
Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design

 , and
, and
Abstract
:1. Introduction: Arrhythmogenic Cardiomyopathy
2. Genetics and Animal Models of Arrhythmogenic Cardiomyopathy
2.1. Desmosomal Genes
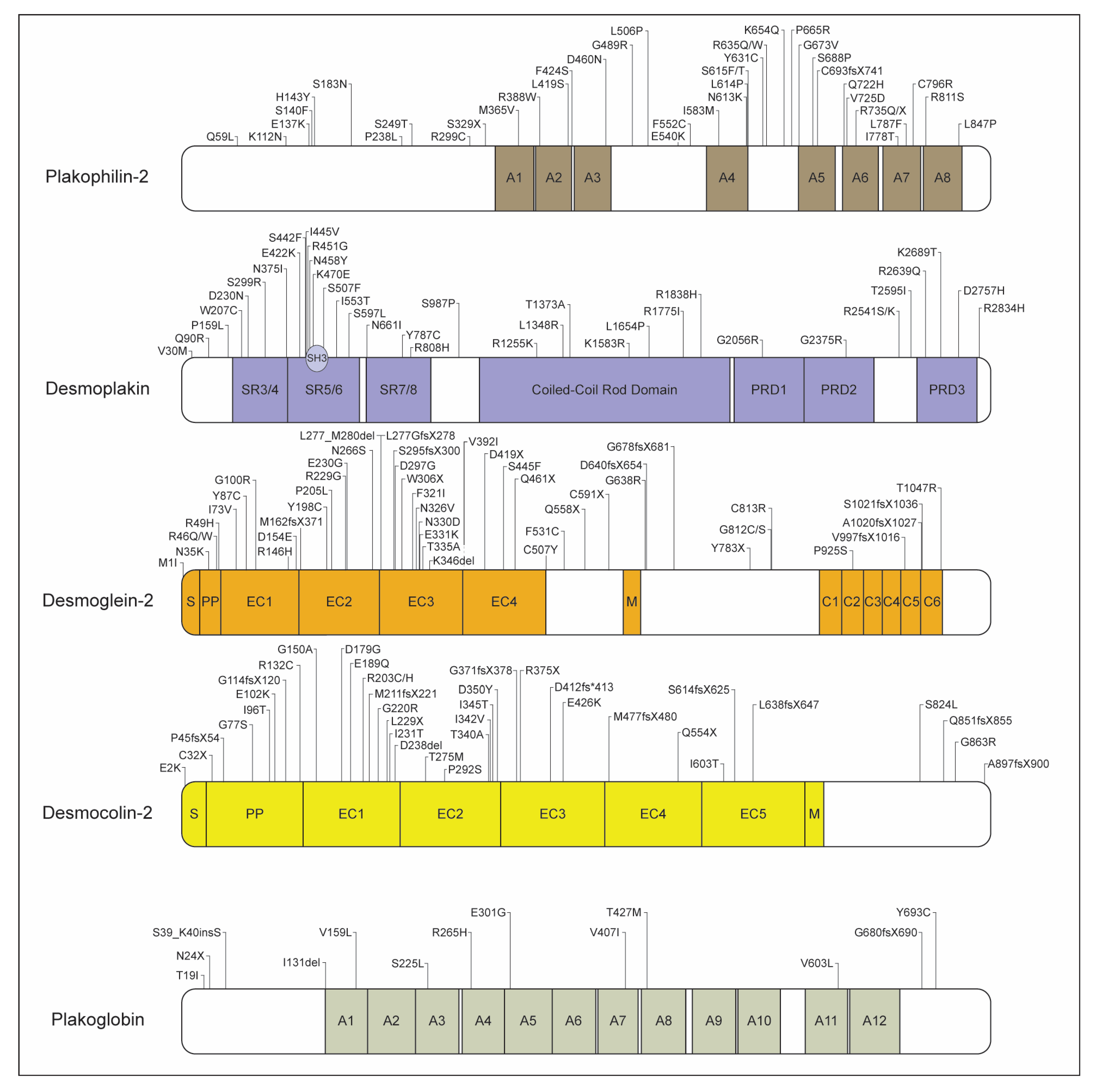
2.1.1. Plakophilin-2
Genetics
Animal Models
2.1.2. Desmoplakin
Genetics
Animal Models
2.1.3. Desmoglein-2
Genetics
Animal Models
2.1.4. Desmocolin-2
Genetics
Animal Models
2.1.5. Junctional Plakoglobin
Genetics
Animal Models
2.2. Non-Desmosomal Genes
2.2.1. α-T-Catenin
2.2.2. Desmin
2.2.3. Lamin A/C
2.2.4. Phospholamban
2.2.5. Transmembrane Protein 43
2.2.6. Titin
2.2.7. Filamin C
2.2.8. Voltage-Gated Sodium Channel Alpha Subunit 5
2.2.9. Tight Junction Protein 1/Zonula Occludens 1
2.2.10. N-Cadherin
2.2.11. Ankyrin-B (ANK2)
2.3. Additional ACM Models
3. Environmental Factors
3.1. Exercise
3.2. Additional Factors
4. Clinical Management of Arrhythmogenic Cardiomyopathy
4.1. Risk Stratification for SCD and Implantable Cardioverter–Defibrillator (ICD) Placement
4.2. Anti-Arrhythmic Medications
4.3. Catheter Ablation
4.4. Sympathectomy
4.5. Heart Transplant
5. Future Implications
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Fontaine, G.; Marcus, F.I.; McKenna, W.J.; Nava, A.; Thiene, G.; Wichter, T. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000, 101. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.G.; Haqqani, H.M.; Berruezo, A.; Della Bella, P.; Marchlinski, F.E.; Hsu, C.J.; Kumar, S. Arrhythmogenic Cardiomyopathy in 2018-2019: ARVC/ALVC or Both? Heart Lung Circ. 2019, 28, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm. Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef] [Green Version]
- McRae, A.A., 3rd; Chung, M.K.; Asher, C.R. Arrhythmogenic right ventricular cardiomyopathy: A cause of sudden death in young people. Clevel. Clin. J. Med. 2001, 68, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Tavora, F.; Zhang, M.; Franco, M.; Oliveira, J.B.; Li, L.; Fowler, D.; Zhao, Z.; Cresswell, N.; Burke, A. Distribution of biventricular disease in arrhythmogenic cardiomyopathy: An autopsy study. Hum. Pathol. 2012, 43, 592–596. [Google Scholar] [CrossRef]
- Herren, T.; Gerber, P.A.; Duru, F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A not so rare “disease of the desmosome” with multiple clinical presentations. Clin. Res. Cardiol. 2009, 98, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.K.; Edwards, W.D.; Hammill, S.C.; Bailey, K.R.; Ballard, D.J.; Gersh, B.J. Sudden unexpected nontraumatic death in 54 young adults: A 30-year population-based study. Am. J. Cardiol. 1995, 76, 148–152. [Google Scholar] [CrossRef]
- Cho, Y. Arrhythmogenic right ventricular cardiomyopathy. J. Arrhythm. 2018, 34, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 1813–1821. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Nava, A.; Bauce, B.; Basso, C.; Muriago, M.; Rampazzo, A.; Villanova, C.; Daliento, L.; Buja, G.; Corrado, D.; Danieli, G.A.; et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2000, 36, 2226–2233. [Google Scholar] [CrossRef] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.W. Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol. 2005, 16, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Saguner, A.M.; Brunckhorst, C.; Duru, F. Arrhythmogenic ventricular cardiomyopathy: A paradigm shift from right to biventricular disease. World J. Cardiol. 2014, 6, 154–174. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Pieperhoff, S. Gene Mutations Resulting in the Development of ARVC/D Could Affect Cells of the Cardiac Conduction System. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.N.; Marchlinski, F.E.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation 2015, 132, 441–453. [Google Scholar] [CrossRef]
- Silvano, M.; Mastella, G.; Zorzi, A.; Migliore, F.; Pilichou, K.; Bauce, B.; Rigato, I.; Perazzolo Marra, M.; Iliceto, S.; Thiene, G.; et al. Management of arrhythmogenic right ventricular cardiomyopathy. Minerva Med. 2016, 107, 194–216. [Google Scholar] [PubMed]
- Migliore, F.; Zorzi, A.; Silvano, M.; Rigato, I.; Basso, C.; Thiene, G.; Corrado, D. Clinical management of arrhythmogenic right ventricular cardiomyopathy: An update. Curr. Pharm. Des. 2010, 16, 2918–2928. [Google Scholar] [CrossRef] [PubMed]
- Marcus, G.M.; Glidden, D.V.; Polonsky, B.; Zareba, W.; Smith, L.M.; Cannom, D.S.; Estes, N.N., III; Marcus, F.; Scheinman, M.M.; Multidisciplinary Study of Right Ventricular Dysplasia Investigators. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: A report from the North American ARVC Registry. J. Am. Coll. Cardiol. 2009, 54, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.; Murray, B.; te Riele, A.S.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, F.H.M.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Lekanne Deprez, R.H.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ. Genom. Precis. Med. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Luscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- Karmouch, J.; Protonotarios, A.; Syrris, P. Genetic basis of arrhythmogenic cardiomyopathy. Curr. Opin. Cardiol. 2018, 33, 276–281. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
