXRD of the samples prepared in the presence of EA, DEA and TEA with 1 and 7 days of HT treatment. (▪) ZnO phase.

Figure 4.
FT-IR spectra of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and with MW and HT treatments.
Figure 4.
FT-IR spectra of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and with MW and HT treatments.

Figure 5.
TG curves, in O2 atmosphere, of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and tracking of the characteristic m/z signals.
Figure 5.
TG curves, in O2 atmosphere, of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and tracking of the characteristic m/z signals.

Figure 6.
DTA curves, in O2 atmosphere, of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and with MW and HT treatments.
Figure 6.
DTA curves, in O2 atmosphere, of the samples prepared in the presence of EA, DEA and TEA with no hydrothermal treatment and with MW and HT treatments.

Figure 7.
Nitrogen adsorption-desorption isotherms for HT treated samples prepared in the presence of EA, DEA and TEA.
Figure 7.
Nitrogen adsorption-desorption isotherms for HT treated samples prepared in the presence of EA, DEA and TEA.

Figure 8.
Nitrogen adsorption-desorption isotherms for MW treated samples prepared in the presence of EA and DEA.
Figure 8.
Nitrogen adsorption-desorption isotherms for MW treated samples prepared in the presence of EA and DEA.

Figure 9.
Particle size distribution before (black) and after (red) sonication in water suspension of synthetized samples (a) ZA2EASTH, (b) ZA2DEASTH, (c) ZA2TEASTH, (d) ZA2EAMW300, (e) ZA2DEAMW300, (f) ZA2TEAMW300, (g) ZA2EAHT7, (h) ZA2DEAHT7, (i) ZA2TEAHT7.
Figure 9.
Particle size distribution before (black) and after (red) sonication in water suspension of synthetized samples (a) ZA2EASTH, (b) ZA2DEASTH, (c) ZA2TEASTH, (d) ZA2EAMW300, (e) ZA2DEAMW300, (f) ZA2TEAMW300, (g) ZA2EAHT7, (h) ZA2DEAHT7, (i) ZA2TEAHT7.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Element chemical analysis results and the chemical formulae of each sample.
| Sample | Al a | Zn a | Zn/Al b | x c | Formulae |
|---|---|---|---|---|---|
| ZA2EASTH | 8.32 | 40.27 | 2.00 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.44 H2O |
| ZA2EAMW60 | 7.98 | 38.63 | 2.00 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.48 H2O |
| ZA2EAMW300 | 7.96 | 38.30 | 1.99 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.50 H2O |
| ZA2EAHT1 | 9.84 | 46.51 | 1.95 | 0.34 | LDH * + ZnO + Al2O3 |
| ZA2EAHT7 | 11.05 | 52.01 | 1.94 | 0.34 | LDH * + ZnO + Al2O3 |
| ZA2DEASTH | 7.51 | 38.44 | 2.11 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.57 H2O |
| ZA2DEAMW60 | 7.67 | 38.87 | 2.09 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.55 H2O |
| ZA2DEAMW300 | 7.64 | 38.31 | 2.07 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.55 H2O |
| ZA2DEAHT1 | 7.73 | 38.47 | 2.05 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.51 H2O |
| ZA2DEAHT7 | 8.37 | 41.88 | 2.07 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.42 H2O |
| ZA2TEASTH | 7.64 | 37.56 | 2.03 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.55 H2O |
| ZA2TEAMW60 | 7.62 | 37.62 | 2.04 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.51 H2O |
| ZA2TEAMW300 | 7.60 | 37.39 | 2.03 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.52 H2O |
| ZA2TEAHT1 | 7.67 | 37.82 | 2.04 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.50 H2O |
| ZA2TEAHT7 | 8.13 | 41.16 | 2.09 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.43 H2O |
a Mass percentage. b Molar ratio. c Al/(Al + Zn) molar ratio. * It was not possible to determine the chemical formula of the LDH phase.
Table 2.
ZnO content in the Zn and Al samples prepared in the presence of EA, DEA and TEA with HT treatment.
Table 2.
ZnO content in the Zn and Al samples prepared in the presence of EA, DEA and TEA with HT treatment.
