
Genome-Wide Identification and Analysis of Nilaparvata lugens microRNAs during Challenge with the Entomopathogenic Fungus Metarhizium anisopliae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Insect and Sample Collection
2.2. Small RNA (sRNA) Extraction and Sequencing
2.3. N. lugens miRNA Prediction
2.4. Differential Expression Analysis and Functional Annotation
2.5. Validation by RT-qPCR
3. Results
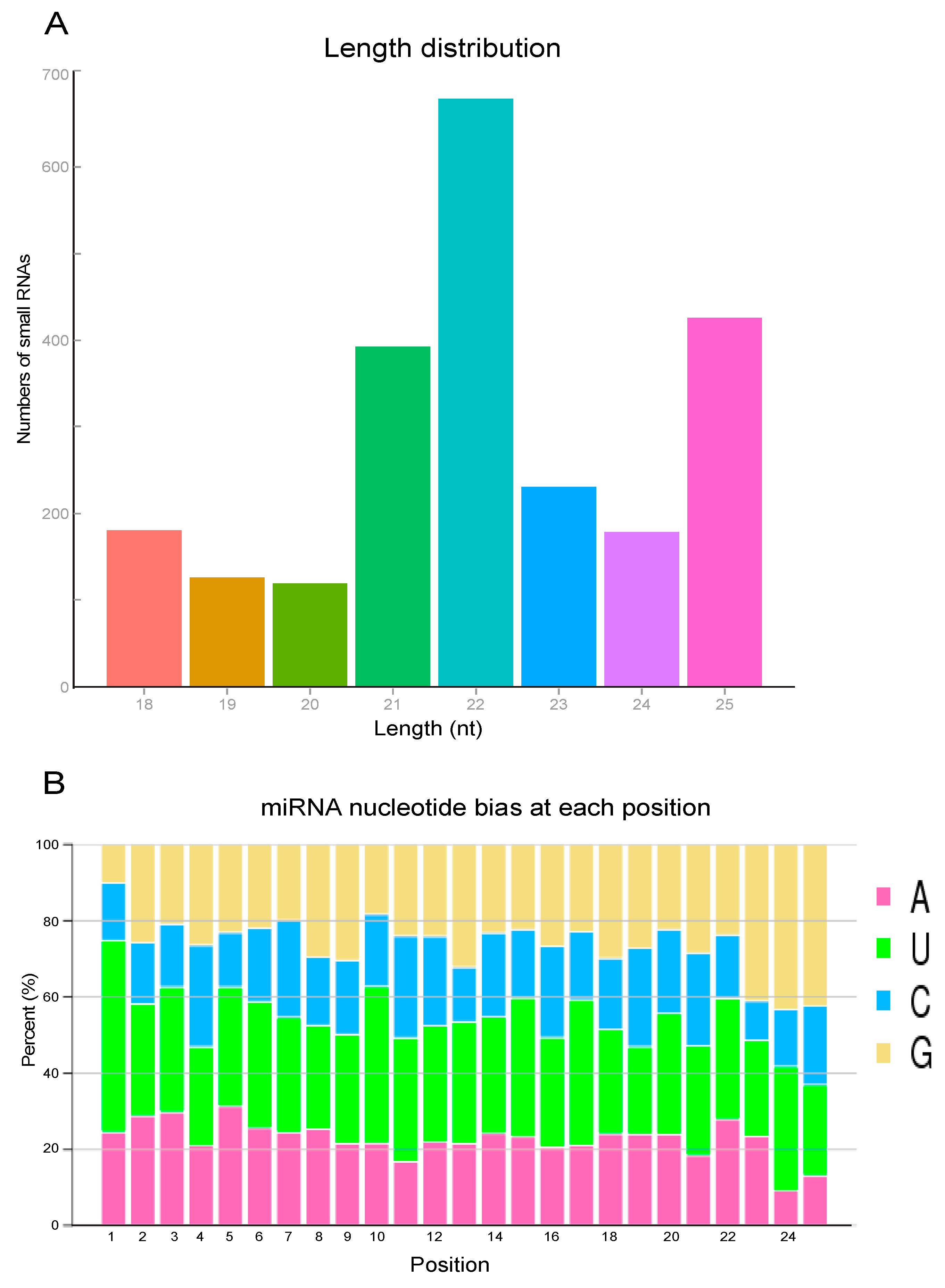
3.1. Overall and Size Distribution of N. lugens Total miRNAs
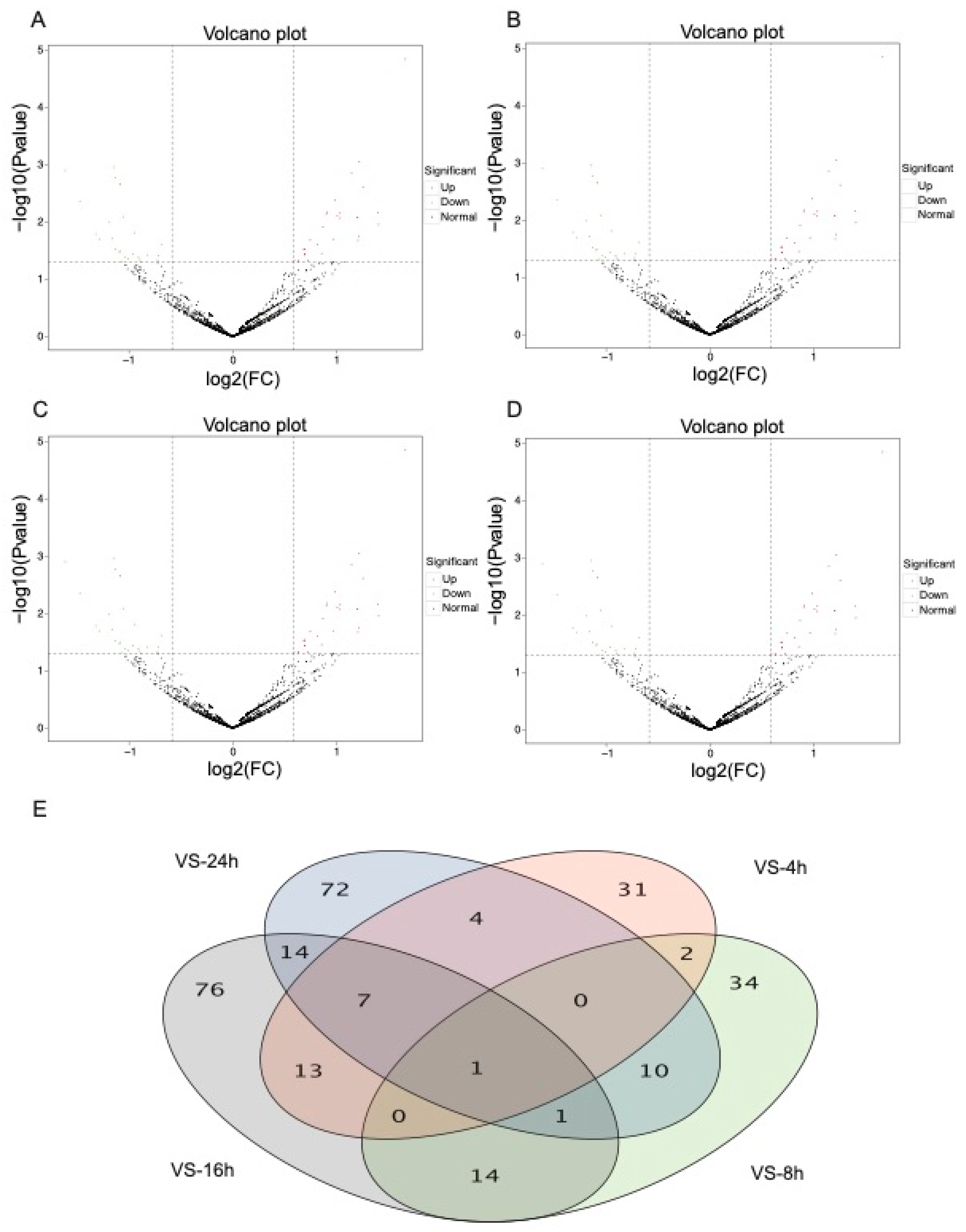
3.2. DEmiRNA Analysis of N. lugens after Fungal Treatment
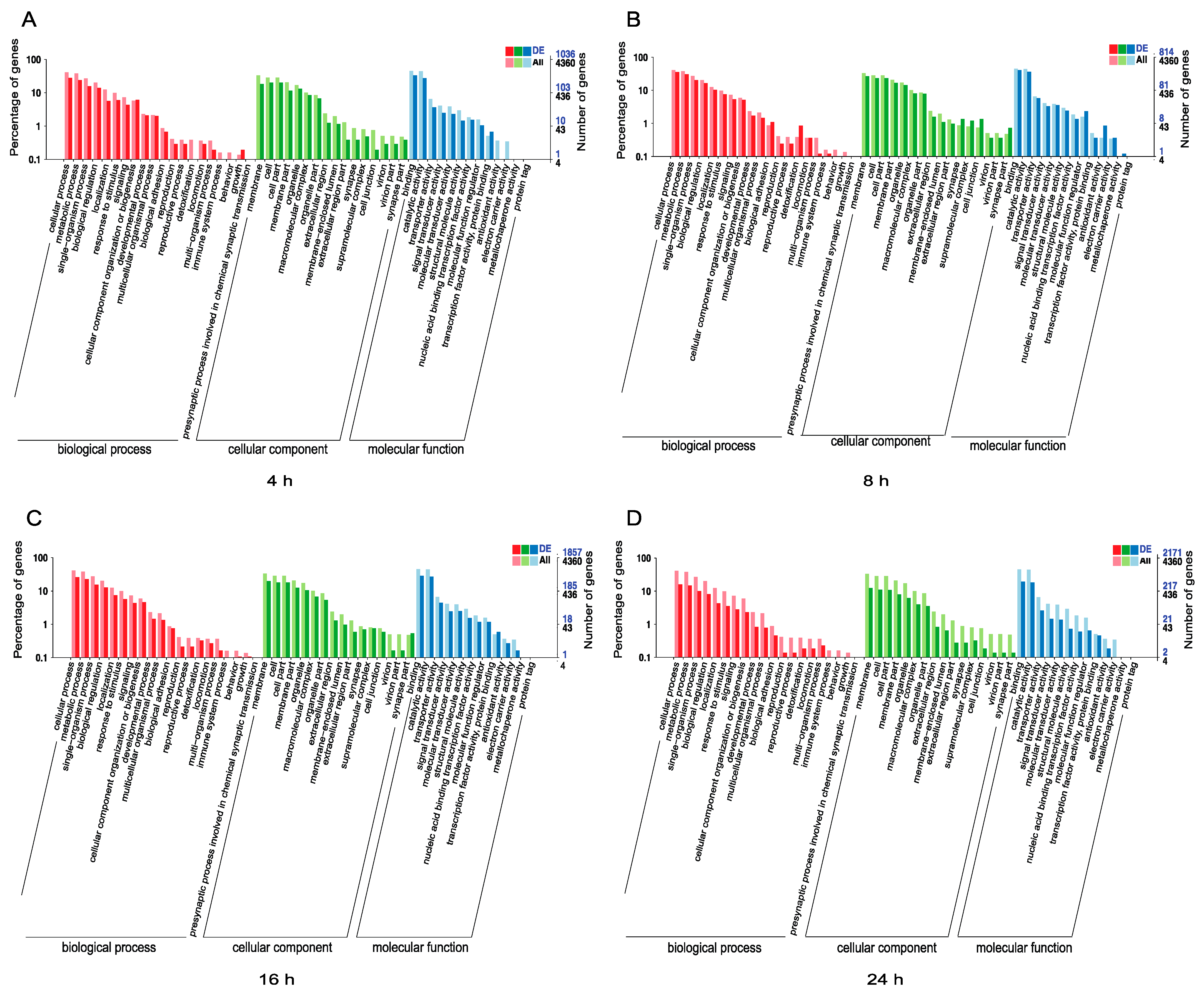
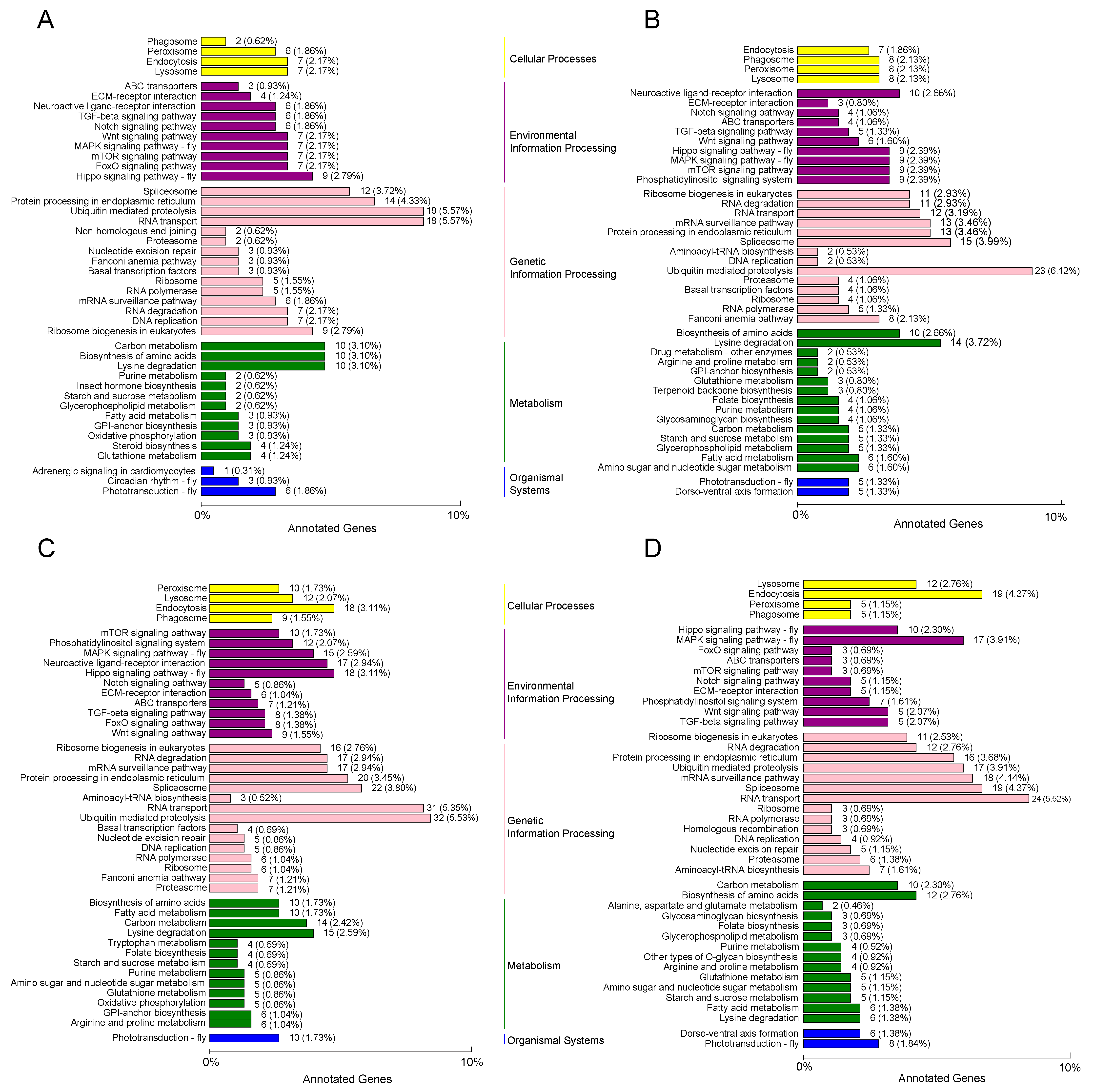
3.3. Functional Analysis of DEmiRNAs in N. lugens
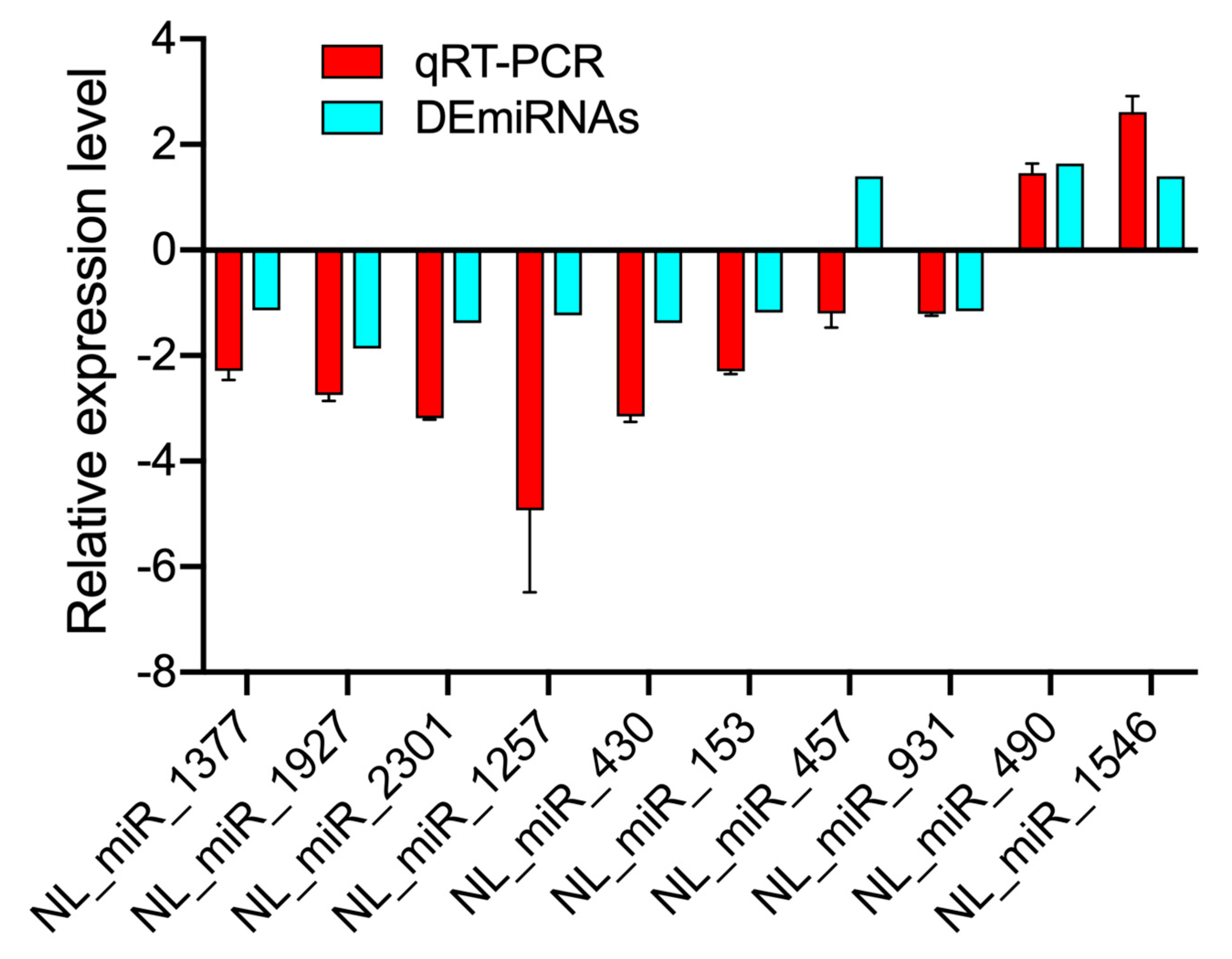
3.4. Validation of DEmiRNAs by RT-qPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lou, Y.G.; Zhang, G.R.; Zhang, W.Q.; Hu, Y.; Zhang, J. Biological control of rice insect pests in China. Biol. Control. 2013, 67, 8–20. [Google Scholar] [CrossRef]
- Cheng, J. Rice planthopper problems and relevant causes in China. Rice 2009, 1, 1–32. [Google Scholar]
- Wang, Y.; Tang, M.; Hao, P.; Yang, Z.; Zhu, L.; He, G. Penetration into rice tissues by brown planthopper and fine structure of the salivary sheaths. Entomol. Exp. Appl. 2010, 129, 295–307. [Google Scholar] [CrossRef]
- Hibino, H. Biology and epidemiology of rice viruses. Annu. Rev. Phytopathol. 1996, 34, 249–274. [Google Scholar] [CrossRef]