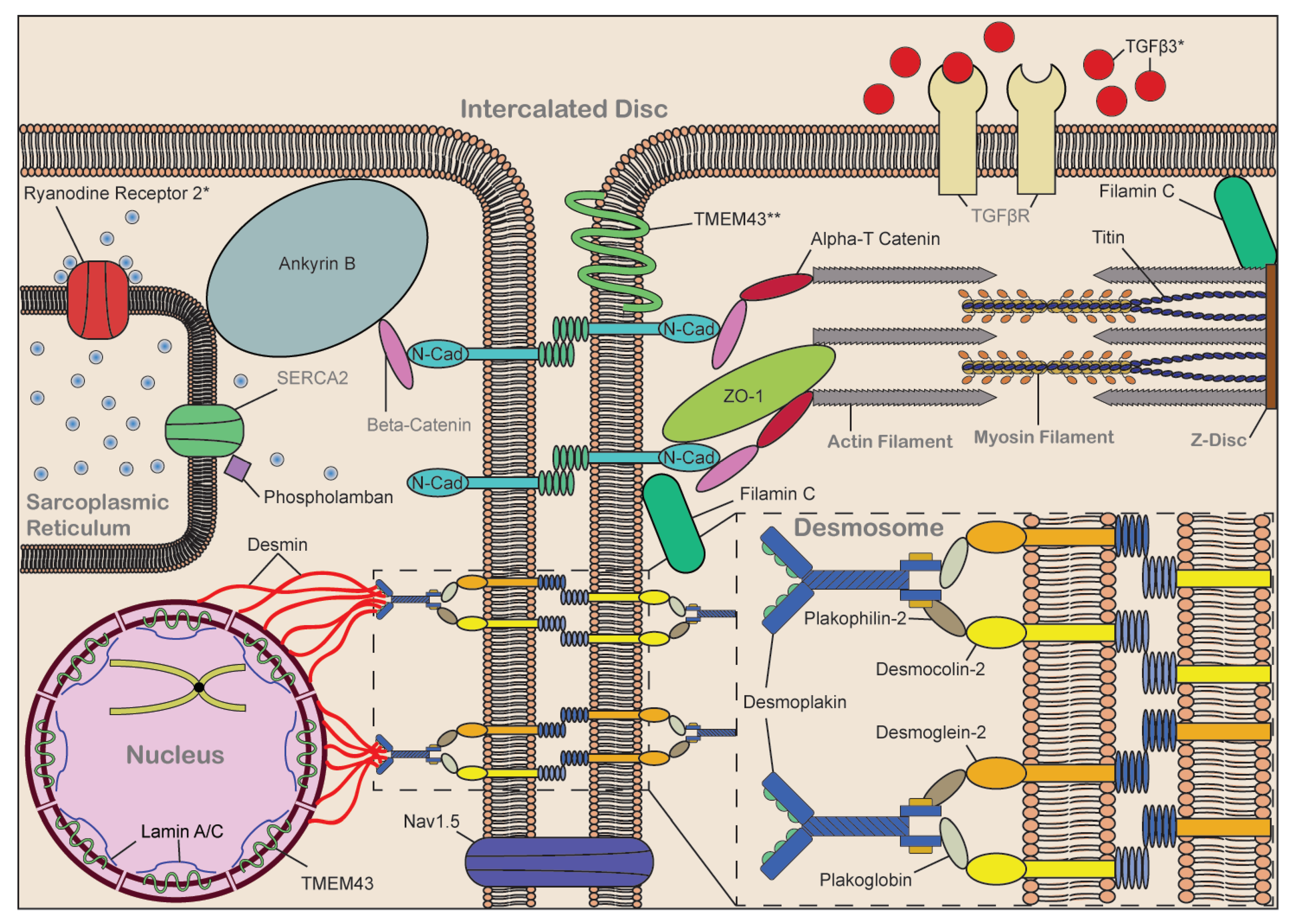
- Calore, M.; Lorenzon, A.; De Bortoli, M.; Poloni, G.; Rampazzo, A. Arrhythmogenic cardiomyopathy: A disease of intercalated discs. Cell Tissue Res. 2015, 360, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, A.; Calore, M.; van Hengel, J.; van Roy, F. Intercalated discs and arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.C. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Swope, D.; Raess, N.; Cheng, L.; Muller, E.J.; Radice, G.G. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol. Cell Biol. 2011, 31, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.A. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; Jongbloed, J.D.; van den Berg, M.P.; van der Smagt, J.J.; Jongbloed, R.; Bikker, H.; Hofstra, R.M.; van Tintelen, J.J. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum. Mutat. 2009, 30, 1278–1283. [Google Scholar] [CrossRef]
- Van der Velde, K.J.; Imhann, F.; Charbon, B.; Pang, C.; van Enckevort, D.; Slofstra, M.; Barbieri, R.; Alberts, R.; Hendriksen, D.; Kelpin, F.; et al. MOLGENIS research: Advanced bioinformatics data software for non-bioinformaticians. Bioinformatics 2019, 35, 1076–1078. [Google Scholar] [CrossRef]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 2010, 12, 861–868. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Landstrom, A.P.; Salisbury, B.A.; Callis, T.E.; Pollevick, G.D.; Tester, D.J.; Cox, M.G.; Bhuiyan, Z.; Bikker, H.; Wiesfeld, A.C.; et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 2011, 57, 2317–2327. [Google Scholar] [CrossRef] [Green Version]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.P. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.R.; Wang, J.Z.; Yao, Y.; Wang, Y.L.; Fan, X.H.; Sun, K.; Zhang, S.; Hui, R.T.; Song, L. Screening of pathogenic genes in Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Chin. Med. J. 2013, 126, 4238–4241. [Google Scholar] [PubMed]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Li Mura, I.E.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Otten, E.; Asimaki, A.; Maass, A.; van Langen, I.M.; van der Wal, A.; de Jonge, N.; van den Berg, M.P.; Saffitz, J.E.; Wilde, A.A.; Jongbloed, J.D.; et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010, 7, 1058–1064. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Takahashi, N.; Fujii, Y.; Umehara, A.; Nishiuchi, S.; Makiyama, T.; Ohno, S.; Horie, M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J. Cardiol. 2016, 68, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Wang, J.; Yao, Y.; Wang, Y.; Fan, X.; Sun, K.; He, D.S.; Marcus, F.I.; Zhang, S.; Hui, R.; et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations are Associated with Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations are Associated with High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm 2014, 11, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.J. TJP1 Mutations in Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.M. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Murphy, N.P.; Hamilton, R.M.; Lubbers, E.R.; James, C.A.; Kline, C.F.; Gollob, M.H.; Krahn, A.D.; Sturm, A.C.; Musa, H.; et al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J. Clin. Investig. 2019, 129, 3171–3184. [Google Scholar] [CrossRef]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.C.; Healey, J.S.; Hamilton, R.; Spears, D.; Gollob, M.H.; Mellor, G.; Steinberg, C.; Sanatani, S.; Laksman, Z.W.; Krahn, A.A. Phospholamban cardiomyopathy: A Canadian perspective on a unique population. Neth. Heart J. 2019, 27, 208–213. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020. [Google Scholar] [CrossRef] [PubMed]
- Vermij, S.H.; Abriel, H.; van Veen, T.T. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.W. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- Bass-Zubek, A.E.; Hobbs, R.P.; Amargo, E.V.; Garcia, N.J.; Hsieh, S.N.; Chen, X.; Wahl, J.K., III; Denning, M.F.; Green, K.K. Plakophilin 2: A critical scaffold for PKC alpha that regulates intercellular junction assembly. J. Cell Biol. 2008, 181, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bonne, S.; Hatzfeld, M.; van Roy, F.; Green, K.K. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J. Biol. Chem. 2002, 277, 10512–10522. [Google Scholar] [CrossRef] [Green Version]
