
Accessing Dietary Effects on the Rumen Microbiome: Different Sequencing Methods Tell Different Stories
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Protocol
2.2. Genomic DNA Isolation from Rumen Content
2.3. Amplicon Library Preparation
2.4. Metagenomic Library Preparation
2.5. Bioinformatic Analysis
2.6. Statistical Analyses
3. Results and Discussion
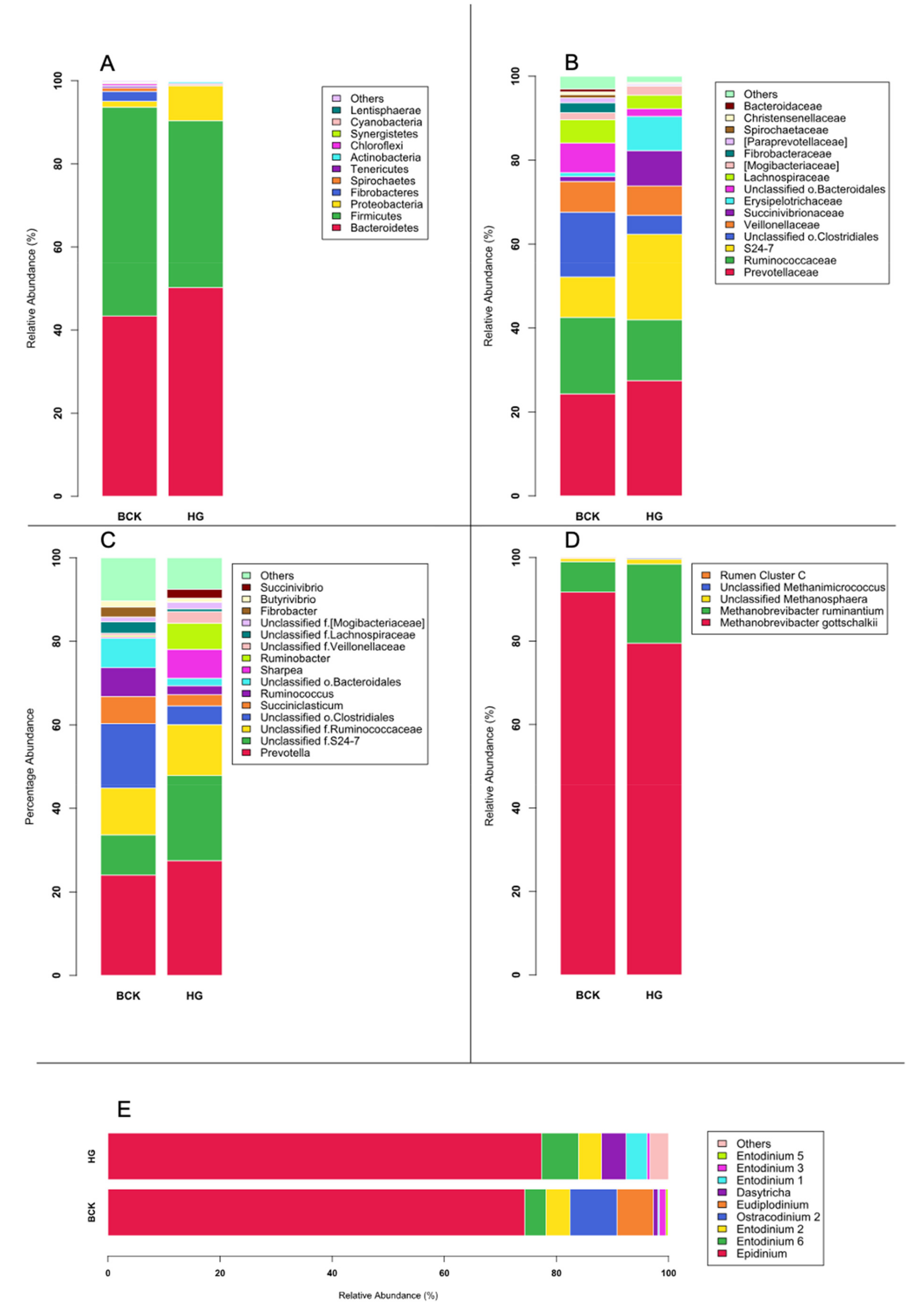
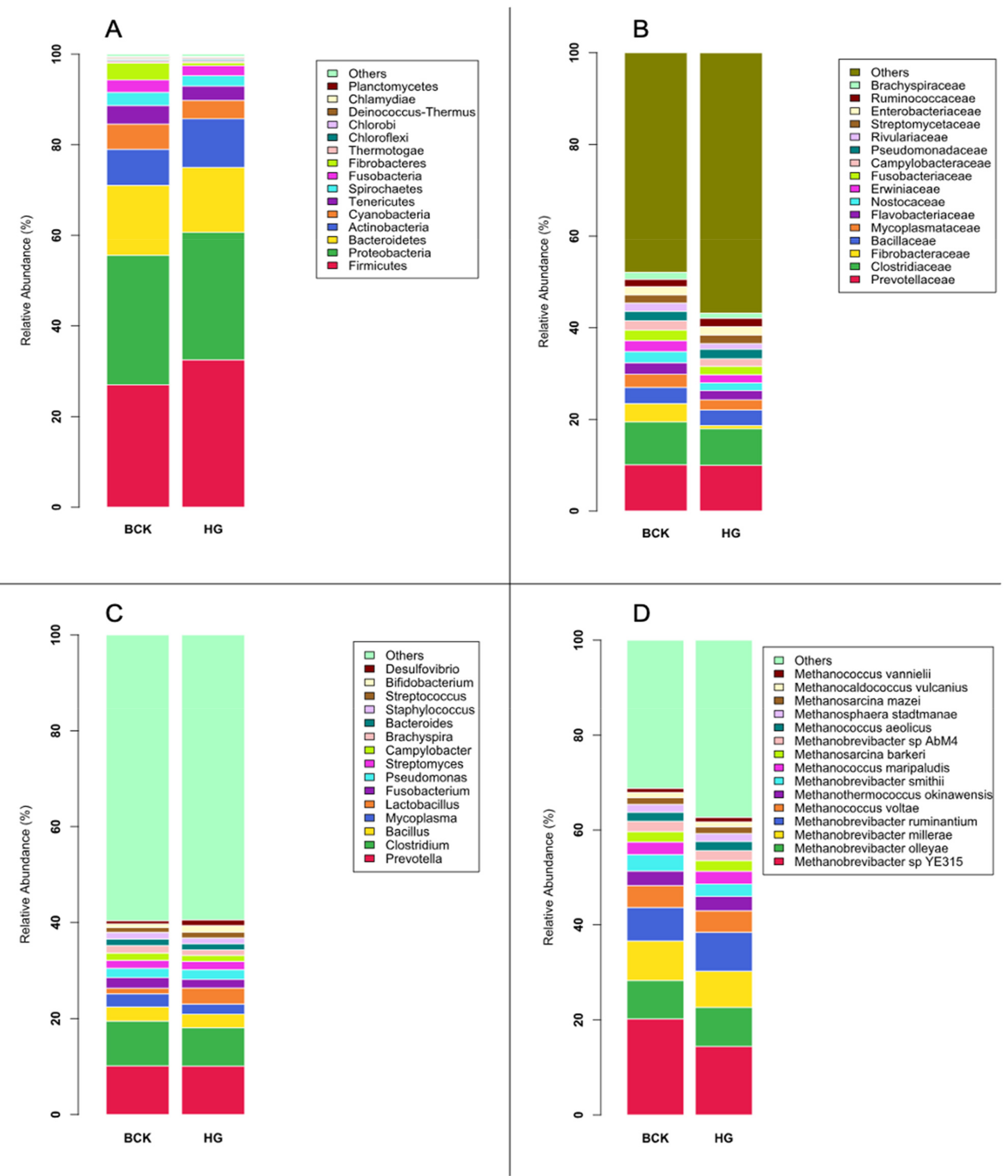
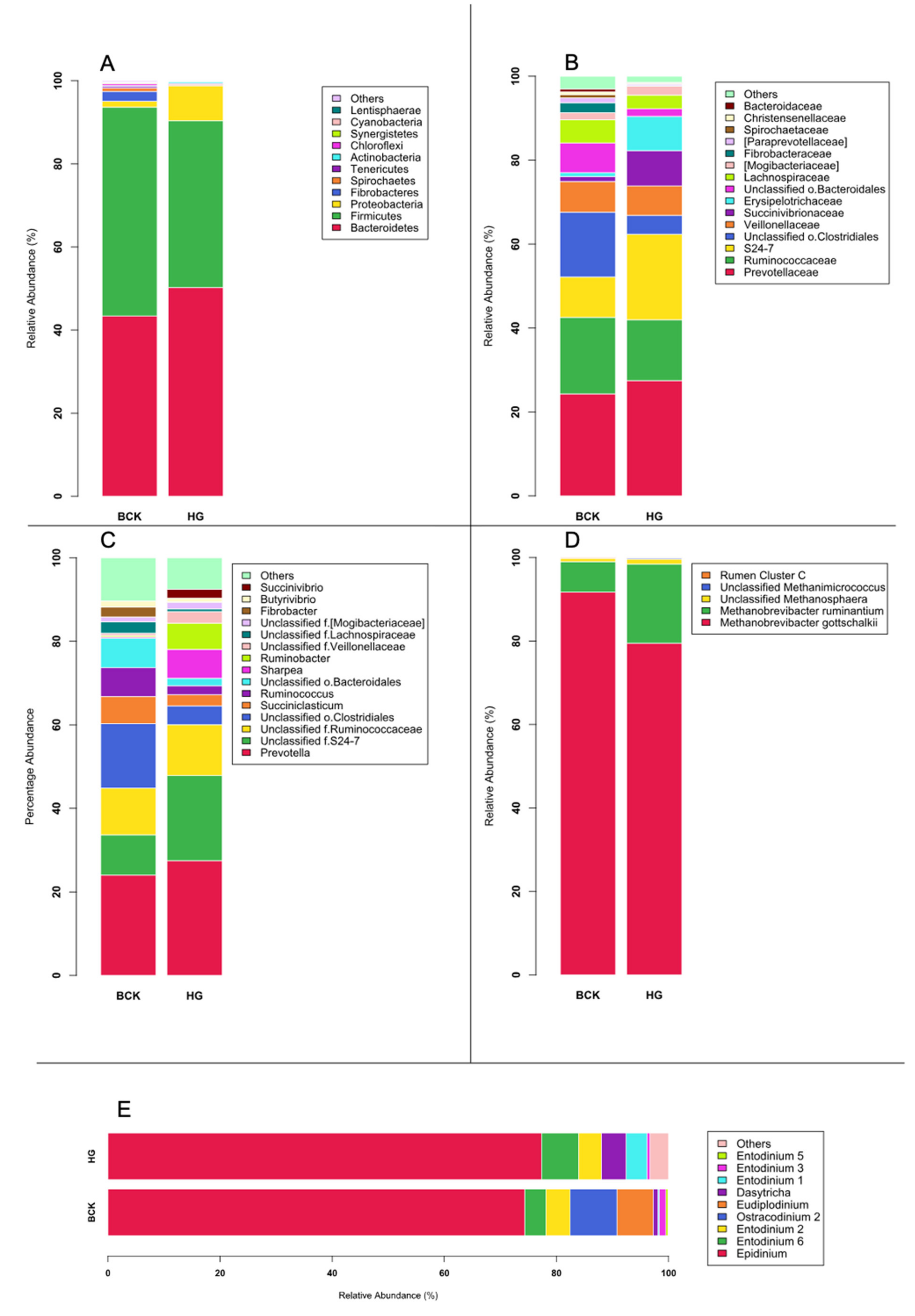
3.1. Microbial Profiles Generated Using Metagenomic Shotgun Sequencing
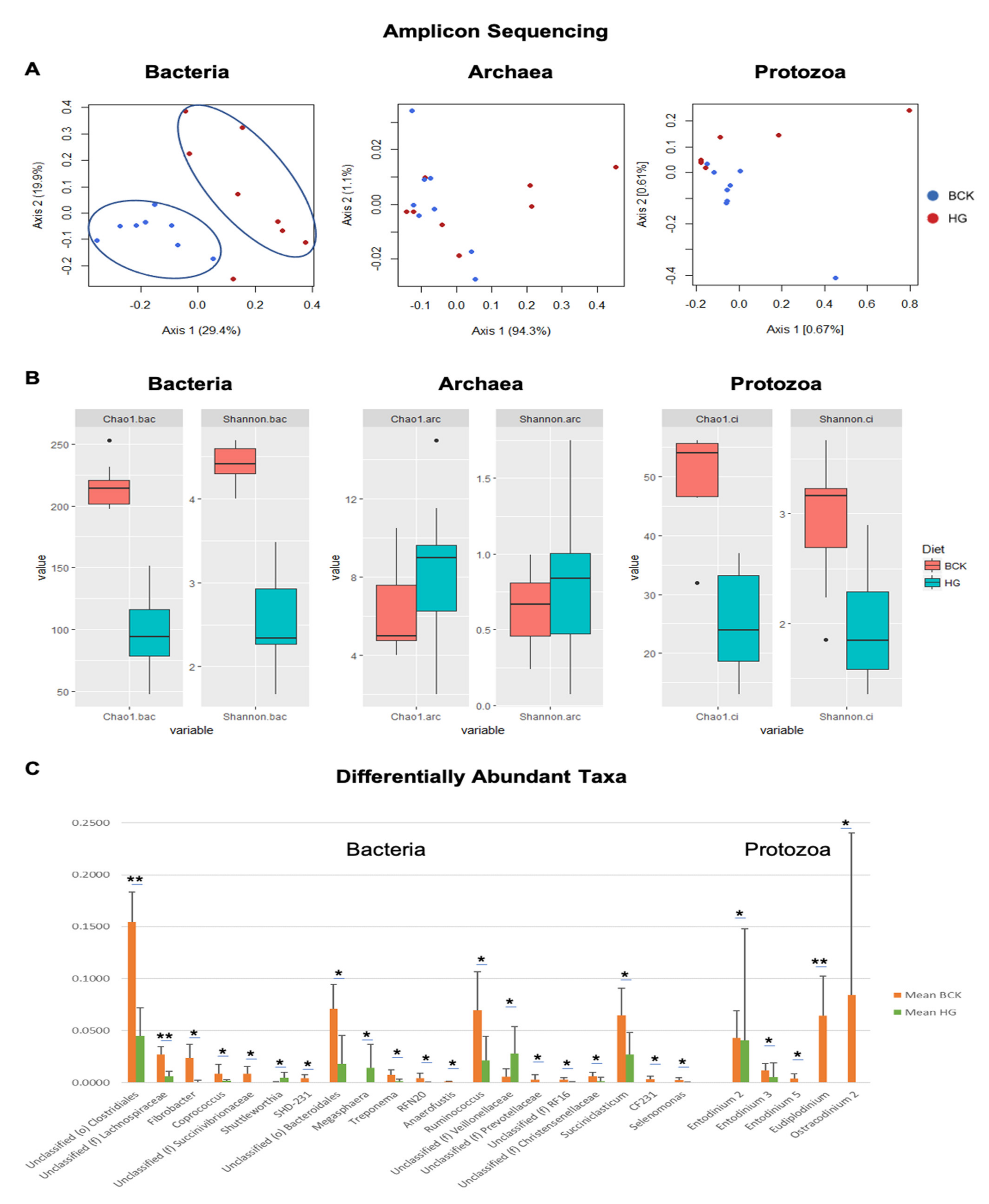
3.2. Microbial Profiles Generated Using Amplicon Sequencing
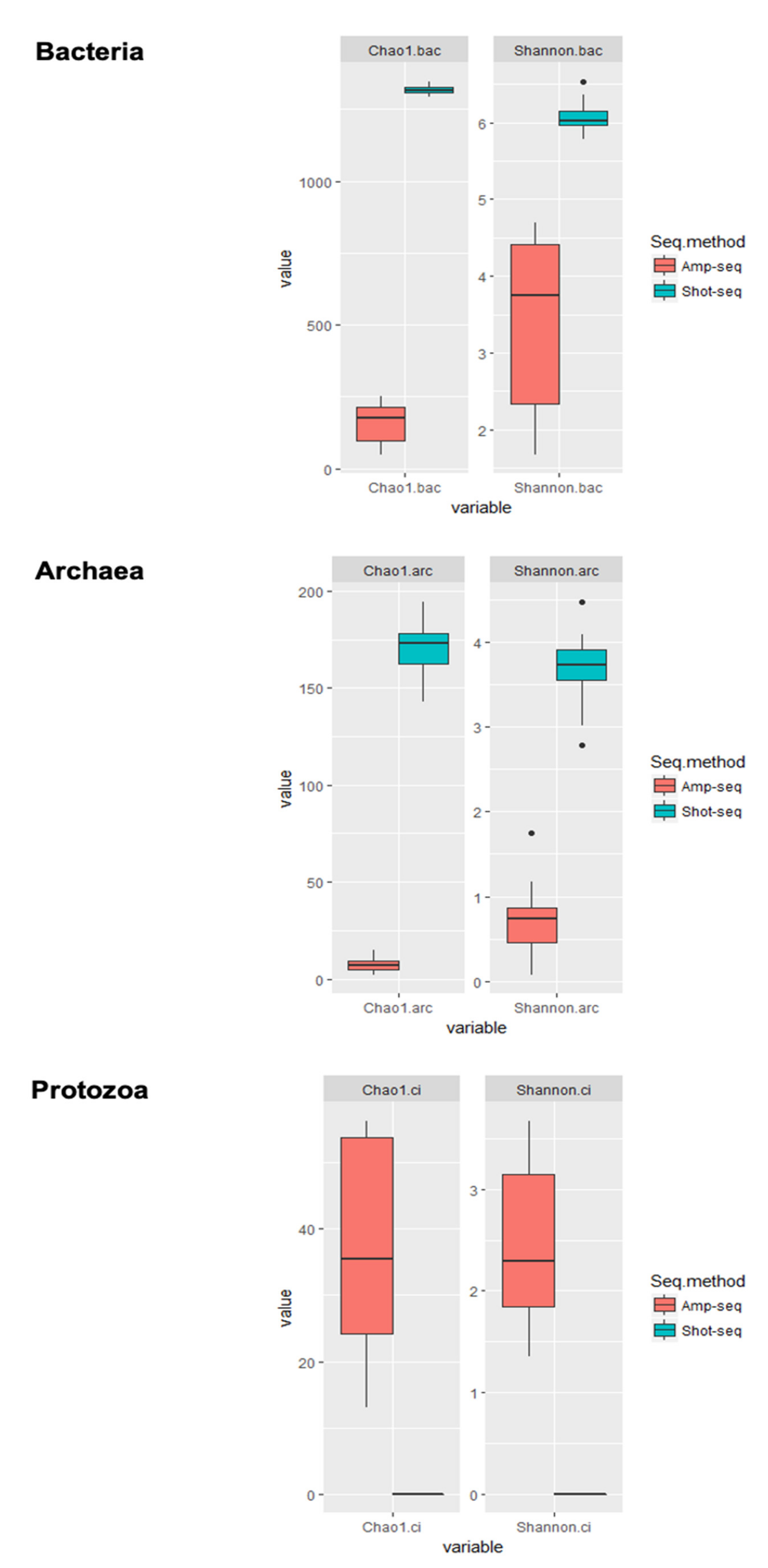
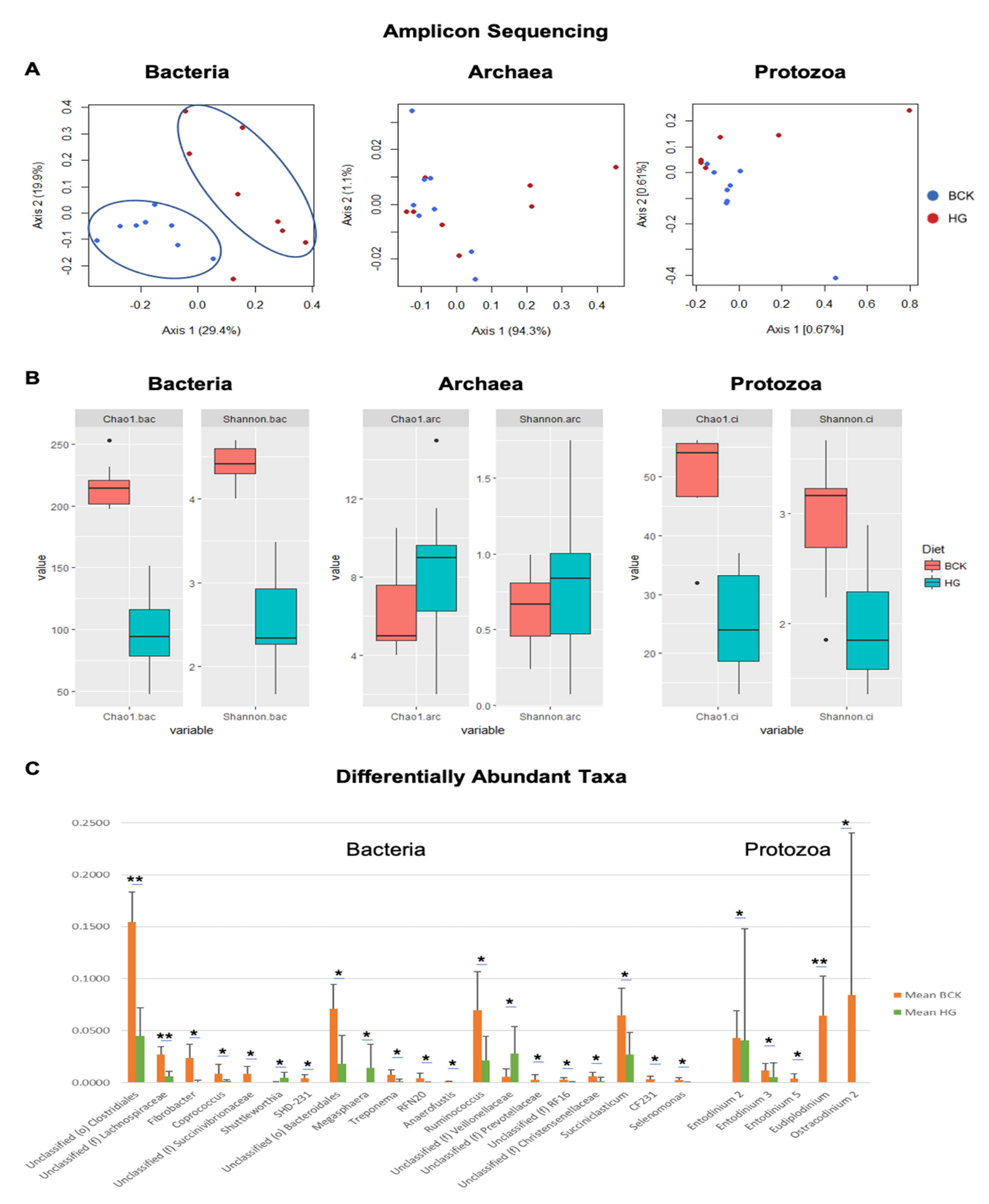
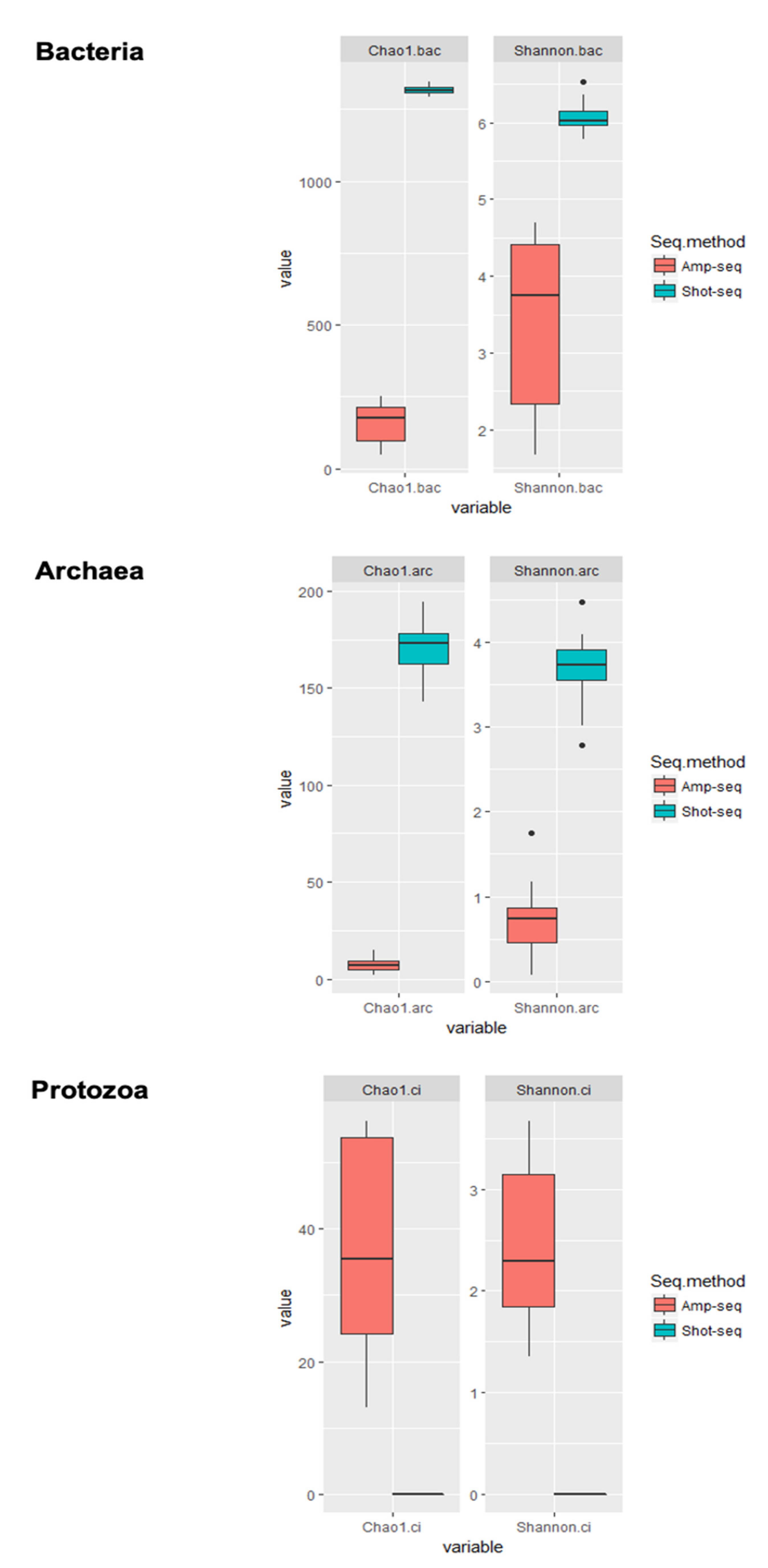
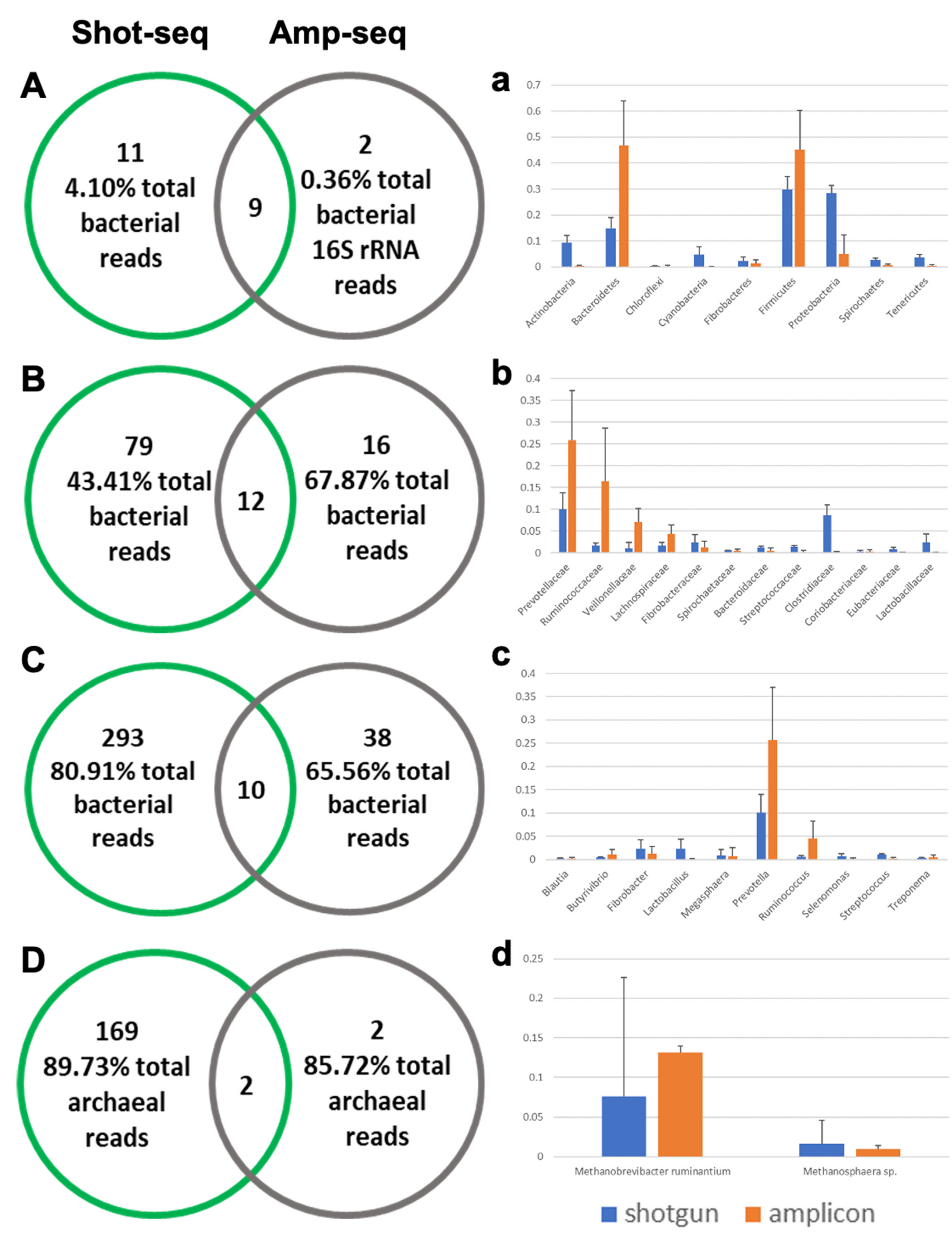
3.3. Shotgun-Seq and Amplicon-Seq Revealed Different Rumen Microbial Communities in Bull Cattle
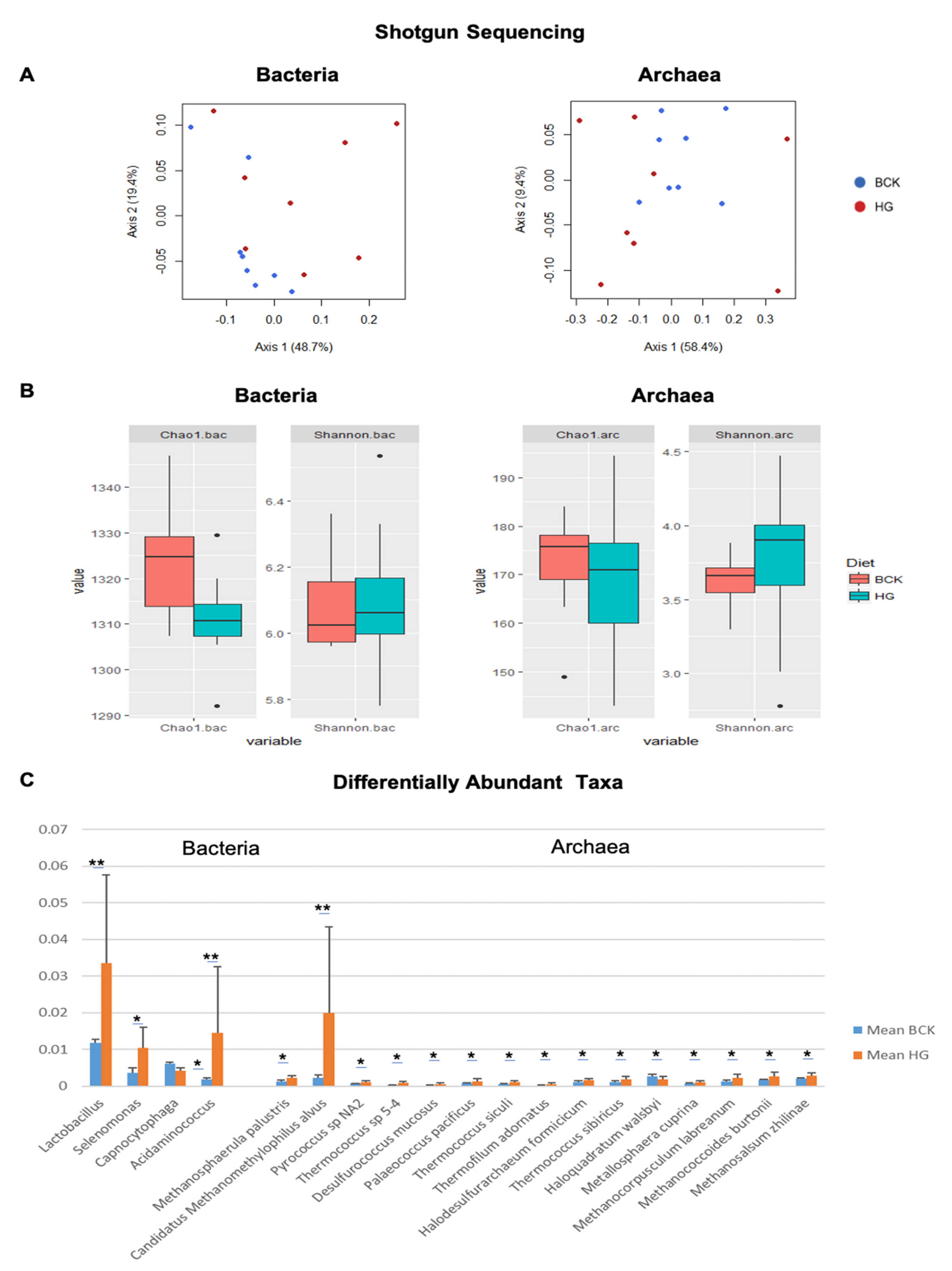
3.4. Dietary Effect on Rumen Microbiota Revealed by Shotgun-Seq and Amplicon-Seq
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guan, L.L.; Nkrumah, J.D.; Basarab, J.A.; Moore, S.S. Linkage of microbial ecology to phenotype: Correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol. Lett. 2008, 288, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Guan, L.L. Metatranscriptomic Profiling Reveals Linkages between the Active Rumen Microbiome and Feed Efficiency in Beef Cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, E.; Kenny, D.A.; McCabe, M.S.; Fitzsimons, C.; McGee, M.; Kelly, A.K.; Waters, S.M. 16S rRNA Sequencing Reveals Relationship Between Potent Cellulolytic Genera and Feed Efficiency in the Rumen of Bulls. Front. Microbiol. 2018, 9, 1842. [Google Scholar] [CrossRef]
- Jami, E.; Whit, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Collaborators, G.R.C.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Report 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Svartstrom, O.; Alneberg, J.; Terrapon, N.; Lombard, V.; de Bruijn, I.; Malmsten, J.; Dalin, A.-M.; Muller, E.E.L.; Shah, P.; Wilmes, P.; et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 2017, 11, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.T.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Myer, P.R.; Kim, M.; Freetly, H.C.; Smith, R.P. Evaluation of 16S rRNA amplicon sequencing using two next-generation sequencing technologies for phylogenetic analysis of the rumen bacterial community in steers. J. Microbiol. Methods 2016, 127, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehe, R.; DewHurst, R.J.; Cuthie, C.