- Desneux, N.; Decourtye, A.; Delpuech, J.M. The sublethal effects of pesticides on beneficial arthropods. Annu. Rev. Entomol. 2007, 52, 81–106. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Liu, Y.; Hou, M.; Babendreier, D.; Zhang, F.; Song, K. Toxicity of insecticides targeting rice planthoppers to adult and immature stages of Trichogramma chilonis (Hymenoptera: Trichogrammatidae). J. Econ. Entomol. 2015, 108, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Zeng, B.; Zheng, C.; Mu, X.C.; Zhang, Y.; Hu, J.; Zhang, S.; Gao, C.F.; Shen, J.L. The evolution of insecticide resistance in the brown planthopper (Nilaparvata lugens Stål) of China in the period 2012–2016. Sci. Rep. 2018, 8, 4586. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hu, R.; Rozelle, S.; Pray, C. Insect-resistant GM rice in farmers’ fields: Assessing productivity and health effects in China. Science 2005, 308, 688–690. [Google Scholar] [CrossRef] [Green Version]
- Estruch, J.J.; Carozzi, N.B.; Desai, N.; Duck, N.B.; Warren, G.W.; Koziel, M.G. Transgenic plants: An emerging approach to pest control. Nat. Biotechnol. 1997, 15, 137–141. [Google Scholar] [CrossRef]
- Price, D.R.; Gatehouse, J.A. RNAi-mediated crop protection against insects. Trends Biotechnol. 2008, 26, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Hamiduzzaman, M.M.; Sinia, A.; Guzman-Novoa, E.; Goodwin, P.H. Entomopathogenic fungi as potential biocontrol agents of the ecto-parasitic mite, Varroa destructor, and their effect on the immune response of honey bees (Apis mellifera L.). J. Invertebr. Pathol. 2012, 111, 237–243. [Google Scholar] [CrossRef]
- Zimmermann, G. The entomopathogenic fungus Metarhizium anisopliae and its potential as a biocontrol agent. Pest Manag. Sci. 2010, 37, 375–379. [Google Scholar] [CrossRef]
