Production of Human Pluripotent Stem Cell-Derived Hepatic Cell Lineages and Liver Organoids: Current Status and Potential Applications



Abstract
:1. Introduction
2. Hepatogenesis: Origin and Fates of Hepatic Cells
3. Differentiation of Hepatic Cell Lineages from Human Pluripotent Stem Cells
3.1. Hepatocytes
3.2. Cholangiocytes
3.3. Other Non-Parenchymal Cells
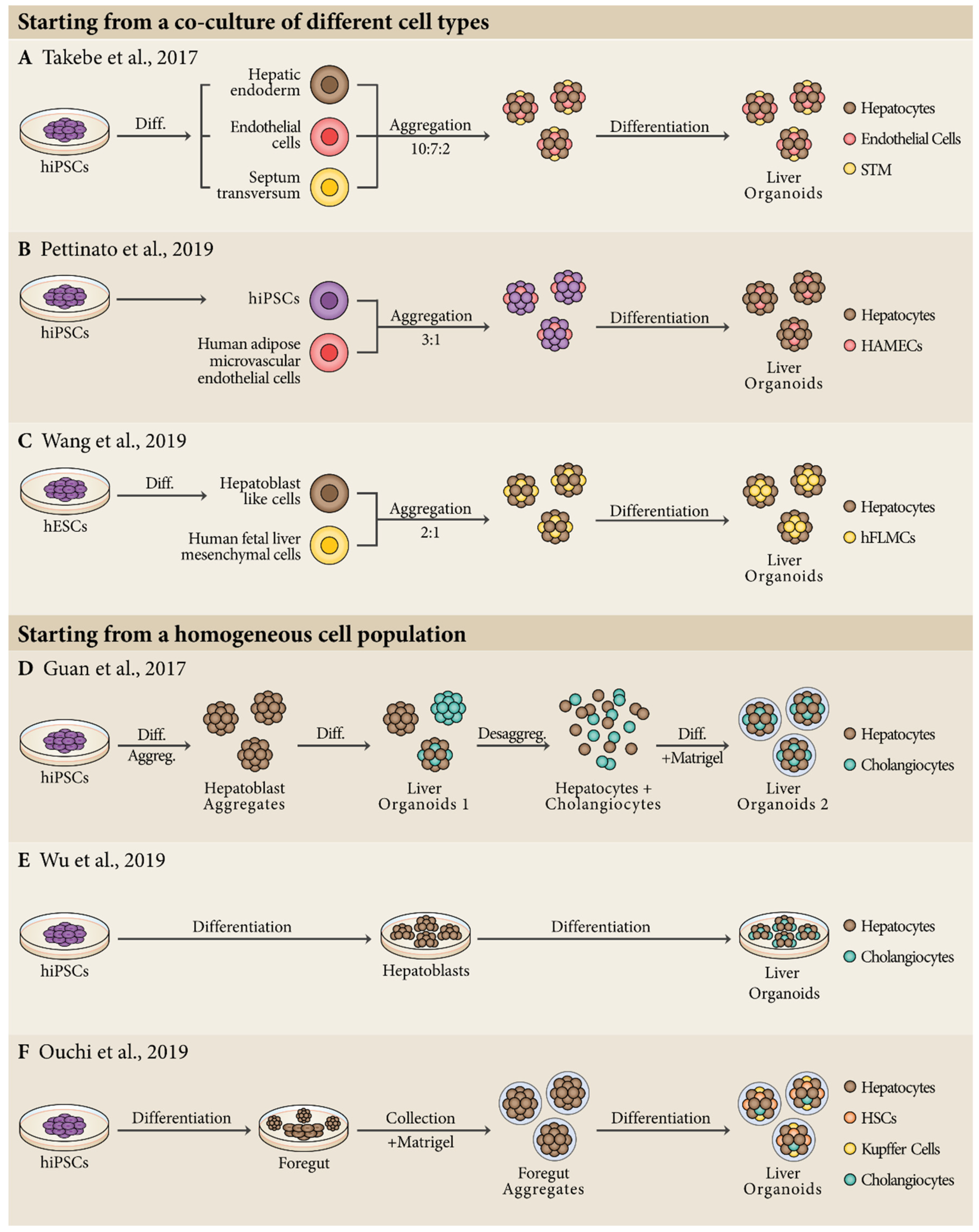
4. Production of Liver Organoids from Human Pluripotent Stem Cells
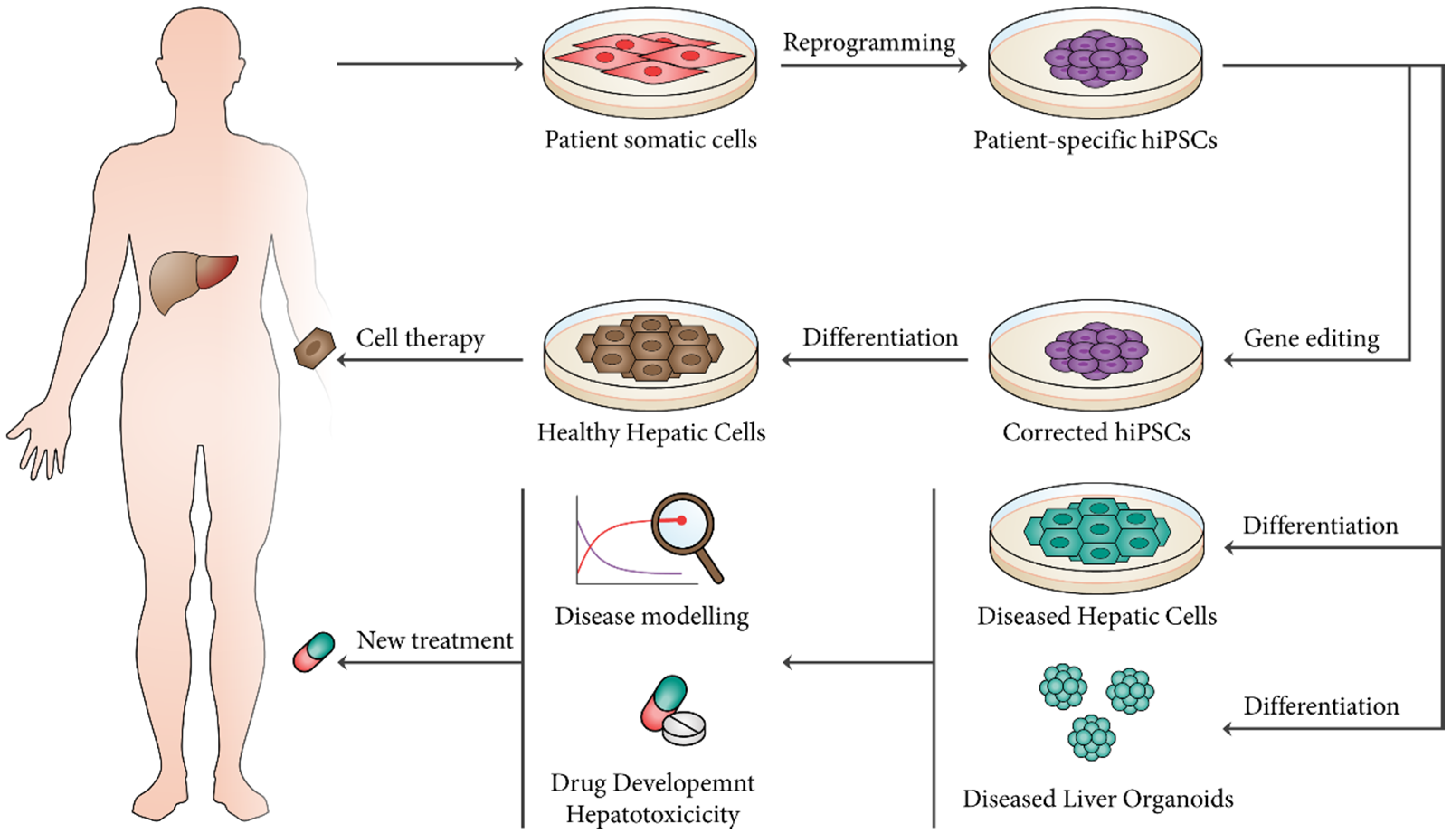
5. Applications of hPSC-Derived Hepatic Cell Lineages and Liver Organoids
5.1. Regenerative Medicine
5.2. Disease Modeling
5.3. Drug Discovery and Hepatotoxicity
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Si-Tayeb, K.; Lemaigre, F.P.; Duncan, S.A. Organogenesis and Development of the Liver. Dev. Cell 2010, 18, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.N.; Underhill, G.H.; Zaret, K.S.; Fox, I.J. Cell and tissue engineering for liver disease. Sci. Transl. Med. 2014, 6, 245sr2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Fleming, H.E.; Khetani, S.R.; Bhatia, S.N. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol. Adv. 2014, 32, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannoun, Z.; Steichen, C.; Dianat, N.; Weber, A.; Dubart-Kupperschmitt, A. The potential of induced pluripotent stem cell derived hepatocytes. J. Hepatol. 2016, 65, 182–199. [Google Scholar] [CrossRef] [Green Version]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 1–12. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Cotovio, J.P.; Fernandes, T.G.; Diogo, M.M.; Cabral, J.M.S. Pluripotent stem cell biology and engineering. In Engineering Strategies for Regenerative Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–31. [Google Scholar]
- Lawson, K.A.; Meneses, J.J.; Pedersen, R.A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 1991, 113, 891–911. [Google Scholar]
- Burdsal, C.A.; Damsky, C.H.; Pedersen, R.A. The role of E-cadherin and integrins in mesoderm differentiation and migration at the mammalian primitive streak. Development 1993, 118, 829–844. [Google Scholar] [PubMed]
- Kwon, G.S.; Viotti, M.; Hadjantonakis, A.K. The Endoderm of the Mouse Embryo Arises by Dynamic Widespread Intercalation of Embryonic and Extraembryonic Lineages. Dev. Cell 2008, 15, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viotti, M.; Nowotschin, S.; Hadjantonakis, A.K. SOX17 links gut endoderm morphogenesis and germ layer segregation. Nat. Cell Biol. 2014, 16, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, L.A.; Yamada, S.; Kuehn, M.R. Genetic dissection of nodal function in patterning the mouse embryo. Development 2001, 128, 1831–1843. [Google Scholar]
- Vincent, S.D.; Dunn, N.R.; Hayashi, S.; Norris, D.P.; Robertson, E.J. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003, 17, 1646–1662. [Google Scholar] [CrossRef] [Green Version]
- Faial, T.; Bernardo, A.S.; Mendjan, S.; Diamanti, E.; Ortmann, D.; Gentsch, G.E.; Mascetti, V.L.; Trotter, M.W.B.; Smith, J.C.; Pedersen, R.A. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 2015, 142, 2121–2135. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, K.D.; Zaret, K.S. Distinct populations of endoderm cells converge to generate the embryonic liver bud and ventral foregut tissues. Dev. Biol. 2005, 280, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Gordillo, M.; Evans, T.; Gouon-Evans, V. Orchestrating liver development. Development 2015, 142, 2094–2108. [Google Scholar] [CrossRef] [Green Version]