- Delmar, M.; McKenna, W.W. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.; Li, S.; Li, H.; Creason, V.; Wahl, J.J., 3rd. Arrhythmogenic right ventricular cardiomyopathy plakophilin-2 mutations disrupt desmosome assembly and stability. Cell Commun. Adhes. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, F.; Schuetz, A.; Boldt, L.H.; Martens, K.; Dittmar, G.; Haverkamp, W.; Thierfelder, L.; Heinemann, U.; Gerull, B. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ. Cardiovasc. Genet. 2012, 5, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 2004, 167, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Noorman, M.; Lin, X.; Chkourko, H.; Liang, F.X.; van der Nagel, R.; Hund, T.; Birchmeier, W.; Mohler, P.; van Veen, T.A.; et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 2012, 95, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Guasch, E.; Sanz-de la Garza, M.; Casado, M.; Garcia, N.A.; Mont, L.; Sitges, M.; Knoll, R.; Buyandelger, B.; Campuzano, O.; et al. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Hum. Mol. Genet. 2016, 25, 3676–3688. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.K. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J. Cell Biol. 2014, 206, 779–797. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.P.; Bornslaeger, E.A.; Borgwardt, J.E.; Palka, H.L.; Dhaliwal, A.S.; Corcoran, C.M.; Denning, M.F.; Green, K.K. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 1997, 139, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Kam, C.Y.; Dubash, A.D.; Magistrati, E.; Polo, S.; Satchell, K.J.F.; Sheikh, F.; Lampe, P.D.; Green, K.K. Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J. Cell Biol. 2018, 217, 3219–3235. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Al-Jassar, C.; Bikker, H.; Overduin, M.; Chidgey, M. Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 2013, 425, 4006–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daday, C.; Kolsek, K.; Grater, F. The mechano-sensing role of the unique SH3 insertion in plakin domains revealed by Molecular Dynamics simulations. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 2006, 99, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Gallicano, G.I.; Bauer, C.; Fuchs, E. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development 2001, 128, 929–941. [Google Scholar]
- Agarwal, U.; Ghalayini, W.; Dong, F.; Weber, K.; Zou, Y.R.; Rabbany, S.Y.; Rafii, S.; Penn, M.M. Role of cardiac myocyte CXCR4 expression in development and left ventricular remodeling after acute myocardial infarction. Circ. Res. 2010, 107, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Tandri, H.; Huang, H.; Halushka, M.K.; Gautam, S.; Basso, C.; Thiene, G.; Tsatsopoulou, A.; Protonotarios, N.; McKenna, W.J.; et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 2009, 360, 1075–1084. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.A. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Dieding, M.; Debus, J.D.; Kerkhoff, R.; Gaertner-Rommel, A.; Walhorn, V.; Milting, H.; Anselmetti, D. Arrhythmogenic cardiomyopathy related DSG2 mutations affect desmosomal cadherin binding kinetics. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.H.; Andersen, C.B.; Wassilew, K.; Svendsen, J.H.; Bundgaard, H.; Brand, S.M.; Schmitz, B. Rare non-coding Desmoglein-2 variant contributes to Arrhythmogenic right ventricular cardiomyopathy. J. Mol. Cell Cardiol. 2019, 131, 164–170. [Google Scholar] [CrossRef]
- Bauce, B.; Nava, A.; Beffagna, G.; Basso, C.; Lorenzon, A.; Smaniotto, G.; De Bortoli, M.; Rigato, I.; Mazzotti, E.; Steriotis, A. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2010, 7, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Kaneko, Y.; Irie, T.; Takahashi, R.; Kato, T.; Iijima, T.; Iso, T.; Kurabayashi, M. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular cardiomyopathy in Japanese patients. Circ. J. 2012, 76, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pilichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, Q.; Zhong, Z.A.; Xu, Z.; He, S.; Rao, F.; Liu, Y.; Tang, J.; Wang, F.; Liu, H.; et al. Whole Genome Sequence Identified a Rare Homozygous Pathogenic Mutation of the DSG2 Gene in a Familial Arrhythmogenic Cardiomyopathy Involving Both Ventricles. Cardiology 2017, 138, 41–54. [Google Scholar] [CrossRef]
- Hermida, A.; Fressart, V.; Hidden-Lucet, F.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Heart Fail. 2019, 21, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.R. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 2002, 81, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Krusche, C.A.; Holthofer, B.; Hofe, V.; van de Sandt, A.M.; Eshkind, L.; Bockamp, E.; Merx, M.W.; Kant, S.; Windoffer, R.; Leube, R.R. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res. Cardiol. 2011, 106, 617–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Krull, P.; Eisner, S.; Leube, R.E.; Krusche, C.C. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012, 348, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.C. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, A.H.; Benn, M.; Bundgaard, H.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J.J. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2010, 47, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beffagna, G.; De Bortoli, M.; Nava, A.; Salamon, M.; Lorenzon, A.; Zaccolo, M.; Mancuso, L.; Sigalotti, L.; Bauce, B.; Occhi, G.; et al. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med. Genet. 2007, 8. [Google Scholar] [CrossRef]