- Jiang, W.; Peng, Y.; Ye, J.; Wen, Y.; Liu, G.; Xie, J. Effects of the entomopathogenic fungus Metarhizium anisopliae on the mortality and immune response of Locusta migratoria. Insects 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, S.J.; Baker, D.K.; Leemon, D.M. Development of mycoinsecticide formulations with Beauveria bassiana and Metarhizium anisopliae for the control of lesser mealworm, Alphitobius diaperinus, in chicken broiler houses. Biocontrol 2019, 64, 489–500. [Google Scholar] [CrossRef]
- Bordalo, M.D.; Gravato, C.; Beleza, S.; Campos, D.; Lopes, I.; Pestana, J.L.T. Lethal and sublethal toxicity assessment of Bacillus thuringiensis var. israelensis and Beauveria bassiana based bioinsecticides to the aquatic insect Chironomus riparius. Sci. Total Environ. 2020, 698, 134155. [Google Scholar] [CrossRef]
- Fite, T.; Tefera, T.; Negeri, M.; Damte, T.; Sori, W. Evaluation of Beauveria bassiana, Metarhizium anisopliae, and Bacillus thuringiensis for the management of Helicoverpa armigera (Hubner) (Lepidoptera: Noctuidae) under laboratory and field conditions. Biocontrol Sci. Technol. 2019, 30, 278–295. [Google Scholar] [CrossRef]
- Knols, B.G.; Bukhari, T.; Farenhorst, M. Entomopathogenic fungi as the next-generation control agents against malaria mosquitoes. Future Microbiol. 2010, 5, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Xia, Y. Integration of an insecticidal scorpion toxin (BjαIT) gene into Metarhizium acridum enhances fungal virulence towards Locusta migratoria manilensis. Pest Manag. Sci. 2015, 71, 58–64. [Google Scholar] [CrossRef]
- Fan, Y.; Borovsky, D.; Hawkings, C.; Ortiz-Urquiza, A.; Keyhani, N.O. Exploiting host molecules to augment mycoinsecticide virulence. Nat. Biotechnol. 2012, 30, 35–37. [Google Scholar] [CrossRef]