- Gualdi, R.; Bossard, P.; Zheng, M.; Hamada, Y.; Coleman, J.R.; Zaret, K.S. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev. 1996, 10, 1670–1682. [Google Scholar] [CrossRef] [Green Version]
- Rossi, J.M.; Dunn, N.R.; Hogan, B.L.M.; Zaret, K.S. Distinct mesodermal signals, including BMPs from the septum, transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001, 15, 1998–2009. [Google Scholar] [CrossRef] [Green Version]
- Serls, A.E.; Doherty, S.; Parvatiyar, P.; Wells, J.M.; Deutsch, G.H. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development 2005, 132, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calmont, A.; Wandzioch, E.; Tremblay, K.D.; Minowada, G.; Kaestner, K.H.; Martin, G.R.; Zaret, K.S. An FGF Response Pathway that Mediates Hepatic Gene Induction in Embryonic Endoderm Cells. Dev. Cell 2006, 11, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Yoshitomi, H.; Rossant, J.; Zaret, K.S. Liver organogenesis promoted by endothelial cells prior to vascular function. Science 2001, 294, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bort, R.; Signore, M.; Tremblay, K.; Pedro, J.; Barbera, M.; Zaret, K.S. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev. Biol. 2006, 290, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa-Pineda, B.; Wigle, J.T.; Oliver, G. Hepatocyte migration during liver development requires Prox1. Nat. Genet. 2000, 25, 254–255. [Google Scholar] [CrossRef] [PubMed]
- Lüdtke, T.H.W.; Christoffels, V.M.; Petry, M.; Kispert, A. Tbx3 promotes liver bud expansion during mouse development by suppression of cholangiocyte differentiation. Hepatology 2009, 49, 969–978. [Google Scholar] [CrossRef]
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296. [Google Scholar] [CrossRef]
- Clotman, F.; Jacquemin, P.; Plumb-Rudewiez, N.; Pierreux, C.E.; Van der Smissen, P.; Dietz, H.C.; Courtoy, P.J.; Rousseau, G.G.; Lemaigre, F.P. Control of liver cell fate decision by a gradient of TGF beta signaling modulated by Onecut transcription factors. Genes Dev. 2005, 19, 1849–1854. [Google Scholar] [CrossRef] [Green Version]
- Ober, E.A.; Lemaigre, F.P. Development of the liver: Insights into organ and tissue morphogenesis. J. Hepatol. 2018, 68, 1049–1062. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.; Tian, S.; Nie, J.; Jonsdottir, G.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.A.; Cotovio, J.P.; Rodrigues, C.A.V.; Vaz, S.H.; Fernandes, T.G.; Moreira, L.M.; Cabral, J.M.S.; Diogo, M.M. Transcriptomic analysis of 3D Cardiac Differentiation of Human Induced Pluripotent Stem Cells Reveals Faster Cardiomyocyte Maturation Compared to 2D Culture. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Ang, L.T.; Tan, A.K.Y.; Autio, M.I.; Goh, S.H.; Choo, S.H.; Lee, K.L.; Tan, J.; Pan, B.; Lee, J.J.H.; Lum, J.J.; et al. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep. 2018, 22, 2190–2205. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Niwa, H.; Iwase, K.; Takiguchi, M.; Mori, M.; Abé, S.I.; Abe, K.; Yamamura, K.I. Endoderm-specific gene expression in embryonic stem cells differentiated to embryoid bodies. Exp. Cell Res. 1996, 229, 27–34. [Google Scholar] [CrossRef]