- Gehmlich, K.; Asimaki, A.; Cahill, T.J.; Ehler, E.; Syrris, P.; Zachara, E.; Re, F.; Avella, A.; Monserrat, L.; Saffitz, J.E.; et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm 2010, 7, 1446–1453. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.Y.; Jain, R.; den Haan, A.D.; Chen, Y.; Dalal, D.; Tandri, H.; Amat-Alarcon, N.; Daly, A.; Tichnell, C.; James, C.; et al. Shared desmosome gene findings in early and late onset arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Cardiovasc. Transl. Res. 2010, 3, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Bhuiyan, Z.A.; Jongbloed, J.D.; van der Smagt, J.; Lombardi, P.M.; Wiesfeld, A.C.; Nelen, M.; Schouten, M.; Jongbloed, R.; Cox, M.G.; van Wolferen, M.; et al. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: Results from a multicenter study. Circ. Cardiovasc. Genet. 2009, 2, 418–427. [Google Scholar] [CrossRef]
- Gehmlich, K.; Syrris, P.; Peskett, E.; Evans, A.; Ehler, E.; Asimaki, A.; Anastasakis, A.; Tsatsopoulou, A.; Vouliotis, A.I.; Stefanadis, C.; et al. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc. Res. 2011, 90, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.P.; Hatzfeld, M.; Bornslaeger, E.A.; Kopp, D.S.; Borgwardt, J.E.; Corcoran, C.M.; Settler, A.; Green, K.K. The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes—Implications for cutaneous disease. J. Biol. Chem. 1999, 274, 18145–18148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004, 1, 3–11. [Google Scholar] [CrossRef]
- Hariharan, V.; Asimaki, A.; Michaelson, J.E.; Plovie, E.; MacRae, C.A.; Saffitz, J.E.; Huang, H. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovasc. Res. 2014, 104, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Krusche, C.A.; Gaertner, A.; Milting, H.; Leube, R.R. Loss of plakoglobin immunoreactivity in intercalated discs in arrhythmogenic right ventricular cardiomyopathy: Protein mislocalization versus epitope masking. Cardiovasc. Res. 2016, 109, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.O. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Gunthert, U.; Franke, W.W.; et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 1996, 135, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [Green Version]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.G. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol. Cell Biol. 2012, 32, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.A. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milting, H.; Lukas, N.; Klauke, B.; Korfer, R.; Perrot, A.; Osterziel, K.J.; Vogt, J.; Peters, S.; Thieleczek, R.; Varsanyi, M. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Res. 2006, 71, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; van Tintelen, J.P.; Vink, A.; van der Wal, A.C.; de Boer, R.A.; van den Berg, M.P.; Suurmeijer, A.A. Phospholamban p.Arg14del cardiomyopathy is characterized by phospholamban aggregates, aggresomes, and autophagic degradation. Histopathology 2016, 69, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Bar, H.; Goudeau, B.; Walde, S.; Casteras-Simon, M.; Mucke, N.; Shatunov, A.; Goldberg, Y.P.; Clarke, C.; Holton, J.L.; Eymard, B.; et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 2007, 28, 374–386. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Bar, H.; Fischer, D.; Goudeau, B.; Kley, R.A.; Clemen, C.S.; Vicart, P.; Herrmann, H.; Vorgerd, M.; Schroder, R. Pathogenic effects of a novel heterozygous R350P desmin mutation on the assembly of desmin intermediate filaments in vivo and in vitro. Hum. Mol. Genet. 2005, 14, 1251–1260. [Google Scholar] [CrossRef]
- Park, K.Y.; Dalakas, M.C.; Goebel, H.H.; Ferrans, V.J.; Semino-Mora, C.; Litvak, S.; Takeda, K.; Goldfarb, L.L. Desmin splice variants causing cardiac and skeletal myopathy. J. Med. Genet. 2000, 37, 851–857. [Google Scholar] [CrossRef]
- Ariza, A.; Coll, J.; Fernandez-Figueras, M.T.; Lopez, M.D.; Mate, J.L.; Garcia, O.; Fernandez-Vasalo, A.; Navas-Palacios, J.J. Desmin myopathy: A multisystem disorder involving skeletal, cardiac, and smooth muscle. Hum. Pathol. 1995, 26, 1032–1037. [Google Scholar] [CrossRef]
- Liang, J.J.; Grogan, M.; Ackerman, M.J.; Goodsell, K. LMNA-Mediated Arrhythmogenic Right Ventricular Cardiomyopathy and Charcot-Marie-Tooth Type 2B1: A Patient-Discovered Unifying Diagnosis. J. Cardiovasc. Electrophysiol. 2016, 27, 868–871. [Google Scholar] [CrossRef]