- Pan, C.Y.; Cai, Y.J.; Li, T.C.; Zhang, W.Q. Control effect of Metarhizium anisopliae and its mixture with dsRNA on the brown planthopper. J. Environ. Entomol. 2016, 38, 1071–1077. [Google Scholar]
- Tang, J.F.; Liu, X.Y.; Ding, Y.C.; Jiang, W.J.; Xie, J.Q. Evaluation of Metarhizium anisopliae for rice planthopper control and its synergy with selected insecticides. Crop Prot. 2019, 121, 132–138. [Google Scholar] [CrossRef]
- Peng, Y.; Tang, J.; Hong, M.; Xie, J. Suppression of rice planthopper populations by the entomopathogenic fungus Metarhizium anisopliae without affecting the rice microbiota. Appl. Environ. Microb. 2020, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Xie, J.; Guo, R.; Keyhani, N.O.; Zeng, D.; Yang, P.; Xia, Y. Long-term field evaluation and large-scale application of a Metarhizium anisopliae strain for controlling major rice pests. J. Pest Sci. 2021, 1–12. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, E.G. MicroRNAs: Hidden in the Genome. Curr. Biol. 2002, 12, R138–R140. [Google Scholar] [CrossRef] [Green Version]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 296, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.; Chen, X. The regulation of genes and genomes by small RNAs. Development 2007, 134, 1635–1641. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Vilcinskas, A. Development and immunity-related microRNAs of the lepidopteran model host Galleria mellonella. BMC Genom. 2014, 15, 705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, T.; Tang, G. Identification and characterization of conserved and novel miRNAs in different development stages of Atrijuglans hetaohei Yang (Lepidoptera: Gelechioidea). J. Asia-Pac. Entomol. 2018, 21, 9–18. [Google Scholar] [CrossRef]