- Rambhatla, L.; Chiu, C.P.; Kundu, P.; Peng, Y.; Carpenter, M.K. Generation of hepatocyte-like cells from human embryonic stem cells. Cell Transplant. 2003, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Cai, J.; Liu, Y.; Zhao, D.; Yong, J.; Duo, S.; Song, X.; Guo, Y.; Zhao, Y.; Qin, H.; et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Shinozaki, K.; Shannon, J.M.; Kouskoff, V.; Kennedy, M.; Woo, S.; Fehling, H.J.; Keller, G. Development of definitive endoderm from embryonic stem cells in culture. Development 2004, 131, 1651–1662. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Soto-Gutierrez, A.; Baptista, P.M.; Spee, B. Biotechnology Challenges to In Vitro Maturation of Hepatic Stem Cells. Gastroenterology 2018, 154, 1258–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, A.; Kinoshita, T.; Miyajima, A. Oncostatin M and hepatocyte growth factor induce hepatic maturation via distinct signaling pathways. FEBS Lett. 2001, 492, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, A. Fetal liver development requires a paracrine action of oncostatin M through the gp130 signal transducer. EMBO J. 1999, 18, 2127–2136. [Google Scholar] [CrossRef]
- Siller, R.; Greenhough, S.; Naumovska, E.; Sullivan, G.J. Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells. Stem Cell Rep. 2015, 4, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Vosough, M.; Omidinia, E.; Kadivar, M.; Shokrgozar, M.A.; Pournasr, B.; Aghdami, N.; Baharvand, H. Generation of functional hepatocyte-like cells from human pluripotent stem cells in a scalable suspension culture. Stem Cells Dev. 2013, 22, 2693–2705. [Google Scholar] [CrossRef]
- Farzaneh, Z.; Najarasl, M.; Abbasalizadeh, S.; Vosough, M.; Baharvand, H. Developing a Cost-Effective and Scalable Production of Human Hepatic Competent Endoderm from Size-Controlled Pluripotent Stem Cell Aggregates. Stem Cells Dev. 2018, 27, 262–274. [Google Scholar] [CrossRef]
- Yamashita, T.; Takayama, K.; Sakurai, F.; Mizuguchi, H. Billion-scale production of hepatocyte-like cells from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2018, 496, 1269–1275. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, Y.; Liu, Y.; Ye, F.; Song, Z.; Qin, H.; Meng, S.; Chen, Y.; Zhou, R.; Song, X.; et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology 2007, 45, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Chen, S.; Cai, J.; Guo, Y.; Song, Z.; Che, J.; Liu, C.; Wu, C.; Ding, M.; Deng, H. Derivation and characterization of hepatic progenitor cells from human embryonic stem cells. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianat, N.; Dubois-Pot-Schneider, H.; Steichen, C.; Desterke, C.; Leclerc, P.; Raveux, A.; Combettes, L.; Weber, A.; Corlu, A.; Dubart-Kupperschmitt, A. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology 2014, 60, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Ogawa, S.; Bear, C.E.; Ahmadi, S.; Chin, S.; Li, B.; Grompe, M.; Keller, G.; Kamath, B.M.; Ghanekar, A. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; De Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Assuncao, T.M.; Sun, Y.; Jalan-Sakrikar, N.; Drinane, M.C.; Huang, B.Q.; Li, Y.; Davila, J.I.; Wang, R.; O’Hara, S.P.; Lomberk, G.A.; et al. Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes. Lab. Investig. 2015, 95, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver sinusoidal endothelial cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar]