| Sample | (101) Peak Area (ZnO) a | (003) Peak Area (LDH) a | Area Ratio (101)/(003) | ZnO Content b |
|---|---|---|---|---|
| ZA2EAHT1 | 1807.0 | 1424.0 | 1.268961 | 67 |
| ZA2EAHT7 | 2776.0 | - | - | ≈100 |
| ZA2DEAHT1 | 122.1 | 18,339.8 | 0.006659 | 0.32 |
| ZA2DEAHT7 | 790.7 | 19,232.1 | 0.041114 | 2.14 |
| ZA2TEAHT1 | - | - | - | - |
| ZA2TEAHT7 | 502.6 | 26,627.0 | 0.018876 | 0.97 |
a a.u. b Mass percentage.
Table 3.
Lattice parameters c and a, average crystal size D and number of stacked layers for the samples obtained.
Table 3.
Lattice parameters c and a, average crystal size D and number of stacked layers for the samples obtained.
| Sample | c (Å) | a (Å) | D (Å) | Number of Stacked Layers |
|---|---|---|---|---|
| ZA2EASTH | 26.80 | 3.0697 | 111 | 12 |
| ZA2EAMW60 | 26.80 | 3.0697 | 118 | 13 |
| ZA2EAMW300 | 26.80 | 3.0720 | 135 | 15 |
| ZA2EAHT1 | - | - | - | - |
| ZA2EAHT7 | - | - | - | - |
| ZA2DEASTH | 26.67 | 3.0673 | 126 | 14 |
| ZA2DEAMW60 | 26.80 | 3.0697 | 148 | 17 |
| ZA2DEAMW300 | 26.80 | 3.0720 | 210 | 23 |
| ZA2DEAHT1 | 26.67 | 3.0743 | 284 | 32 |
| ZA2DEAHT7 | 26.80 | 3.0766 | 334 | 37 |
| ZA2TEASTH | 26.80 | 3.0697 | 150 | 17 |
| ZA2TEAMW60 | 26.80 | 3.0697 | 142 | 16 |
| ZA2TEAMW300 | 26.80 | 3.0697 | 218 | 24 |
| ZA2TEAHT1 | 26.80 | 3.0766 | 319 | 36 |
| ZA2TEAHT7 | 26.67 | 3.0743 | 322 | 36 |
Table 4.
Total weight loss and H2O molecules per chemical formula calculated for each sample.
| Sample | Weight Loss (%) | H2O Molecules Per Chemical Formula (n) |
|---|---|---|
| ZA2EASTH | 36.6 | 0.44 |
| ZA2EAMW60 | 36.7 | 0.48 |
| ZA2EAMW300 | 38.0 | 0.50 |
| ZA2EAHT1 | 22.6 | - |
| ZA2EAHT7 | 14.6 | - |
| ZA2DEASTH | 38.1 | 0.57 |
| ZA2DEAMW60 | 37.8 | 0.55 |
| ZA2DEAMW300 | 38.2 | 0.55 |
| ZA2DEAHT1 | 37.3 | 0.51 |
| ZA2DEAHT7 | 31.0 | 0.42 |
| ZA2TEASTH | 38.7 | 0.55 |
| ZA2TEAMW60 | 38.3 | 0.51 |
| ZA2TEAMW300 | 38.5 | 0.52 |
| ZA2TEAHT1 | 37.9 | 0.50 |
| ZA2TEAHT7 | 32.8 | 0.43 |
Table 5.
BET specific surface area, pore volume and average pore diameter of the samples prepared.
| Sample | SBET (m2/g) | Vpore (mm3/g) | BJH Desorption Average Pore Diameter (nm) |
|---|---|---|---|
| ZA2EASTH | 6.1 | 14.9 | 9.1 |
| ZA2EAMW60 | 3.4 | 9.8 | 9.6 |
| ZA2EAMW300 | 4.5 | 11.2 | 9.6 |
| ZA2EAHT1 | 74.3 | 73.7 | 4.6 |
| ZA2EAHT7 | 118.5 | 83.2 | 3.0 |
| ZA2DEASTH | - | - | - |
| ZA2DEAMW60 | - | - | - |
| ZA2DEAMW300 | 6.4 | 16.0 | 6.9 |
| ZA2DEAHT1 | 15.3 | 32.2 | 7.6 |
| ZA2DEAHT7 | 28.2 | 42.2 | 5.3 |
| ZA2TEASTH | - | - | - |
| ZA2TEAMW60 | - | - | - |
| ZA2TEAMW300 | - | - | - |
| ZA2TEAHT1 | 9.0 | 23.3 | 9.9 |
| ZA2TEAHT7 | 21.0 | 32.4 | 5.7 |
Table 6.
Volume-weighted mean particle diameter, D[4,3], values of prepared samples.
| Sample | D[4,3] Before Sonication | D[4,3] After Sonication |
|---|---|---|
| ZA2EASTH | 428 | 399 |
| ZA2EAMW60 | 280 | 258 |
| ZA2EAMW300 | 298 | 248 |
| ZA2EAHT1 | 327 | 230 |
| ZA2EAHT7 | 114 | 16 |
| ZA2DEASTH | 347 | 324 |
| ZA2DEAMW60 | 268 | 263 |
| ZA2DEAMW300 | 275 | 210 |
| ZA2DEAHT1 | 272 | 94 |
| ZA2DEAHT7 | 232 | 22 |
| ZA2TEASTH | 274 | 212 |
| ZA2TEAMW60 | 221 | 219 |
| ZA2TEAMW300 | 201 | 112 |
| ZA2TEAHT1 | 253 | 79 |
| ZA2TEAHT7 | 231 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Misol, A.; Jiménez, A.; Labajos, F.M. Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides. ChemEngineering 2022, 6, 53. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040053
AMA Style
Misol A, Jiménez A, Labajos FM. Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides. ChemEngineering. 2022; 6(4):53. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040053
Chicago/Turabian StyleMisol, Alexander, Alejandro Jiménez, and Francisco M. Labajos. 2022. "Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides" ChemEngineering 6, no. 4: 53. https://0-doi-org.brum.beds.ac.uk/10.3390/chemengineering6040053