- Ylla, G.; Piulachs, M.-D.; Belles, X. Comparative analysis of miRNA expression during the development of insects of different metamorphosis modes and germ-band types. BMC Genom. 2017, 18, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Tariq, K.; Xie, J.; Zhang, H. Identification and characterization of sex-biased MicroRNAs in Bactrocera dorsalis (Hendel). PLoS ONE 2016, 11, e0159591. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, F.; Coates, B.; Zhang, Y.; Zhou, X.; Cheng, D. Comparative profiling of microRNAs in the winged and wingless English grain aphid, Sitobion avenae (F.) (Homoptera: Aphididae). Sci. Rep. 2016, 6, 35668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zhang, J.; Zhan, A.; Wang, Y.; Ma, X.; Jie, W.; Cao, Z.; Omar, M.A.A.; He, K.; Li, F. Identification and analysis of MicroRNAs associated with wing polyphenism in the brown planthopper, Nilaparvata lugens. Int. J. Mol. Sci. 2020, 21, 9754. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L.; Sharp, P.A. MicroRNAfunctions in stress responses. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Galiano, M.J.; Sentandreu, V.; Martínez-Ramírez, A.C.; Rausell, C.; Real, M.D.; Camañes, G.; Ruiz-Rivero, O.; Crespo-Salvador, O.; García-Robles, I. Stress associated microRNAs in Solanum lycopersicum by high-throughput sequencing. Genes 2019, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakeel, M.; Xu, X.; Xu, J.; Li, S.; Yu, J.; Zhou, X.; Xu, X.; Hu, Q.; Yu, X.; Jin, F. Genome-Wide identification of destruxin A-responsive immunity-related microRNAs in diamondback moth, Plutella xylostella. Front. Immunol. 