- Koui, Y.; Kido, T.; Ito, T.; Oyama, H.; Chen, S.W.; Katou, Y.; Shirahige, K.; Miyajima, A. An In Vitro Human Liver Model by iPSC-Derived Parenchymal and Non-parenchymal Cells. Stem Cell Rep. 2017, 9, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Sakurai, T.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Iinuma, N.; Iesato, Y.; Koyama, T.; Yoshizawa, T.; Uetake, R.; Yamauchi, A.; et al. Induction of LYVE-1/stabilin-2-positive liver sinusoidal endothelial-like cells from embryoid bodies by modulation of adrenomedullin-RAMP2 signaling. Peptides 2011, 32, 1855–1865. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.M.; Wu, J.C. Generation of Endothelial Cells From Human Pluripotent Stem Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; He, J.; Zhang, C.; Xu, J.; Wang, Y. Strategies for derivation of endothelial lineages from human stem cells. Stem Cell Res. Ther. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, F.; Xing, J.; Huang, X.; Mo, S.; Wei, X.; Tan, M.H.; Yu, H. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 2019, 192, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Asahina, K.; Zhou, B.; Pu, W.T.; Tsukamoto, H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 2011, 53, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdú, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef] [Green Version]
- Hay, D.C.; Fletcher, J.; Payne, C.; Terrace, J.D.; Gallagher, R.C.J.; Snoeys, J.; Black, J.R.; Wojtacha, D.; Samuel, K.; Hannoun, Z.; et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 12301–12306. [Google Scholar] [CrossRef] [Green Version]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, G.J.; Hay, D.C.; Park, I.-H.; Fletcher, J.; Hannoun, Z.; Payne, C.M.; Dalgetty, D.; Black, J.R.; Ross, J.A.; Samuel, K.; et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology 2010, 51, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touboul, T.; Hannan, N.R.F.; Corbineau, S.; Martinez, A.; Martinet, C.; Branchereau, S.; Mainot, S.; Strick-Marchand, H.; Pedersen, R.; Di Santo, J.; et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology 2010, 51, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, M.; Aoi, T.; Okita, K.; Takahashi, R.; Inoue, H.; Takayama, N.; Endo, H.; Eto, K.; Toguchida, J.; Uemoto, S.; et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12538–12543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Silva, T.P.; Cotovio, J.P.; Bekman, E.; Carmo-Fonseca, M.; Cabral, J.M.S.; Fernandes, T.G. Design Principles for Pluripotent Stem Cell-Derived Organoid Engineering. Stem Cells Int. 2019, 2019, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulos, G.K.; Bowen, W.C.; Mulè, K.; Stolz, D.B. Histological organization in hepatocyte organoid cultures. Am. J. Pathol. 2001, 159, 1877–1887. [Google Scholar] [CrossRef] [Green Version]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van De Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Aihara, E.; Watson, C.; Mourya, R.; Mizuochi, T.; Shivakumar, P.; Phelan, K.; Mayhew, C.; Helmrath, M.; Takebe, T.; et al. Paracrine signals regulate human liver organoid maturation from induced pluripotent stem cells. Development 2017, 144, 1056–1064. [Google Scholar] [CrossRef] [Green Version]
- Pettinato, G.; Lehoux, S.; Ramanathan, R.; Salem, M.M.; He, L.X.; Muse, O.; Flaumenhaft, R.; Thompson, M.T.; Rouse, E.A.; Cummings, R.D.; et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with Endothelial Cells. Sci. Rep. 2019, 9, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, X.; Tan, Z.; Su, Y.; Liu, J.; Chang, M.; Yan, F.; Chen, J.; Chen, T.; Li, C.; et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019, 29, 1009–1026. [Google Scholar] [CrossRef]
