
Transcriptome Profiles of Sporisorium reilianum during the Early Infection of Resistant and Susceptible Maize Isogenic Lines


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Axenic Culture Samples of S. reilianum for RNA Sequencing (RNA-seq)
2.2. Infected Maize Mesocotyls for RNA-seq
2.3. Preparation of RNA-seq Libraries and Sequencing
2.4. Differential Gene Expression Analysis
2.5. Transcript Annotation and Pathway Mapping
3. Results
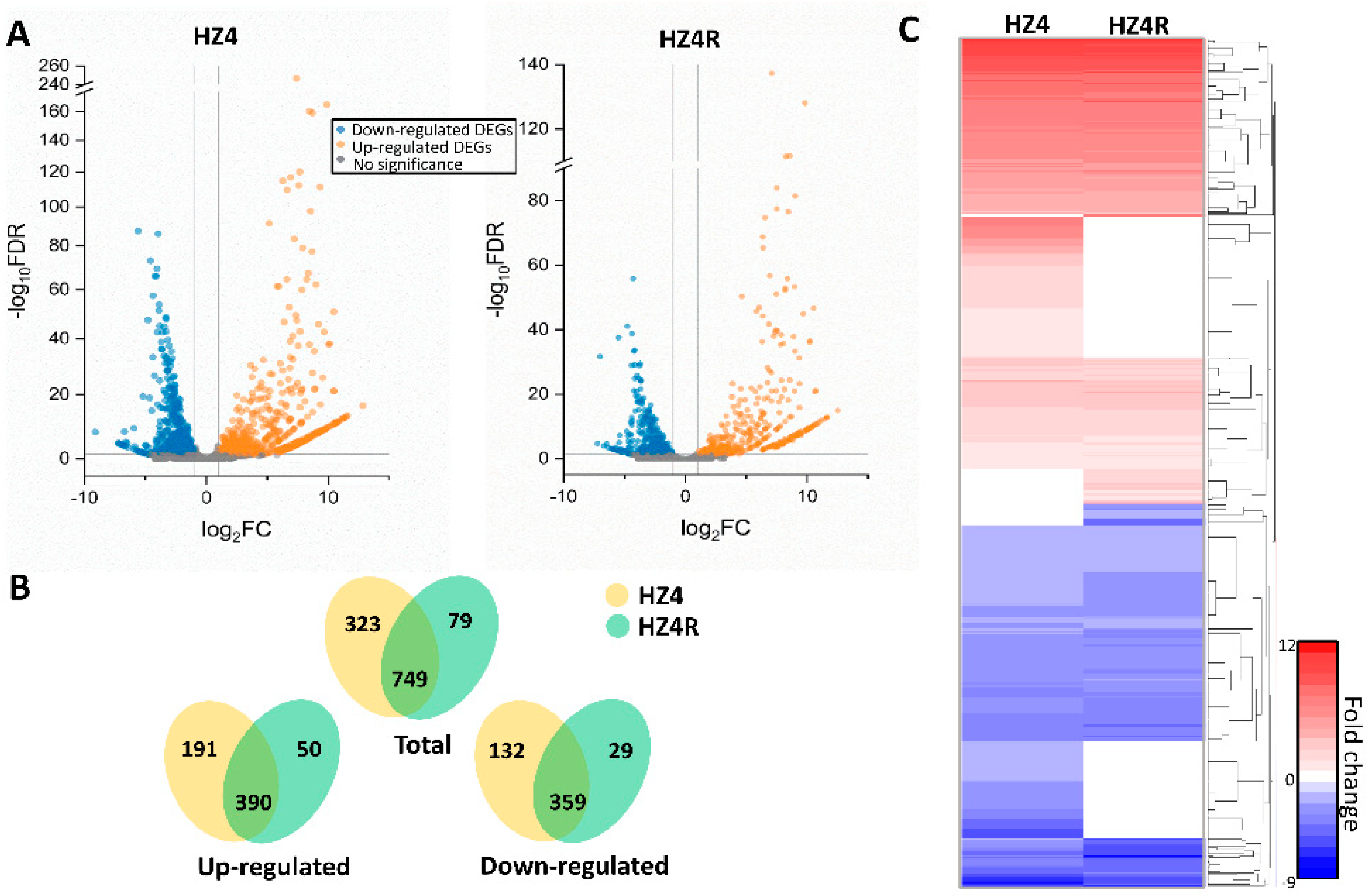
3.1. Global Gene Regulation of S. reilianum during Biotrophy
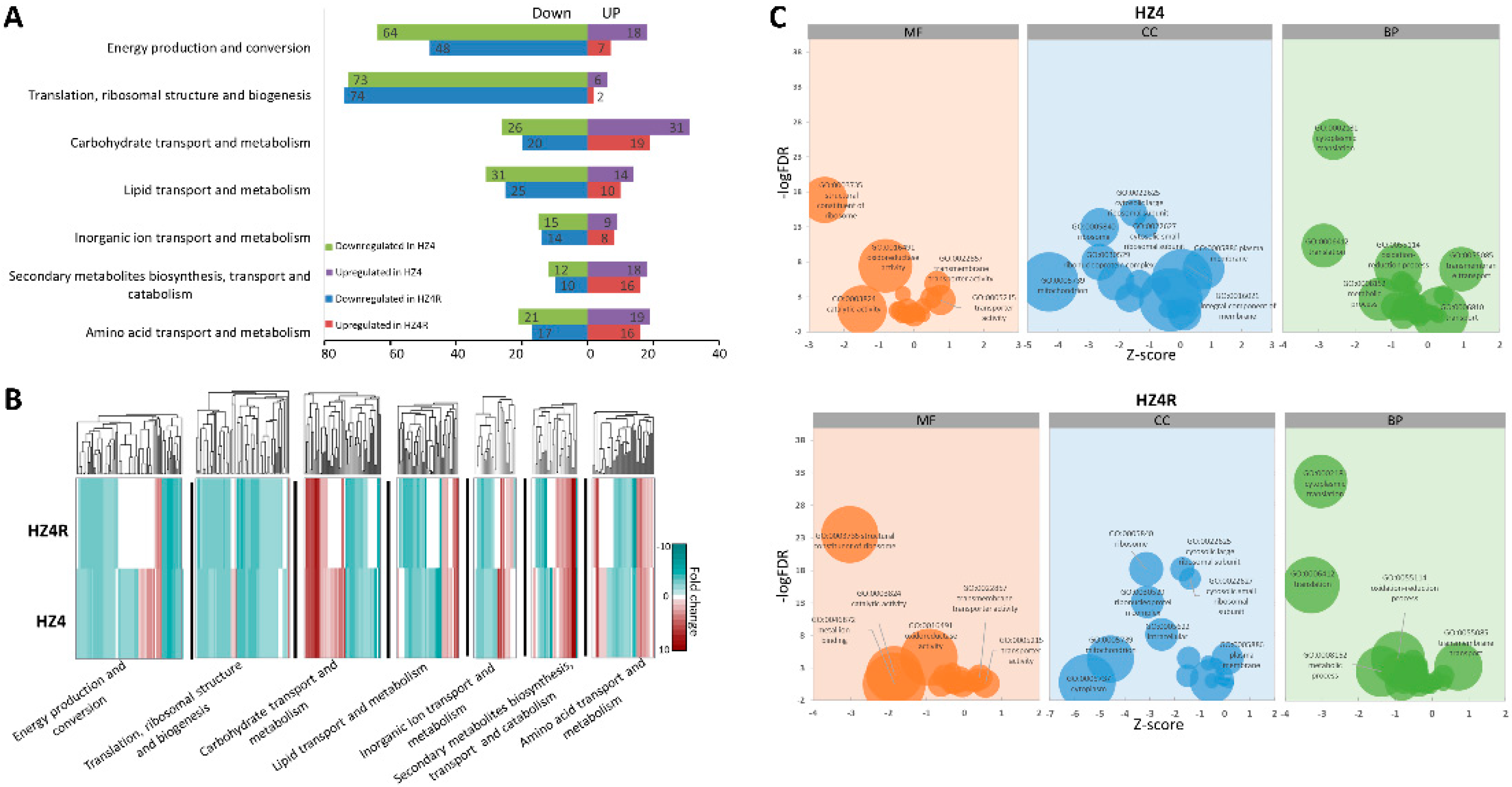
3.2. Functional KOG and GO Terms Enriched with DEGs
3.3. The Secretome of S. reilianum Was Activated during Biotrophy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stromberg, E.L. Head smut of maize, a new disease in Minnesota. Phytopathology 1981, 71, 906. [Google Scholar]
- Jin, Q.; Wang, X.; Wang, Z.; Sha, H.; Li, H.; Song, S. The Epidemiological Factors and Control Tactics of Head Smut in Spring Corn Area of Northeast of China. J. Maize Sci. 2003, 11, 86–87. [Google Scholar]
- Wang, Z.; Jiang, Y.; Wang, L.; Jin, Y.; Li, X.; Shi, H. Research Advance on Head Smut Disease in Maize. J. Maize Sci. 2002, 10, 61–64. [Google Scholar]
- Bernardo, R.; Bourrier, M.; Olivier, J. Generation means analysis of resistance to head smut in maize. Agronomie 1992, 12, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Martinez, C.; Roux, C.; Jauneau, A.; Dargent, R. The biological cycle of Sporisorium reilianum f.sp. zeae: An overview using microscopy. Mycologia 2002, 94, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ye, J.; Wei, L.; Zhang, N.; Xing, Y.; Zuo, W.; Chao, Q.; Tan, G.; Xu, M. Inhibition of the spread of endophytic Sporisorium reilianum renders maize resistance to head smut. Crop J. 2015, 3, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Ghareeb, H.; Becker, A.; Iven, T.; Feussner, I.; Schirawski, J. Sporisorium reilianum Infection Changes Inflorescence and Branching Architectures of Maize. Plant Physiol. 2011, 156, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromberg, E.; Stienstra, W.; Kommedahl, T.; Matyac, C.; Windels, C.; Geadelmann, J. Smut expression and resistance of corn to Sphacelotheca reiliana in Minnesota. Plant Dis. 1984, 68, 880–884. [Google Scholar] [CrossRef]
- Matyac, C. Histological development of Sphacelotheca reiliana on Zea mays. Phytopathology 1985, 75, 924–929. [Google Scholar] [CrossRef]