- Petillo, R.; D’Ambrosio, P.; Torella, A.; Taglia, A.; Picillo, E.; Testori, A.; Ergoli, M.; Nigro, G.; Piluso, G.; Nigro, V.; et al. Novel mutations in LMNA A/C gene and associated phenotypes. Acta Myol. 2015, 34, 116–119. [Google Scholar]
- Van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.G., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Rijdt, W.P.; Asimaki, A.; Jongbloed, J.D.H.; Hoorntje, E.T.; Lazzarini, E.; van der Zwaag, P.A.; de Boer, R.A.; van Tintelen, J.P.; Saffitz, J.E.; van den Berg, M.P.; et al. Distinct molecular signature of phospholamban p.Arg14del arrhythmogenic cardiomyopathy. Cardiovasc. Pathol. 2019, 40, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, W.W.; Dorflinger, Y.; Kuhn, C.; Zimbelmann, R.; Winter-Simanowski, S.; Frey, N.; Heid, H. Protein LUMA is a cytoplasmic plaque constituent of various epithelial adherens junctions and composite junctions of myocardial intercalated disks: A unifying finding for cell biology and cardiology. Cell Tissue Res. 2014, 357, 159–172. [Google Scholar] [CrossRef]
- Padron-Barthe, L.; Villalba-Orero, M.; Gomez-Salinero, J.M.; Dominguez, F.; Roman, M.; Larrasa-Alonso, J.; Ortiz-Sanchez, P.; Martinez, F.; Lopez-Olaneta, M.; Bonzon-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3beta. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- AbdelWahab, A.; Gardner, M.; Parkash, R.; Gray, C.; Sapp, J. Ventricular tachycardia ablation in arrhythmogenic right ventricular cardiomyopathy patients with TMEM43 gene mutations. J. Cardiovasc. Electrophysiol. 2018, 29, 90–97. [Google Scholar] [CrossRef]
- Hodgkinson, K.A.; Connors, S.P.; Merner, N.; Haywood, A.; Young, T.L.; McKenna, W.J.; Gallagher, B.; Curtis, F.; Bassett, A.S.; Parfrey, P.P. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin. Genet. 2013, 83, 321–331. [Google Scholar] [CrossRef]
- Aalbaek Kjaergaard, K.; Kristensen, J.; Molgaard, H.; Cosedis Nielsen, J.; Jensen, H.H. Failure of ICD therapy in lethal arrhythmogenic right ventricular cardiomyopathy type 5 caused by the TMEM43 p.Ser358Leu mutation. HeartRhythm Case Rep. 2016, 2, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Bogomolovas, J.; Fleming, J.R.; Anderson, B.R.; Williams, R.; Lange, S.; Simon, B.; Khan, M.M.; Rudolf, R.; Franke, B.; Bullard, B.; et al. Exploration of pathomechanisms triggered by a single-nucleotide polymorphism in titin’s I-band: The cardiomyopathy-linked mutation T2580I. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Franaszczyk, M.; Chmielewski, P.; Truszkowska, G.; Stawinski, P.; Michalak, E.; Rydzanicz, M.; Sobieszczanska-Malek, M.; Pollak, A.; Szczygiel, J.; Kosinska, J.; et al. Titin Truncating Variants in Dilated Cardiomyopathy—Prevalence and Genotype-Phenotype Correlations. PLoS ONE 2017, 12, e0169007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Zorzi, A. Filamin C: A New Arrhythmogenic Cardiomyopathy-Causing Gene? JACC Clin. Electrophysiol. 2018, 4, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, A.; Sonnenberg, A. Structural and functional aspects of filamins. Biochim. Biophys. Acta 2001, 1538, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, J.; Dai, X.; Cao, Q.; Xiong, Q.; Liu, X.; Liu, X.; Shen, Y.; Chen, Q.; Hua, W.; et al. SCN5A mutation in Chinese patients with arrhythmogenic right ventricular dysplasia. Herz 2014, 39, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Sun, A.; Paajanen, V.; Wang, S.; Su, C.; Yang, Z.; Li, Y.; Wang, S.; Jia, J.; Wang, K.; et al. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2008, 1, 83–92. [Google Scholar] [CrossRef] [Green Version]
- McNair, W.P.; Sinagra, G.; Taylor, M.R.; Di Lenarda, A.; Ferguson, D.A.; Salcedo, E.E.; Slavov, D.; Zhu, X.; Caldwell, J.H.; Mestroni, L.; et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol. 2011, 57, 2160–2168. [Google Scholar] [CrossRef] [Green Version]
- Erkapic, D.; Neumann, T.; Schmitt, J.; Sperzel, J.; Berkowitsch, A.; Kuniss, M.; Hamm, C.W.; Pitschner, H.H. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace 2008, 10, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Gessner, G.; Runge, S.; Koenen, M.; Heinemann, S.H.; Koenen, M.; Haas, J.; Meder, B.; Thomas, D.; Katus, H.A.; Schweizer, P.P. ANK2 functionally interacts with KCNH2 aggravating long QT syndrome in a double mutation carrier. Biochem. Biophys. Res. Commun. 2019, 512, 845–851. [Google Scholar] [CrossRef]