- Guan, Y.; Xu, D.; Garfin, P.M.; Ehmer, U.; Hurwitz, M.; Enns, G.; Michie, S.; Wu, M.; Zheng, M.; Nishimura, T.; et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; Van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; Van Wenum, M.; Fuchs, S.A.; De Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijk, E.W.; Rasmussen, S.; Blokzijl, F.; Huch, M.; Gehart, H.; Toonen, P.; Begthel, H.; Clevers, H.; Geurts, A.M.; Cuppen, E. Generation and characterization of rat liver stem cell lines and their engraftment in a rat model of liver failure. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kruitwagen, H.S.; Oosterhoff, L.A.; Vernooij, I.G.W.H.; Schrall, I.M.; van Wolferen, M.E.; Bannink, F.; Roesch, C.; van Uden, L.; Molenaar, M.R.; Helms, J.B.; et al. Long-Term Adult Feline Liver Organoid Cultures for Disease Modeling of Hepatic Steatosis. Stem Cell Rep. 2017, 8, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Nantasanti, S.; Spee, B.; Kruitwagen, H.S.; Chen, C.; Geijsen, N.; Oosterhoff, L.A.; Van Wolferen, M.E.; Pelaez, N.; Fieten, H.; Wubbolts, R.W.; et al. Disease modeling and gene therapy of copper storage disease in canine hepatic organoids. Stem Cell Rep. 2015, 5, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 2016, 16, 115–130. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Lanza, R. Current status of pluripotent stem cells: moving the first therapies to the clinic. Nat. Rev. Drug Discov. 2015, 14, 681–692. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchová, Z.; Benvenisty, N.; Kerem, B. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Iansante, V.; Mitry, R.R.; Filippi, C.; Fitzpatrick, E.; Dhawan, A. Human hepatocyte transplantation for liver disease: current status and future perspectives. Pediatr. Res. 2018, 83, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matas, A.J.; Sutherland, D.E.R.; Steffes, M.W.; Michael Mauer, S.; Lowe, A.; Simmons, R.L.; Najarian, J.S. Hepatocellular transplantation for metabolic deficiencies: Decrease of plasma bilirubin in Gunn rats. Science 1976, 192, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Mito, M.; Kusano, M. Hepatocyte Transplantation in Man. Cell Transplant. 1993, 2, 65–74. [Google Scholar] [CrossRef]
- Carpentier, A.; Tesfaye, A.; Chu, V.; Nimgaonkar, I.; Zhang, F.; Lee, S.B.; Thorgeirsson, S.S.; Feinstone, S.M.; Liang, T.J. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J. Clin. Investig. 2014, 124, 4953–4964. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, Y.; Wang, X.; Zhang, W.; Sauer, V.; Chang, C.J.; Han, B.; Tchaikovskaya, T.; Avsar, Y.; Tafaleng, E.; et al. Amelioration of Hyperbilirubinemia in Gunn Rats after Transplantation of Human Induced Pluripotent Stem Cell-Derived Hepatocytes. Stem Cell Rep. 2015, 5, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, L.; Caron, J.; Hannoun, Z.; Antoni, M.; López, S.; Burks, D.; Castell, J.V.; Weber, A.; Gomez-Lechon, M.J.; Dubart-Kupperschmitt, A. Transplantation of hESC-derived hepatocytes protects mice from liver injury. Stem Cell Res. Ther. 2015, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Nagamoto, Y.; Takayama, K.; Ohashi, K.; Okamoto, R.; Sakurai, F.; Tachibana, M.; Kawabata, K.; Mizuguchi, H. Transplantation of a human iPSC-derived hepatocyte sheet increases survival in mice with acute liver failure. J. Hepatol. 2016, 64, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, H.; Luu, N.T.; Alwahsh, S.M.; Ginai, M.; Alhaque, S.; Dong, H.; Tomaz, R.A.; Vernay, B.; Vigneswara, V.; Hallett, J.M.; et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch. Toxicol. 2018, 92, 3117–3129. [Google Scholar] [CrossRef] [Green Version]