- Martinez, C.; Roux, C.; Jauneau, A.; Bécard, G.; Dargent, R. Effect of water potential on the development of an haploid strain of Sporisorium reilianum f.sp. zeae. Plant Soil 2003, 251, 65–71. [Google Scholar] [CrossRef]
- Schirawski, J.; Mannhaupt, G.; Münch, K.; Brefort, T.; Schipper, K.; Doehlemann, G.; Stasio, M.D.; Rössel, N.; Mendoza-Mendoza, A.; Pester, D. Pathogenicity Determinants in Smut Fungi Revealed by Genome Comparison. Science 2010, 330, 1546–1548. [Google Scholar] [CrossRef]
- Schuster, M.; Schweizer, G.; Kahmann, R. Comparative analyses of secreted proteins in plant pathogenic smut fungi and related basidiomycetes. Fungal Genet. Biol. 2018, 112, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, G.; Münch, K.; Mannhaupt, G.; Schirawski, J.; Kahmann, R.; Dutheil, J.Y. Positively Selected Effector Genes and Their Contribution to Virulence in the Smut Fungus Sporisorium reilianum. Genome Biol. Evol. 2018, 10, 629–645. [Google Scholar] [CrossRef] [Green Version]
- Ghareeb, H.; Drechsler, F.; Löfke, C.; Teichmann, T.; Schirawski, J. Suppressor of Apical Dominance1 of Sporisorium reilianum Modulates Inflorescence Branching Architecture in Maize and Arabidopsis. Plant Physiol. 2015, 169, 2789–2804. [Google Scholar] [PubMed] [Green Version]
- Mueller, A.N.; Ziemann, S.; Treitschke, S.; Aßmann, D.; Doehlemann, G. Compatibility in the Ustilago maydis–Maize Interaction Requires Inhibition of Host Cysteine Proteases by the Fungal Effector Pit2. PLoS Pathog. 2013, 9, e1003177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misas Villamil, J.C.; Mueller, A.N.; Demir, F.; Meyer, U.; Ökmen, B.; Schulze Hüynck, J.; Breuer, M.; Dauben, H.; Win, J.; Huesgen, P.F.; et al. A fungal substrate mimicking molecule suppresses plant immunity via an inter-kingdom conserved motif. Nat. Commun. 2019, 10, 1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, D.; Agrawal, N.; Ghareeb, H.; Schirawski, J. Screening of Secreted Proteins of Sporisorium reilianum f. sp. zeae for Cell Death Suppression in Nicotiana benthamiana. Front. Plant Sci. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghareeb, H.; Zhao, Y.; Schirawski, J. Sporisorium reilianum possesses a pool of effector proteins that modulate virulence on maize. Mol. Plant Pathol. 2018, 20, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112. [Google Scholar] [CrossRef] [Green Version]
- Lübberstedt, T.; Xia, X.C.; Tan, G.; Liu, X.; Melchinger, A.E. QTL mapping of resistance to Sporisorium reiliana in maize. Theor. Appl. Genet. 1999, 99, 593. [Google Scholar] [CrossRef]