- Cunha, S.R.; Hund, T.J.; Hashemi, S.; Voigt, N.; Li, N.; Wright, P.; Koval, O.; Li, J.; Gudmundsson, H.; Gumina, R.J.; et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 2011, 124, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Le Scouarnec, S.; Bhasin, N.; Vieyres, C.; Hund, T.J.; Cunha, S.R.; Koval, O.; Marionneau, C.; Chen, B.; Wu, Y.; Demolombe, S.; et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. USA 2008, 105, 15617–15622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, Y.; Takashima, S.; Asakura, M.; Shintani, Y.; Liao, Y.; Minamino, T.; Asanuma, H.; Sanada, S.; Kim, J.; Ogai, A.; et al. Lamr1 functional retroposon causes right ventricular dysplasia in mice. Nat. Genet. 2004, 36, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S. Loss of cardiac Wnt/β-catenin signalling in Desmoplakin-deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovasc. Res. 2018, 114, 1082–1097. [Google Scholar] [CrossRef]
- Basso, C.; Fox, P.R.; Meurs, K.M.; Towbin, J.A.; Spier, A.W.; Calabrese, F.; Maron, B.J.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: A new animal model of human disease. Circulation 2004, 109, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Fox, P.R.; Maron, B.J.; Basso, C.; Liu, S.K.; Thiene, G. Spontaneously occurring arrhythmogenic right ventricular cardiomyopathy in the domestic cat: A new animal model similar to the human disease. Circulation 2000, 102, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Sommariva, E.; Stadiotti, I.; Perrucci, G.L.; Tondo, C.; Pompilio, G. Cell models of arrhythmogenic cardiomyopathy: Advances and opportunities. Dis. Model. Mech. 2017, 10, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarska, E.K.; Konka, M.; Zaleska, T.; Ploski, R.; Cedro, K.; Pucilowska, B.; Bekiesinska-Figatowska, M.; Rydlewska-Sadowska, W.; Ruzyllo, W.; Hoffman, P. Arrhythmogenic right ventricular cardiomyopathy in two pairs of monozygotic twins. Int. J. Cardiol. 2005, 105, 126–133. [Google Scholar] [CrossRef]
- Furlanello, F.; Bertoldi, A.; Dallago, M.; Furlanello, C.; Fernando, F.; Inama, G.; Pappone, C.; Chierchia, S. Cardiac arrest and sudden death in competitive athletes with arrhythmogenic right ventricular dysplasia. Pacing Clin. Electrophysiol. 1998, 21, 331–335. [Google Scholar] [CrossRef]
- Gemayel, C.; Pelliccia, A.; Thompson, P.P. Arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2001, 38, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Saberniak, J.; Hasselberg, N.E.; Borgquist, R.; Platonov, P.G.; Sarvari, S.I.; Smith, H.J.; Ribe, M.; Holst, A.G.; Edvardsen, T.; Haugaa, K.K. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur. J. Heart Fail. 2014, 16, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.W. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Huang, H.; Asimaki, A.; Lo, D.; McKenna, W.; Saffitz, J. Disparate effects of different mutations in plakoglobin on cell mechanical behavior. Cell Motil. Cytoskelet. 2008, 65, 964–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlipp, A.; Schinner, C.; Spindler, V.; Vielmuth, F.; Gehmlich, K.; Syrris, P.; McKenna, W.J.; Dendorfer, A.; Hartlieb, E.; Waschke, J. Desmoglein-2 interaction is crucial for cardiomyocyte cohesion and function. Cardiovasc. Res. 2014, 104, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finocchiaro, G.; Papadakis, M.; Robertus, J.L.; Dhutia, H.; Steriotis, A.K.; Tome, M.; Mellor, G.; Merghani, A.; Malhotra, A.; Behr, E.; et al. Etiology of Sudden Death in Sports: Insights From a United Kingdom Regional Registry. J. Am. Coll. Cardiol. 2016, 67, 2108–2115. [Google Scholar] [CrossRef]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schafers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.H.; Haskell, W.; Snell, P.; Van Camp, S.S. Task Force 8: Classification of sports. J. Am. Coll. Cardiol. 2005, 45, 1364–1367. [Google Scholar] [CrossRef] [Green Version]
- Tabib, A.; Loire, R.; Chalabreysse, L.; Meyronnet, D.; Miras, A.; Malicier, D.; Thivolet, F.; Chevalier, P.; Bouvagnet, P. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003, 108, 3000–3005. [Google Scholar] [CrossRef] [Green Version]
- La Gerche, A.; Robberecht, C.; Kuiperi, C.; Nuyens, D.; Willems, R.; de Ravel, T.; Matthijs, G.; Heidbuchel, H. Lower than expected desmosomal gene mutation prevalence in endurance athletes with complex ventricular arrhythmias of right ventricular origin. Heart 2010, 96, 1268–1274. [Google Scholar] [CrossRef]
- Taggart, P.; Boyett, M.R.; Logantha, S.; Lambiase, P.P. Anger, emotion, and arrhythmias: From brain to heart. Front. Physiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, M.G.; van der Zwaag, P.A.; van der Werf, C.; van der Smagt, J.J.; Noorman, M.; Bhuiyan, Z.A.; Wiesfeld, A.C.; Volders, P.G.; van Langen, I.M.; Atsma, D.D. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011, 123, 2690–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, C.; Corrado, D.; Bauce, B.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2012, 5, 1233–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar]