-A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M. Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.-W.; Chokhawala, H.; Schroth, G.; Luo, S.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic Discovery of Biomass-Degrading Genes and Genomes from Cow Rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies. Appl. Environ. Microbiol. 2009, 75, 6524–6533. [Google Scholar] [CrossRef] [Green Version]
- Tymensen, L.; Barkley, C.; McAllister, T.A. Relative diversity and community structure analysis of rumen protozoa according to T-RFLP and microscopic methods. J. Microbiol. Methods 2012, 88, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Marume, U.; Kusina, N.T.; Hamudikuwanda, H.; Ndengu, M.; Nyoni, O. Effect of dry season nutritional supplementation on fertility in bulls in Sanyati smallholder farming area, Zimbabwe. Afr. J. Agri. Res. 2011, 9, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap short read aligner, and other bioinformatic tools. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 10 January 2017).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar]
- Clark, K.R. Non-parametric multivariate analysis of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Scharen, M.; Drong, C.; Kiri, K.; Riede, S.; Gardener, M.; Meyer, U.; Hummel, J.; Urich, T.; Breves, G.; Danicke, S. Differential effects of monensin and a blend of essential oils on rumen microbiota composition of transition dairy cows. J. Dairy Sci. 2017, 100, 2765–2783. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, E.; Kelly, A.; McCabe, M.S.; Kenny, D.A.; Guan, L.L.; Waters, S.M. Effect of a butyrate-fortified milk replacer on gastrointestinal microbiota and products of fermentation in artificially reared dairy calves at weaning. Sci. Rep. 2018, 8, 14901. [Google Scholar] [CrossRef]
- Soo, R.M.; Skennerton, C.T.; Sekiguchi, Y.; Imelfort, M.; Paech, S.J.; Dennis, P.G.; Steen, J.A.; Parks, D.H.; Tyson, G.W.; Hugenholtz, P. An Expanded Genomic Representation of the Phylum Cyanobacteria. Genome Biol. Evol. 2014, 6, 1031–1045. [Google Scholar] [CrossRef] [Green Version]