- Li, X.H.; Wang, Z.H.; Gao, S.R.; Shi, H.L.; Zhang, S.H.; Mlc, G.; Li, M.S.; Xie, C.X. Analysis of QTL for resistance to head smut (Sporisorium reiliana) in maize. Field Crops Res. 2008, 106, 148–155. [Google Scholar] [CrossRef]
- Chen, Y.; Chao, Q.; Tan, G.; Zhao, J.; Zhang, M.; Ji, Q.; Xu, M. Identification and fine-mapping of a major QTL conferring resistance against head smut in maize. Theor. Appl. Genet. 2008, 117, 1241. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Chao, Q.; Zhang, N.; Ye, J.; Tan, G.; Li, B.; Xing, Y.; Zhang, B.; Liu, H.; Fengler, K.A. A maize wall-associated kinase confers quantitative resistance to head smut. Nat. Genet. 2015, 47, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tan, G.; Xing, Y.; Wei, L.; Chao, Q.; Zuo, W.; Lübberstedt, T.; Xu, M. Marker-assisted introgression of qHSR1 to improve maize resistance to head smut. Mol. Breed. 2012, 30, 1077–1088. [Google Scholar] [CrossRef]
- Thatcher, L.F.; Gardiner, D.M.; Kazan, K.; Manners, J.M. A highly conserved effector in Fusarium oxysporum is required for full virulence on Arabidopsis. Mol. Plant-Microbe Interact. 2012, 25, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poloni, A.; Schirawski, J. Host specificity in Sporisorium reilianum is determined by distinct mechanisms in maize and sorghum. Mol. Plant Pathol. 2016, 17, 741. [Google Scholar] [CrossRef] [PubMed]
- Lanver, D.; Müller, A.N.; Happel, P.; Schweizer, G.; Haas, F.B.; Franitza, M.; Pellegrin, C.; Reissmann, S.; Altmüller, J.; Rensing, S.A.; et al. The biotrophic development of Ustilago maydis studied by RNAseq analysis. Plant Cell 2018, 30, 300–323. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahmann, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. eLife 2014, 3, e01355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, B.; Zuo, W.; Xing, Y.; Konlasuk, S.; Tan, G.; Zhang, Q.; Ye, J.; Xu, M. Cytological and Molecular Characterization of ZmWAK-Mediated Head-Smut Resistance in Maize. Mol. Plant-Microbe Interact. 2017, 30, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef]
- Beißbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]
- Al-Shahrour, F.; Díaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Zeeberg, B.R.; Feng, W.; Wang, G.; Wang, M.D.; Fojo, A.T.; Sunshine, M.; Narasimhan, S.; Kane, D.W.; Reinhold, W.C.; Lababidi, S.; et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003, 4, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnabas, L.; Ashwin, N.M.R.; Kaverinathan, K.; Trentin, A.R.; Pivato, M.; Sundar, A.R.; Malathi, P.; Viswanathan, R.; Carletti, P.; Arrigoni, G.; et