2018, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Etebari, K.; Hussain, M.; Asgari, S. Identification of microRNAs from Plutella xylostella larvae associated with parasitization by Diadegma semiclausum. Insect Biochem. Mol. Biol. 2013, 43, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Piyaphongkul, J.; Pritchard, J.; Bale, J. Heat stress impedes development and lowers fecundity of the brown planthopper Nilaparvata lugens. PLoS ONE 2012, 7, e47413. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Zhu, Y.C.; Ma, C.; Shen, J. Susceptibility to neonicotinoids and risk of resistance development in the brown planthopper, Nilaparvata lugens (Stål) (Homoptera: Delphacidae). Pest Manag. Sci. 2010, 64, 1278–1284. [Google Scholar] [CrossRef]
- Zha, W.; Zhou, L.; Li, S.; Liu, K.; Yang, G.; Chen, Z.; Liu, K.; Xu, H.; Li, P.; Hussain, S.; et al. Characterization and comparative profiling of the small RNA transcriptomes in the Hemipteran insect Nilaparvata lugens. Gene 2016, 595, 83–91. [Google Scholar] [CrossRef]
- Xue, J.; Zhou, X.; Zhang, C.X.; Yu, L.L.; Fan, H.W.; Wang, Z.; Xu, H.J.; Xi, Y.; Zhu, Z.R.; Zhou, W.W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids. Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Vikram, A.; Bell, G.W.; Jin-Wu, N.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Gemome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Liao, X.; Xu, P.F.; Gong, P.P.; Wan, H.; Li, J.H. Current susceptibilities of brown planthopper Nilaparvata lugens to triflumezopyrim and other frequently used insecticides in China. Insect Sci. 2021, 28, 115–126. [Google Scholar] [CrossRef]