- Utami, Y.D.; Kuwahara, H.; Murakami, T.; Morikawa, T.; Sugaya, K.; Kihara, K.; Yuki, M.; Lo, N.; Deevong, P.; Hasin, S.; et al. Phylogenetic Diversity and Single-Cell Genome Analysis of “Melainabacteria”, a Non-Photosynthetic Cyanobacterial Group, in the Termite Gut. Microbes Environ. 2018, 33, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Brulc, J.M.; Antonopoulos, D.A.; Miller, M.E.B.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J.; Rooke, J.A.; McKain, N.; Duthie, C.-A.; Hyslop, J.J.; Ross, D.W.; Waterhouse, A.; Watson, M.; Roehe, R. The rumen microbial metagenome associated with high methane production in cattle. BMC Genom. 2015, 16, 839. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Wei, H.; et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaliannan, K.; Robertson, R.C.; Murphy, K.; Stanton, C.; Kang, C.; Wang, B.; Hao, L.; Bhan, A.K.; Kang, J.X. Estrogen-mediated gut microbiome alterations influence sexual dimorphism in metabolic syndrome in mice. Microbiome 2018, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.F.; Muthusamy, A.; Al-Ghalith, G.A.; Knights, D.; Guo, B.; Wu, B.; Remmel, R.P.; Schladt, D.P.; Alegre, M.-L.; Oetting, W.S.; et al. Urinary microbiome associated with chronic allograft dysfunction in kidney transplant recipients. Clin. Transplant. 2018, 32, e13436. [Google Scholar] [CrossRef] [PubMed]
- Auffret, M.D.; Stewart, R.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; Wallace, R.J.; Freeman, T.C.; Snelling, T.J.; Watson, M.; Roeche, R. Identification, comparison, and validation of robust rumen microbial biomarkers for methane emissions using diverse Bos taurus breeds and basal diets. Front. Microbiol. 2017, 8, 2642. [Google Scholar] [CrossRef] [Green Version]
- Danman, S.E.; Fernandea, G.M.; Shinkai, T.; Mitsumori, M.; McSweeney, C.S. Metagenomic analysis of the rumen microbial community following inhibition of methane formation by a halogenated methane analog. Front. Microbiol. 2015, 6, 1087. [Google Scholar] [CrossRef]
- Popova, M.; McGovern, E.; McCabe, M.S.; Martin, C.; Doreau, M.; Arbre, M.; Meale, S.; Morgavi, D.; Waters, S.M. The Structural and Functional Capacity of Ruminal and Cecal Microbiota in Growing Cattle Was Unaffected by Dietary Supplementation of Linseed Oil and Nitrate. Front. Microbiol. 2017, 8, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.; Ouwerkerk, D.; Klieve, A. Archaeaphage Therapy to Control Rumen Methanogens. Meat and Livestock Australia Final Project Report (B.CCH.1007). 2011. Available online: http://www.mla.com.au/download/finalreports?itemId=1593 (accessed on 12 May 2018).