- Lorvellec, M.; Scottoni, F.; Crowley, C.; Fiadeiro, R.; Maghsoudlou, P.; Pellegata, A.F.; Mazzacuva, F.; Gjinovci, A.; Lyne, A.M.; Zulini, J.; et al. Mouse decellularised liver scaffold improves human embryonic and induced pluripotent stem cells differentiation into hepatocyte-like cells. PLoS ONE 2017, 12, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Minami, T.; Ishii, T.; Yasuchika, K.; Fukumitsu, K.; Ogiso, S.; Miyauchi, Y.; Kojima, H.; Kawai, T.; Yamaoka, R.; Oshima, Y.; et al. Novel hybrid three-dimensional artificial liver using human induced pluripotent stem cells and a rat decellularized liver scaffold. Regen. Ther. 2019, 10, 127–133. [Google Scholar] [CrossRef]
- Blackford, S.J.I.; Ng, S.S.; Segal, J.M.; King, A.J.F.; Austin, A.L.; Kent, D.; Moore, J.; Sheldon, M.; Ilic, D.; Dhawan, A.; et al. Validation of Current Good Manufacturing Practice Compliant Human Pluripotent Stem Cell-Derived Hepatocytes for Cell-Based Therapy. Stem Cells Transl. Med. 2019, 8, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, K.; Akita, N.; Mimura, N.; Akahira, R.; Taniguchi, Y.; Ikeda, M.; Sakurai, F.; Ohara, O.; Morio, T.; Sekiguchi, K.; et al. Generation of safe and therapeutically effective human induced pluripotent stem cell-derived hepatocyte-like cells for regenerative medicine. Hepatol. Commun. 2017, 1, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.Z.; Zheng, Y.W.; Ogawa, M.; Miyagi, E.; Taniguchi, H. Human liver organoids generated with single donor-derived multiple cells rescue mice from acute liver failure. Stem Cell Res. Ther. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 1. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef]
- Nie, Y.Z.; Zheng, Y.W.; Miyakawa, K.; Murata, S.; Zhang, R.R.; Sekine, K.; Ueno, Y.; Takebe, T.; Wakita, T.; Ryo, A.; et al. Recapitulation of hepatitis B virus–host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine 2018, 35, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Brenner, D.A. The Crosstalk between Hepatocytes, Hepatic Macrophages, and Hepatic Stellate Cells Facilitates Alcoholic Liver Disease. Cell Metab. 2019, 30, 850–852. [Google Scholar] [CrossRef]
- Choi, W.M.; Kim, H.H.; Kim, M.H.; Cinar, R.; Yi, H.S.; Eun, H.S.; Kim, S.H.; Choi, Y.J.; Lee, Y.S.; Kim, S.Y.; et al. Glutamate Signaling in Hepatic Stellate Cells Drives Alcoholic Steatosis. Cell Metab. 2019, 30, 877–889.e7. [Google Scholar] [CrossRef]
- Miranda, C.C.; Fernandes, T.G.; Pinto, S.N.; Prieto, M.; Diogo, M.M.; Cabral, J.M.S. A scale out approach towards neural induction of human induced pluripotent stem cells for neurodevelopmental toxicity studies. Toxicol. Lett. 2018, 294, 51–60. [Google Scholar] [CrossRef]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells from Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, S.; Tasoglu, S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends Biotechnol. 2016, 34, 681–682. [Google Scholar] [CrossRef]