- Haugaa, K.H.; Basso, C.; Badano, L.P.; Bucciarelli-Ducci, C.; Cardim, N.; Gaemperli, O.; Galderisi, M.; Habib, G.; Knuuti, J.; Lancellotti, P. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyopathy—An expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 237–253. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, 301–372. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.; MacLeod, H.; Dellefave-Castillo, L. Arrhythmogenic right ventricular cardiomyopathy. In GeneReviews®; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.J.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, O.H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Orgeron, G.M.; Te Riele, A.; Tichnell, C.; Wang, W.; Murray, B.; Bhonsale, A.; Judge, D.P.; Kamel, I.R.; Zimmerman, S.L.; Tandri, H. Performance of the 2015 International Task Force Consensus Statement risk stratification algorithm for implantable cardioverter-defibrillator placement in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2018, 11. [Google Scholar] [CrossRef]
- Sagawa, Y.; Nagata, Y.; Yamaguchi, T.; Mitsui, K.; Nagamine, T.; Yamaguchi, J.; Hijikata, S.; Watanabe, K.; Masuda, R.; Miyazaki, R.; et al. Long-Term Performance of Right Ventricular Implantable Cardioverter-Defibrillator Leads in Arrhythmogenic Right Ventricular Cardiomyopathy and Hypertrophic Cardiomyopathy. Int. Heart J. 2020, 61, 39–45. [Google Scholar] [CrossRef]
- Wang, W.; Orgeron, G.; Tichnell, C.; Murray, B.; Crosson, J.; Monfredi, O.; Cadrin-Tourigny, J.; Tandri, H.; Calkins, H.; James, C.C. Impact of exercise restriction on arrhythmic risk among patients with arrhythmogenic right ventricular cardiomyopathy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Leoni, L.; Link, M.S.; Bella, P.D.; Gaita, F.; Curnis, A.; Salerno, J.U.; Igidbashian, D.; Raviele, A.; Disertori, M. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003, 108, 3084–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkinson, K.A.; Howes, A.J.; Boland, P.; Shen, X.S.; Stuckless, S.; Young, T.L.; Curtis, F.; Collier, A.; Parfrey, P.S.; Connors, S.S. Long-Term Clinical Outcome of Arrhythmogenic Right Ventricular Cardiomyopathy in Individuals With a p.S358L Mutation in TMEM43 Following Implantable Cardioverter Defibrillator Therapy. Circ. Arrhythm. Electrophysiol. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, S.; Gerstenfeld, E.P.; Svetlichnaya, Y.; Scheinman, M.M. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2017, 14, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.H. Mortality and morbidity in patients receiving encainide, flecainide, or placebo: The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Zareba, W. Clinical Trial: Pilot Randomized Trial wWith Flecainide in ARVC Patients; NCT03685149, currently ongoing (unpublished 2018); University of Rochester Medical Center: Rochester, NY, USA, Estimated completion August 2021.
- Mahida, S.; Venlet, J.; Saguner, A.M.; Kumar, S.; Baldinger, S.H.; AbdelWahab, A.; Tedrow, U.B.; Castelletti, S.; Pantazis, A.; John, R.M.; et al. Ablation compared with drug therapy for recurrent ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy: Results from a multicenter study. Heart Rhythm 2019, 16, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Santangeli, P.; Zado, E.S.; Supple, G.E.; Haqqani, H.M.; Garcia, F.C.; Tschabrunn, C.M.; Callans, D.J.; Lin, D.; Dixit, S.; Hutchinson, M.D.; et al. Long-Term Outcome With Catheter Ablation of Ventricular Tachycardia in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2015, 8, 1413–1421. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.K.; Haugaa, K.H.; Svensson, A.; Gilljam, T.; Madsen, T.; Hansen, J.; Holst, A.G.; Bundgaard, H.; Edvardsen, T.; Svendsen, J.J. Incidence, Predictors, and Success of Ventricular Tachycardia Catheter Ablation in Arrhythmogenic Right Ventricular Cardiomyopathy (from the Nordic ARVC Registry). Am. J. Cardiol. 2020, 125, 803–811. [Google Scholar] [CrossRef]
- Bourke, T.; Vaseghi, M.; Michowitz, Y.; Sankhla, V.; Shah, M.; Swapna, N.; Boyle, N.G.; Mahajan, A.; Narasimhan, C.; Lokhandwala, Y.; et al. Neuraxial Modulation for Refractory Ventricular Arrhythmias Value of Thoracic Epidural Anesthesia and Surgical Left Cardiac Sympathetic Denervation. Circulation 2010, 121, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Te Riele, A.; Ajijola, O.A.; Shivkumar, K.; Tandri, H. Role of Bilateral Sympathectomy in the Treatment of Refractory Ventricular Arrhythmias in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Assis, F.R.; Krishnan, A.; Zhou, X.; James, C.A.; Murray, B.; Tichnell, C.; Berger, R.; Calkins, H.; Tandri, H.; Mandal, K. Cardiac sympathectomy for refractory ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2019, 16, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.A.; Lellouche, N.; Bourke, T.; Tung, R.; Ahn, S.; Mahajan, A.; Shivkumar, K. Bilateral cardiac sympathetic denervation for the management of electrical storm. J. Am. Coll. Cardiol. 