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Characterization of variation in rumen methanogenic communities under different dietary and host feed efficiency conditions, as determined by PCR-denaturing gradient gel electrophoresis analysis. Appl. Environ. Microbiol. 2010, 76, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundset, M.A.; Edwards, J.E.; Cheng, Y.F.; Senosiain, R.S.; Fraile, M.N.; Northwood, K.; Praesteng, K.E.; Glad, T.; Mathiesen, S.D.; Wright, A.-D.G. Rumen microbial diversity in Svalbard reindeer, with particular emphasis on methanogenic archaea. FEMS Microbiol. Ecol. 2009, 70, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.W.; Zhang, Q.; Yang, Y.; Li, L.; Zou, C.; Huang, C.; Lin, B. Comparative study of rumen fermentation and microbial community differences between water buffalo and Jersey cows under similar feeding conditions. J. Appl. Anim. Res. 2018, 46, 740–748. [Google Scholar] [CrossRef]
- Rausch, P.; Rühlemann, M.; Hermes, B.M.; Doms, S.; Dagan, T.; Dierking, K.; Domin, H.; Fraune, S.; Von Frieling, J.; Hentschel, U.; et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019, 7, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hunerberg, M.; Chen, Y.; Reuter, T.; McAllister, T.A.; Evans, F.; Critchley, A.T.; Guan, L.L. Air-dried brown seaweed, Ascophyllum nodosum, alters rumen microbiome in a manner that changes rumen fermentation profiles and lowers the prevalence of foodborne pathgens. mSphere 2018, 3, e00017–e00018. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, R.; Dicksved, J.; Sun, L.; Gonda, H.; Müller, B.; Schnürer, A.; Bertilsson, J. Methane production in dairy cows correlates with rumen methanogenic and bacterial community structure. Front. Microbial. 2017, 8, 226. [Google Scholar] [CrossRef]
- Rolfe, J. Economics of reducing methane emissions from cattle production in central Queensland. In Project Final Report for Meat and Livestock Australia Lid; Meat and Livestock Australia Lid.: North Sydney, NSW, Australia, 2001. [Google Scholar]
- Kittelmann, S.; Devente, S.R.; Kirk, M.R.; Seedorf, H.; Dehority, B.A.; Janssen, P.H. Phylogeny of the intestinal ciliates including first sequences from Charonina ventriculi and comparison of microscopy and 18S rRNA gene pyrosequencing for rumen ciliate community structure analysis. Appl. Environ. Microbiol. 2015, 81, 2433–2444. [Google Scholar] [CrossRef] [Green Version]
- Ng, F.; Kittelmann, S.; Patchett, M.L.; Attwood, G.T.; Janssen, P.H.; Rakonjac, J.; Gagic, D. An adhesin from hydrogen-utilizing rumen methanogenMethanobrevibacter ruminantium M1 binds a broad range of hydrogen-producing microorganisms. Environ. Microbiol. 2016, 18, 3010–3021. [Google Scholar] [CrossRef]
- Ominski, K.H.; Boadi, D.A.; Wittenherg, K.M.; Fulawka, D.L.; Basarab, J.A. Estimates of enteric methane emissions from cattle in Canada using the IPCC Tier-2 methodology. Can. J. Anim. Sci. 2007, 87, 459–467. [Google Scholar] [CrossRef]