- Siramshetty, V.B.; Nickel, J.; Omieczynski, C.; Gohlke, B.O.; Drwal, M.N.; Preissner, R. WITHDRAWN—A resource for withdrawn and discontinued drugs. Nucleic Acids Res. 2016, 44, D1080–D1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, A.; Nimgaonkar, I.; Chu, V.; Xia, Y.; Hu, Z.; Liang, T.J. Hepatic differentiation of human pluripotent stem cells in miniaturized format suitable for high-throughput screen. Stem Cell Res. 2016, 16, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.M.; Kim, Y.; Shim, J.S.; Park, J.T.; Wang, R.H.; Leach, S.D.; Liu, J.O.; Deng, C.; Ye, Z.; Jang, Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013, 57, 2458–2468. [Google Scholar] [CrossRef] [Green Version]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489.e5. [Google Scholar] [CrossRef] [Green Version]
- Jing, R.; Corbett, J.L.; Cai, J.; Beeson, G.C.; Beeson, C.C.; Chan, S.S.; Dimmock, D.P.; Lazcares, L.; Geurts, A.M.; Lemasters, J.J.; et al. A Screen Using iPSC-Derived Hepatocytes Reveals NAD+ as a Potential Treatment for mtDNA Depletion Syndrome. Cell Rep. 2018, 25, 1469–1484.e5. [Google Scholar] [CrossRef] [Green Version]
- Medine, C.N.; Lucendo-Villarin, B.; Storck, C.; Wang, F.; Szkolnicka, D.; Khan, F.; Pernagallo, S.; Black, J.R.; Marriage, H.M.; Ross, J.A.; et al. Developing high-fidelity hepatotoxicity models from pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 505–509. [Google Scholar] [CrossRef]
- Sirenko, O.; Hancock, M.K.; Hesley, J.; Hong, D.; Cohen, A.; Gentry, J.; Carlson, C.B.; Mann, D.A. Phenotypic characterization of toxic compound effects on liver spheroids derived from ipsc using confocal imaging and three-dimensional image analysis. Assay Drug Dev. Technol. 2016, 14, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Ware, B.R.; Berger, D.R.; Khetani, S.R. Prediction of drug-induced liver injury in micropatterned co-cultures containing iPSC-derived human hepatocytes. Toxicol. Sci. 2015, 145, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson-Bouchard, K.; Vunjak-Novakovic, G. Organs-on-a-Chip: A Fast Track for Engineered Human Tissues in Drug Development. Cell Stem Cell 2018, 22, 310–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Miranda, C.C.; Fernandes, T.G.; Diogo, M.M.; Cabral, J.M.S. Towards multi-organoid systems for drug screening applications. Bioengineering 2018, 5, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.L.K.; Edington, C.; Suter, E.; Yu, J.; Velazquez, J.J.; Velazquez, J.G.; Shockley, M.; Large, E.M.; Venkataramanan, R.; Hughes, D.J.; et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng. 2017, 114, 2648–2659. [Google Scholar] [CrossRef] [Green Version]
- Edington, C.D.; Chen, W.L.K.; Geishecker, E.; Kassis, T.; Soenksen, L.R.; Bhushan, B.M.; Freake, D.; Kirschner, J.; Maass, C.; Tsamandouras, N.; et al. Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Study | Media | Molecules | Ref. |
|---|---|---|---|
| Hepatocytes | |||
| Rambhatla et al., 2003 | KO-DMEM+FBS | NaB/DMSO – NaB/HGF | [42] |
| Kubo et al., 2004 | StemPro34 – IMDM+SR | Act A – DEX | [44] |
| Hay et al., 2008 | RPMI+B27 – DMEM+SR+DMSO – L15 | Act A/Wnt3a – HGF/OSM | [71] |
| Song et al., 2009 | RPMI – HCM – N2B27 | Act A – FGF4/BMP2 – HGF/KGF – OSM/DEX | [43] |
| Si-Tayeb et al., 2010 | RPMI+B27 – HCM | Act A – BMP4/FGF2 – HGF – OSM | [72] |
| Sullivan et al., 2010 | RPMI+B27 – DMEM+SR+DMSO – L15 | Act A/Wnt3a – Act A – HGF/OSM | [73] |
| Touboul et al., 2010 | CDM | Act A/Ly/BMP4/FGF2 – FGF10 – FGF10/RA/SB – FGF4/HGF/EGF | [74] |
| Kajiwara et al., 2012 | RPMI+B27 – DMEM+SR+DMSO – HCM | Act A/Wnt3a/NaB – Act A/Wnt3a – HGF/OSM | [75] |
| Siller et al., 2015 | RPMI+B27 – DMEM+SR+DMSO – L15 | CHIR – Dihexa/DEX | [48] |