2012, 59, 91–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseghi, M.; Gima, J.; Kanaan, C.; Ajijola, O.A.; Marmureanu, A.; Mahajan, A.; Shivkumar, K. Cardiac sympathetic denervation in patients with refractory ventricular arrhythmias or electrical storm: Intermediate and long-term follow-up. Heart Rhythm 2014, 11, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseghi, M.; Barwad, P.; Corrales, F.J.M.; Tandri, H.; Mathuria, N.; Shah, R.; Sorg, J.M.; Gima, J.; Mandal, K.; Morales, L.C.S. Cardiac sympathetic denervation for refractory ventricular arrhythmias. J. Am. Coll. Cardiol. 2017, 69, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tedrow, U.B.; Stevenson, W.W. Adjunctive interventional techniques when percutaneous catheter ablation for drug refractory ventricular arrhythmias fail: A contemporary review. Circ. Arrhythm. Electrophysiol. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.J. Molecular mechanisms in the pathogenesis of arrhythmogenic cardiomyopathy. Cardiovasc. Pathol. 2017, 28, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Cox, L.L. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.P.; Binalsheikh, I.M.; Leach, S.B.; Domeier, T.L.; Duan, D.D. Prospect of gene therapy for cardiomyopathy in hereditary muscular dystrophy. Exp. Opin. Orphan Drugs 2016, 4, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Peña, J.R.; Szkudlarek, A.C.; Warren, C.M.; Heinrich, L.S.; Gaffin, R.D.; Jagatheesan, G.; del Monte, F.; Hajjar, R.J.; Goldspink, P.H.; Solaro, R.R. Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 993–1002. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Gene | Protein | Structure or Location | Pathogenic Variants * | ACM Prevalence ** | Phenotype | References |
|---|---|---|---|---|---|---|
| PKP2 | Plakophilin-2 | ID/desmosome | 171 | 23.4–57.6% | RV dominant ACM | [25,26,30,38,39,40,41] |
| DSP | Desmoplakin | ID/desmosome | 86 | 1.6–15.7% | RV, LV, and biventricular ACM, DCM, Carvajal syndrome (recessive), palmoplantar keratoderma, additional cutaneous diseases. | [25,26,30,38,39,40,41] |
| DSG2 | Desmoglein-2 | ID/desmosome | 50 | 4.0–20.4% | RV and biventricular ACM | [25,26,30,38,39,40,41] |
| DSC2 | Desmocollin-2 | ID/desmosome | 42 | 1.0–8.3% | RV, LV, and biventricular ACM | [25,26,30,38,39,40,41] |
| JUP | Junction plakoglobin (PKG) | ID/desmosome | 15 | <1–3.0% | RV dominant ACM, Naxos disease (recessive), palmoplantar keratoderma, woolly hair disease | [25,30,39,41] |
| CTNNA3 | α-T-catenin | ID/area composita | 2 | <2.6% (among ACM cases without common mutation) | RV dominant ACM | [42] |
| DES | Desmin | Intermediate filaments/cytoskeleton | 11 | <1–2.2% | RV and biventricular dominant ACM. DCM, HCM, DRM, muscular dystrophy | [41,43,44,45] |
| LMNA | Lamin A/C | Nuclear envelope | 16 | 3.5–3.7% | RV and biventricular dominant ACM. DCM, HCM, muscular dystrophy | [40,41,46] |
| PLN | Phospholamban | Sarcoplasmic reticulum | 4 | 2.2–12.4% | RV, LV, and biventricular dominant ACM, DCM | [18,25,26,30] |
| TMEM43 | Transmembrane protein 43 (LUMA) | Nuclear envelope/ID | 3 | <1–2.2% | RV, LV, and biventricular dominant ACM, DCM, muscular dystrophy | [25,26,30,41,47] |
| TTN | Titin | Sarcomere | 10 | <1% (18.4% of ACM cases negative for variants in common genes) | RV and biventricular dominant ACM, DCM, HCM, muscular dystrophy | [48,49] |
| FLNC | Filamin C | Sarcomere/ID | <1% (7.5% of ACM cases negative for variants in common genes) | RV, LV, and biventricular dominant ACM, DCM, restrictive cardiomyopathy | [50,51,52] | |
| SCN5A | Sodium channel Nav1.5 | Cell membrane | <1–1.8% | RV dominant ACM, DCM, HCM, Brugada syndrome, long QT syndrome | [26,53,54] | |
| TJP1 | Zonula occludens 1 (ZO1) | ID/tight junction | <1% (<5% of ACM cases negative for variants in common genes) | RV, LV, and biventricular dominant ACM | [26,53,55,56] | |
| CDH2 | N-Cadherin | ID/adherens junction | <1% (2.7% of ACM cases negative for variants in common genes) | RV and biventricular ACM | [55,56,57,58] | |
| ANK2 | Ankyrin B (AnkB) | Z-lines/T-tubules | Unknown | RV dominant ACM, long QT-syndrome, atrial Fibrillation, ankyrin-B syndrome | [57,58,59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, T.L.; Wallace, M.J.; El Refaey, M.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020021
Stevens TL, Wallace MJ, El Refaey M, Roberts JD, Koenig SN, Mohler PJ. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. Journal of Cardiovascular Development and Disease. 2020; 7(2):21. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020021
Chicago/Turabian StyleStevens, Tyler L., Michael J. Wallace, Mona El Refaey, Jason D. Roberts, Sara N. Koenig, and Peter J. Mohler. 2020. "Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design" Journal of Cardiovascular Development and Disease 7, no. 2: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020021