- Pope, P.B.; Mackenzie, A.K.; Gregor, I.; Smith, W.; Sundset, M.A.; McHardy, A.C.; Morrison, M.; Eijsink, V.G.H. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS ONE 2012, 7, e38571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamke, J.; Kittlemann, S.; Sino, P.; Li, Y.; Tavendale, M.; Ganesh, S.; Janssen, P.H.; Shi, W.; Froula, J.; Rubin, E.M.; et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterized by lactic acid formation and utilization. Microbiome 2016, 4, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Reddy, B.; Patel, A.; Panchasara, H.; Parmar, N.; Shah, T.; Bhatt, V.; Joshi, C.; Singh, K.; Reddy, B.; et al. Metagenomic analysis of buffalo rumen microbiome: Effect of roughage diet on Dormancy and Sporulation genes. Meta Gene 2014, 2, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.L.A.; Li, F.; Ghoshal, B.; McAllister, T.; Guan, L.L. Enhancing the resolution of rumen microbial classification from metatranscriptomic data using Kraken and Mothur. Front. Microbiol. 2017, 8, 2445. [Google Scholar] [CrossRef]
- Ibarbalz, F.M.; Perez, M.V.; Figuerola, E.L.M.; Erijman, L. The bias associated with amplicon sequencing does not affect the quantitative assessment of bacterial community dynamics. PLoS ONE. 2014, 9, e99722. [Google Scholar] [CrossRef] [Green Version]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [Green Version]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef]
- McCann, J.C.; Wiley, L.M.; Forbes, T.D.; Rouquette, F.M., Jr.; Tedeschi, L.O. Relationship between the rumen microbiome and residual feed intake-efficiency of Brahman bulls stocked on Bermudagrass pastures. PLoS ONE 2014, 9, e91864. [Google Scholar] [CrossRef] [Green Version]
- Thomas, F.A.; Webb, M.; Ghimire, S.; Blair, A.; Olson, K.; Fenske, G.J.; Fonder, A.; Christopher-Hennings, J.; Brake, D.; Scaria, J. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fernando, S.C.; Purvis, H.T., II; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitta, D.W.; Parmar, N.; Patel, A.K.; Indugu, N.; Kumar, S.; Prajapathi, K.B.; Patel, A.B.; Reddy, B.; Joshi, C. Bacterial Diversity Dynamics Associated with Different Diets and Different Primer Pairs in the Rumen of Kankrej Cattle. PLoS ONE 2014, 9, e111710. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, G.T. Microbial community composition along the digestive tract in forage- and grain-fed bison. BMC Vet. Res. 2017, 13, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messana, J.D.; Carvalho, A.L.E.G.F.; Ribeiro, A.F.; Fiorentini, G.; Castagnino, P.D.S.; Granja-Salcedo, Y.T.; Pires, A.V.; Berchielli, T. Effects of different sources of forage in high-concentrate diets on fermentation parameters, ruminal biohydrogenation and microbiota in Nellore feedlot steers. J. Agric. Sci. 2016, 154, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Piro, V.C.; Matschkowski, M.; Renard, B.Y. MetaMeta: Integrating metagenome analysis tools to improve taxonomic profiling. Microbiome 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom。 2015, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Piro, V.C.; Lindner, M.S.; and Renard, B.Y. DUDes: A top-down taxonomic profiler for metagenomics. Bioinformatics 2016, 32, 2272–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, T.A.K.; Li, P.-E.; Scholz, M.B.; Chain, P.S.G. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, S.; Mende, D.R.; Zeller, G.; Izquierdo-Carrasco, F.; Berger, S.A.; Kultima, J.R.; Coelho, L.P.; Arumugam, M.; Tap, J.; Nielsen, H.B.; et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 2013, 10, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, R.; Leahy, S.C.; Attwood, G.T.; Teh, K.H.; Lambie, S.C.; Cookson, A.L.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.; Hadjithomas, M.; Varghese, N.; et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 2018, 36, 359–367. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Backgrounding (BGK) Diets | High-Grain (HG) Diets | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BGK1 | BGK2 | BGK3 | HG1 | HG2 | HG3 | HG4 | HG5 | HG6 | HG7 | HG8 | |
| Inclusion rate, % dry matter (DM) | |||||||||||
| Barley Silage | 65 | 55 | 45 | 65 | 55 | 45 | 35 | 25 | 20 | 15 | 10 |
| Barley Grain | 25 | 35 | 45 | 25 | 35 | 45 | 55 | 65 | 70 | 75 | 80 |
| Pellet 1 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Days fed | 1 to 7 | 8 to 14 | 15 till slaughter | 1 to 4 | 5 to 8 | 9 to 12 | 13 to 16 | 17 to 20 | 21 to 24 | 24 to 27 | 28 till slaughter |
| Nutrient composition, DM basis | |||||||||||
| CP | 14.0 | 14.1 | 14.1 | 14.0 | 14.0 | 14.1 | 14.2 | 14.3 | 14.3 | 14.4 | 14.4 |
| NDF | 40.9 | 37.1 | 33.4 | 40.9 | 37.2 | 33.4 | 29.7 | 25.9 | 24.0 | 22.1 | 20.3 |
| Starch | 23.2 | 27.8 | 32.5 | 22.9 | 27.6 | 32.3 | 36.9 | 41.6 | 43.9 | 46.3 | 48.6 |
| Ca | 0.8 | 0.7 | 0.7 | 0.8 | 0.8 | 0.8 | 0.7 | 0.7 | 0.7 | 0.7 | 0.6 |
| P | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 |
| Item 1 | Backgrounding | Finishing |
|---|---|---|