al. In vitro secretomic analysis identifies putative pathogenicity-related proteins of Sporisorium scitamineum—The sugarcane smut fungus. Fungal Biol. 2017, 121, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Draper, J.; Rasmussen, S.; Zubair, H. Metabolite Analysis and Metabolomics in the Study of Biotrophic Interactions between Plants and Microbes. Annu. Plant Rev. Online 2018, 25–59. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; de Wit, P.J.G.M. Fungal Effector Proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Gollin, I.; Rössel, N.; Kahmann, R. The functionally conserved effector Sta1 is a fungal cell wall protein required for virulence in Ustilago maydis. New Phytol. 2020, 227, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, D.; Karina, V.D.L.; Daniela, A.; Daniela, S.; Alexander, H.; Amitabh, M.; David, J.; Regine, K. Pep1, a Secreted Effector Protein of Ustilago maydis, Is Required for Successful Invasion of Plant Cells. PLoS Pathog. 2009, 5, e1000290. [Google Scholar]
- Schuler, D.; Wahl, R.; Wippel, K.; Vranes, M.; Sauer, N. Hxt1, a monosaccharide transporter and sensor required for virulence of the maize pathogen Ustilago maydis. New Phytol. 2015, 206, 1086. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Altegoer, F.; Steinchen, W.; Binnebesel, L.; Schuhmacher, J.; Glatter, T.; Giammarinaro, P.I.; Djamei, A.; Rensing, S.A.; Reissmann, S.; et al. A kiwellin disarms the metabolic activity of a secreted fungal virulence factor. Nature 2019, 565, 650–653. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Zhang, N.; Zhang, Q.; Xu, Q.; Zhong, T.; Zhang, K.; Xu, M. Transcriptome Profiles of Sporisorium reilianum during the Early Infection of Resistant and Susceptible Maize Isogenic Lines. J. Fungi 2021, 7, 150. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020150
Zhang B, Zhang N, Zhang Q, Xu Q, Zhong T, Zhang K, Xu M. Transcriptome Profiles of Sporisorium reilianum during the Early Infection of Resistant and Susceptible Maize Isogenic Lines. Journal of Fungi. 2021; 7(2):150. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020150
Chicago/Turabian StyleZhang, Boqi, Nan Zhang, Qianqian Zhang, Qianya Xu, Tao Zhong, Kaiyue Zhang, and Mingliang Xu. 2021. "Transcriptome Profiles of Sporisorium reilianum during the Early Infection of Resistant and Susceptible Maize Isogenic Lines" Journal of Fungi 7, no. 2: 150. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020150