| Ang et al., 2018 | CDM | Act A/CHIR/PI – Act A/LDN – A83/BMP4/FGF2/ATRA – Act A/CHIR/BMP4/Forskolin – BMP4/OSM/DEX/Forsk/Ro/AA/Insulin – DEX/Forskolin/Ro/AA/Insulin | [40] |
| Cholangiocytes | |||
| Dianat et al., 2014 | RPMI | Act A/Ly – Act A/FGF2/BMP4 – FGF4/HGF/EGF/RA – EGF/GH/IL6 | [54] |
| De Assuncao et al., 2015 | RPMI – H69 | Act A/Wnt3a – FGF2/BMP4/SHH – SHH/JAG1 – TGFβ | [57] |
| Ogawa et al., 2015 | RPMI – H16 – H16/Ham’s F12 – H21/Ham’s F12 | Act A/CHIR – FGF2/BMP4 – HGF/OSM/DEX – HGF/EGF/TGFβ/OP9 | [55] |
| Sampaziotis et al., 2015 | CDM – RPMI – William’s E | Act A/Ly/FGF2/BMP4 – Act A – BMP4/SB – Act A/FGF10/RA | [56] |
| Liver Sinusoidal Endothelial Cells | |||
| Koui et al., 2017 | StemPro34 SFM – EGM2 | BMP4 – BMP4/Act A/FGF2 – VEGF/SB/Dorsomorphin – VEGF – A83 | [60] |
| Kupffer Cells | |||
| Tasnim et al., 2019 | mTeSR1 – X-VIVO – PHCM/Adv DMEM | BMP4/VEGF/SCF – MCSF/IL3 – MCSF | [65] |
| Hepatic Stellate Cells | |||
| Koui et al., 2017 | StemPro34 SFM – MSCGM | BMP4 – BMP4/Act A/FGF2 – VEGF/SB/Dorso – ROCKi | [60] |
| Coll et al., 2018 | MCDB 201 | BMP4 – BMP4/FGF1/FGF3 – FGF1/FGF3/PA/Retinol | [70] |
| Study | Route | Cells | Nr of Cells | % Repopulation | Ref. |
|---|---|---|---|---|---|
| Carpentieret al., 2014 | Intrasplenic injection | hPSC-hepatocytes | 4 × 106 | <1–20% | [105] |
| Chen et al., 2015 | Intrasplenic injection | hPSC-hepatocytes | 2 × 106 | 2.5–7.5% | [106] |
| Tolosa et al., 2015 | Intrasplenic injection | hPSC-hepatocytes | 1 × 106 | 10% | [107] |
| Nagamoto et al., 2016 | Sheet transplantation | hPSC-hepatocyte sheet | 8 × 105 | - | [108] |
| Takayama et al., 2017 | Intraperitoneal transplantation | hPSC-hepatocytes | 1 × 106 | - | [113] |
| Nie et al., 2018 | Renal subcapsular space | hPSC-hepatocyte aggregates | 1 × 106 | - | [114] |
| Rashidi et al., 2018 | Intraperitoneal transplantation | hPSC-hepatocyte aggregates | 2 × 106 (aggregates) | - | [109] |
| Subcutaneous transplantation | hPSC-hepatocytes in PCL fibers | - | - | [109] | |
| Blackford et al., 2019 | Intraperitoneal transplantation | hPSC-hepatocyte aggregates | 2 × 103 (aggregates) | - | [112] |
| Study | Disease | Gene/Toxin | Approach | Ref. |
|---|---|---|---|---|
| Genetic Liver Diseases | ||||
| Guan et al., 2017 | Alagille syndrome | JAG1 | Patient-derived/gene editing | [90] |
| Tetralogy of Fallot | JAG1 | Patient-derived | [90] | |
| Ouchi et al., 2019 | Wolman disease | Free fatty acids | Patient-derived/induced | [92] |
| Acquired Liver Diseases | ||||
| Nie et al., 2018 | Hepatitis B | Hepatitis B virus | Induced | [118] |
| Wang et al., 2019 | Alcoholic liver disease | EtOH | Induced | [89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotovio, J.P.; Fernandes, T.G. Production of Human Pluripotent Stem Cell-Derived Hepatic Cell Lineages and Liver Organoids: Current Status and Potential Applications. Bioengineering 2020, 7, 36. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020036
Cotovio JP, Fernandes TG. Production of Human Pluripotent Stem Cell-Derived Hepatic Cell Lineages and Liver Organoids: Current Status and Potential Applications. Bioengineering. 2020; 7(2):36. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020036
Chicago/Turabian StyleCotovio, João P., and Tiago G. Fernandes. 2020. "Production of Human Pluripotent Stem Cell-Derived Hepatic Cell Lineages and Liver Organoids: Current Status and Potential Applications" Bioengineering 7, no. 2: 36. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7020036