| Initial BW, kg | 422 | 419 |
| Start of test BW, kg | 454 | 453 |
| Ending BW, kg | 536 | 558 |
| DMI, kg/d | 13.4 | 13.2 |
| Microbe | Sample | Diet | Assembled Contigs | Kraken Aligned Reads | Chao1 | Shannon |
|---|---|---|---|---|---|---|
| Archaea | 105 | HG | 164,912 | 4587 | 171.50 | 3.97 |
| 107 | HG | 281,557 | 9368 | 194.50 | 3.84 | |
| 131 | HG | 332,247 | 7625 | 170.32 | 4.08 | |
| 147 | HG | 86,704 | 4188 | 142.97 | 3.01 | |
| 155 | HG | 114,590 | 1965 | 174.89 | 4.47 | |
| 217 | BCK | 254,337 | 8301 | 175.12 | 3.59 | |
| 227 | HG | 325,493 | 13,375 | 159.53 | 2.78 | |
| 23 | HG | 251,609 | 5124 | 181.21 | 3.98 | |
| 273 | BCK | 355,723 | 8500 | 170.97 | 3.88 | |
| 297 | HG | 341,391 | 9367 | 160.18 | 3.79 | |
| 343 | BCK | 448,664 | 13,913 | 177.81 | 3.70 | |
| 381Y | BCK | 451,291 | 11,885 | 163.34 | 3.70 | |
| 61 | BCK | 350,872 | 14,375 | 148.97 | 3.62 | |
| 81 | BCK | 214,350 | 6499 | 178.84 | 3.75 | |
| 865 | BCK | 335,964 | 9230 | 176.43 | 3.40 | |
| 87 | BCK | 296,022 | 9432 | 184.03 | 3.30 | |
| Bacteria | 105 | HG | 164,912 | 73,165 | 1310.67 | 6.09 |
| 107 | HG | 281,557 | 141,287 | 1312.52 | 6.11 | |
| 131 | HG | 332,247 | 164,026 | 1305.38 | 6.03 | |
| 147 | HG | 86,704 | 35,030 | 1292.00 | 6.02 | |
| 155 | HG | 114,590 | 46,849 | 1310.63 | 6.33 | |
| 217 | BCK | 254,337 | 105,263 | 1329.69 | 5.96 | |
| 227 | HG | 325,493 | 111,886 | 1320.00 | 6.54 | |
| 23 | HG | 251,609 | 123,501 | 1308.09 | 5.78 | |
| 273 | BCK | 355,723 | 141,330 | 1323.82 | 6.13 | |
| 297 | HG | 341,391 | 145,019 | 1329.56 | 5.92 | |
| 343 | BCK | 448,664 | 190,508 | 1325.74 | 6.02 | |
| 381Y | BCK | 451,291 | 172,049 | 1347.00 | 6.23 | |
| 61 | BCK | 350,872 | 182,443 | 1307.35 | 6.03 | |
| 81 | BCK | 214,350 | 87,436 | 1307.86 | 6.36 | |
| 865 | BCK | 335,964 | 142,313 | 1315.88 | 5.98 | |
| 87 | BCK | 296,022 | 133,750 | 1329.02 | 5.96 |
| Microbe | Sample | Diet | Seqs | Chao1 | Goods Coverage | Shannon |
|---|---|---|---|---|---|---|
| Archaea | 105 | HG | 1453 | 7.00 | 0.99 | 0.95 |
| 107 | HG | 1764 | 9.00 | 0.99 | 0.44 | |
| 131 | HG | 2198 | 15.00 | 0.97 | 0.84 | |
| 147 | HG | 2261 | 9.00 | 1.00 | 1.18 | |
| 155 | HG | 2661 | 11.50 | 0.98 | 1.75 | |
| 217 | BCK | 1233 | 9.33 | 0.99 | 1.00 | |
| 227 | HG | 1659 | 4.00 | 0.98 | 0.83 | |
| 23 | HG | 1459 | 2.00 | 1.00 | 0.07 | |
| 273 | BCK | 1282 | 10.50 | 0.96 | 0.80 | |
| 297 | HG | 1500 | 9.00 | 0.99 | 0.49 | |
| 343 | BCK | 1467 | 4.00 | 0.98 | 0.47 | |
| 381Y | BCK | 1192 | 5.00 | 0.99 | 0.43 | |
| 61 | BCK | 1239 | 5.00 | 0.98 | 0.83 | |