- Whitehorn, P.R.; O’Connor, S.; Wackers, F.L.; Goulson, D. Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science 2012, 336, 351–352. [Google Scholar] [CrossRef] [Green Version]
- Daza, F.F.F.; Roman, G.R.; Rodriguez, M.V.; Vargas, I.A.G.; Heano, H.C.; Cereda, M.P.; Mulet, R.A.C. Spores of Beauveria bassiana and Trichoderma lignorum as a bioinsecticide for the control of Atta cephalotes. Biol. Res. 2019, 52, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, J.; Keyhani, N.O.; Zhang, Z.; Li, S.; Xia, Y. Comparative transcriptomic analysis of immune responses of the migratory locust, Locusta migratoria, to challenge by the fungal insect pathogen, Metarhizium acridum. BMC Genom. 2015, 16, 867. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Annu. Rev. Entomol. 2017, 62, 73. [Google Scholar] [CrossRef]
- Lu, H.L.; St Leger, R.J. Insect immunity to entomopathogenic fungi. Adv. Genet. 2016, 94, 251–285. [Google Scholar]
- Dan, H. Drosophila immunity: Paths and patterns. Curr. Opin. Immunol. 2003, 15, 12–19. [Google Scholar]
- Duressa, T.F.; Vanlaer, R.; Huybrechts, R. Locust cellular defense against infections: Sites of pathogen clearance and hemocyte proliferation. Dev. Comp. Immunol. 2015, 48, 244–253. [Google Scholar] [CrossRef]
- Kounatidis, I.; Ligoxygakis, P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012, 2, 120075. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.S.; St Leger, R.J. The MAD1 adhesin of Metarhizium anisopliae links adhesion with blastospore production and virulence to insects, and the MAD2 adhesin enables attachment to plants. Eukaryot. Cell 2007, 6, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cao, Y.; Xia, Y.; Liu, F. Wright-Giemsa staining to observe phagocytes in Locusta migratoria infected with Metarhizium acridum. J. Invertebr. Pathol. 2016, 139, 19–24. [Google Scholar] [CrossRef]
- Keren, A.; Tamir, Y.; Bengal, E.; Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.; Wang, Z.; Wang, Q.; Pan, Y. The molecular evolutionary patterns of the insulin/FOXO signaling pathway. Evol. Bioinform. 2013, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Xu, X.; Li, S.; Wang, S.; Xu, X.; Zhou, X.; Yu, J.; Yu, X.; Shakeel, M.; Jin, F. Genome-wide profiling of Plutella xylostella immunity-related miRNAs after Isaria fumosorosea infection. Front. Physiol. 2017, 8, 1054. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, J.; Peng, Y.; Xia, Y. Genome-Wide Identification and Analysis of Nilaparvata lugens microRNAs during Challenge with the Entomopathogenic Fungus Metarhizium anisopliae. J. Fungi 2021, 7, 295. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040295
Xie J, Peng Y, Xia Y. Genome-Wide Identification and Analysis of Nilaparvata lugens microRNAs during Challenge with the Entomopathogenic Fungus Metarhizium anisopliae. Journal of Fungi. 2021; 7(4):295. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040295
Chicago/Turabian StyleXie, Jiaqin, Yifan Peng, and Yuxian Xia. 2021. "Genome-Wide Identification and Analysis of Nilaparvata lugens microRNAs during Challenge with the Entomopathogenic Fungus Metarhizium anisopliae" Journal of Fungi 7, no. 4: 295. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040295