| 81 | BCK | 1434 | 4.00 | 0.99 | 0.24 | |
| 865 | BCK | 462 | 7.00 | 0.97 | 0.66 | |
| 87 | BCK | 1323 | 5.00 | 0.99 | 0.67 | |
| Bacteria | 105 | HG | 16,378 | 100.83 | 0.95 | 2.31 |
| 107 | HG | 14,037 | 87.30 | 0.96 | 2.34 | |
| 131 | HG | 16,059 | 144.88 | 0.93 | 3.07 | |
| 147 | HG | 15,974 | 47.43 | 0.97 | 1.67 | |
| 155 | HG | 17,090 | 63.14 | 0.98 | 2.34 | |
| 217 | BCK | 18,187 | 231.29 | 0.85 | 4.38 | |
| 227 | HG | 16,327 | 151.25 | 0.90 | 3.48 | |
| 23 | HG | 16,869 | 83.40 | 0.96 | 2.17 | |
| 273 | BCK | 15,886 | 215.33 | 0.87 | 4.00 | |
| 297 | HG | 13,406 | 106.55 | 0.93 | 2.88 | |
| 343 | BCK | 15,412 | 212.33 | 0.84 | 4.66 | |
| 381Y | BCK | 16,456 | 217.23 | 0.86 | 4.43 | |
| 61 | BCK | 18,355 | 253.09 | 0.85 | 4.70 | |
| 81 | BCK | 19,457 | 197.25 | 0.86 | 4.58 | |
| 865 | BCK | 15,326 | 201.00 | 0.82 | 4.40 | |
| 87 | BCK | 16,137 | 201.68 | 0.87 | 4.07 | |
| Protozoa | 105 | HG | 1006 | 17.75 | 0.98 | 1.35 |
| 107 | HG | 721 | 37.00 | 0.97 | 2.28 | |
| 131 | HG | 1424 | 34.00 | 0.96 | 1.81 | |
| 147 | HG | 1441 | 19.00 | 0.99 | 2.31 | |
| 155 | HG | 1820 | 33.00 | 0.96 | 1.62 | |
| 217 | BCK | 1645 | 55.43 | 0.94 | 2.84 | |
| 227 | HG | 779 | 13.00 | 1.00 | 2.90 | |
| 23 | HG | 1222 | 24.33 | 0.97 | 1.47 | |
| 273 | BCK | 1827 | 32.00 | 0.96 | 1.85 | |
| 297 | HG | 1434 | 23.50 | 0.98 | 1.88 | |
| 343 | BCK | 1537 | 46.67 | 0.95 | 3.19 | |
| 381Y | BCK | 1039 | 56.14 | 0.94 | 3.13 | |
| 61 | BCK | 1808 | 53.36 | 0.94 | 3.21 | |
| 81 | BCK | 1357 | 54.60 | 0.94 | 3.29 | |
| 865 | BCK | 1128 | 46.38 | 0.95 | 3.67 | |
| 87 | BCK | 1862 | 56.20 | 0.94 | 2.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; O’Hara, E.; Tang, S.; Chen, Y.; Walpole, M.E.; Górka, P.; Penner, G.B.; Guan, L.L. Accessing Dietary Effects on the Rumen Microbiome: Different Sequencing Methods Tell Different Stories. Vet. Sci. 2021, 8, 138. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8070138
Zhou M, O’Hara E, Tang S, Chen Y, Walpole ME, Górka P, Penner GB, Guan LL. Accessing Dietary Effects on the Rumen Microbiome: Different Sequencing Methods Tell Different Stories. Veterinary Sciences. 2021; 8(7):138. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8070138
Chicago/Turabian StyleZhou, Mi, Eóin O’Hara, Shaoxun Tang, Yanhong Chen, Matthew E. Walpole, Paweł Górka, Gregory B. Penner, and Le Luo Guan. 2021. "Accessing Dietary Effects on the Rumen Microbiome: Different Sequencing Methods Tell Different Stories" Veterinary Sciences 8, no. 7: 138. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8070138