
Haplotype-Resolved Genome Analyses Reveal Genetically Distinct Nuclei within a Commercial Cultivar of Lentinula edodes

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. L. edodes Strains and DNA Preparation
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation
2.4. Comparative Genomic Analysis
2.5. Identification of Mat-Genes
2.6. Data Availability and Image Analysis
3. Results
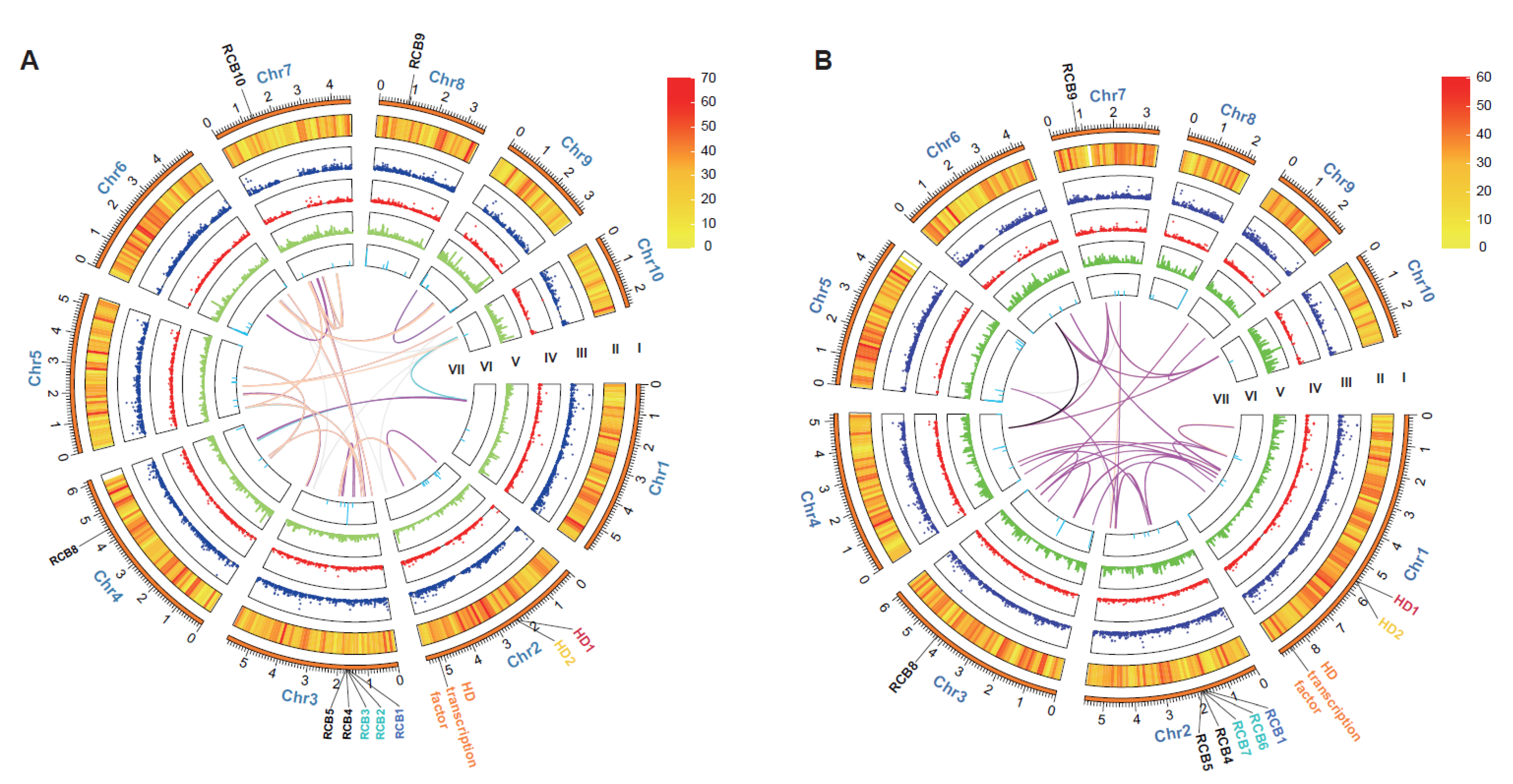
3.1. Genome Sequencing, Assembly, and Annotation
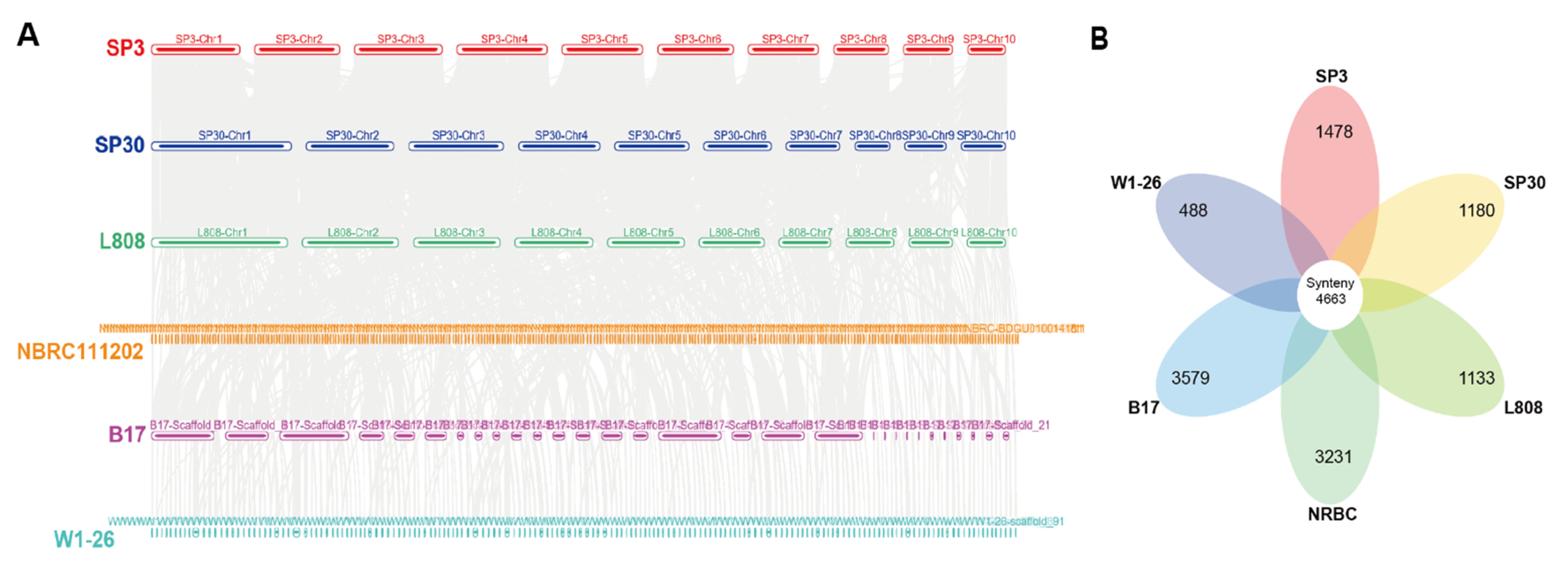
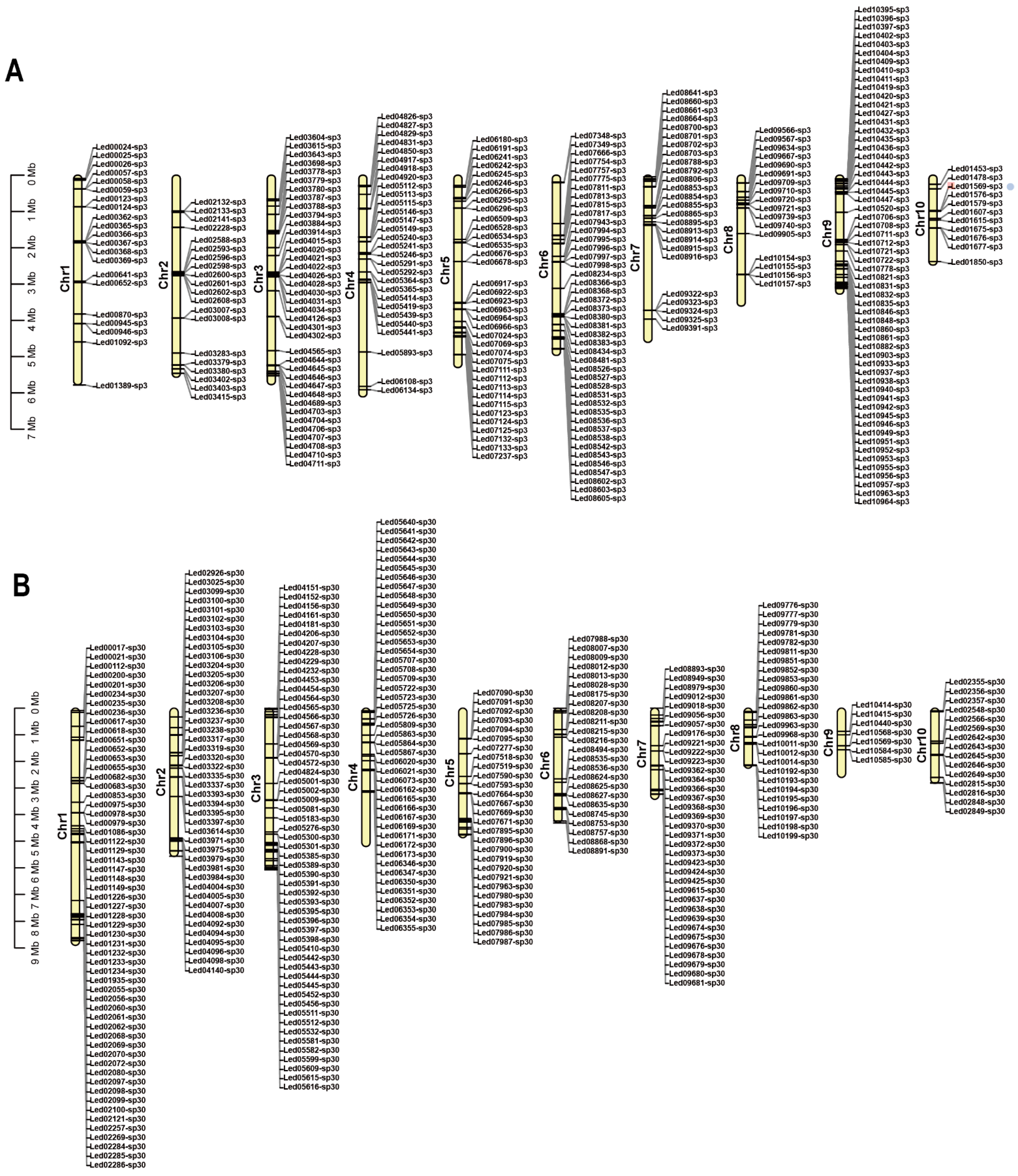
3.2. Collinearity Analysis of L. edodes Strains
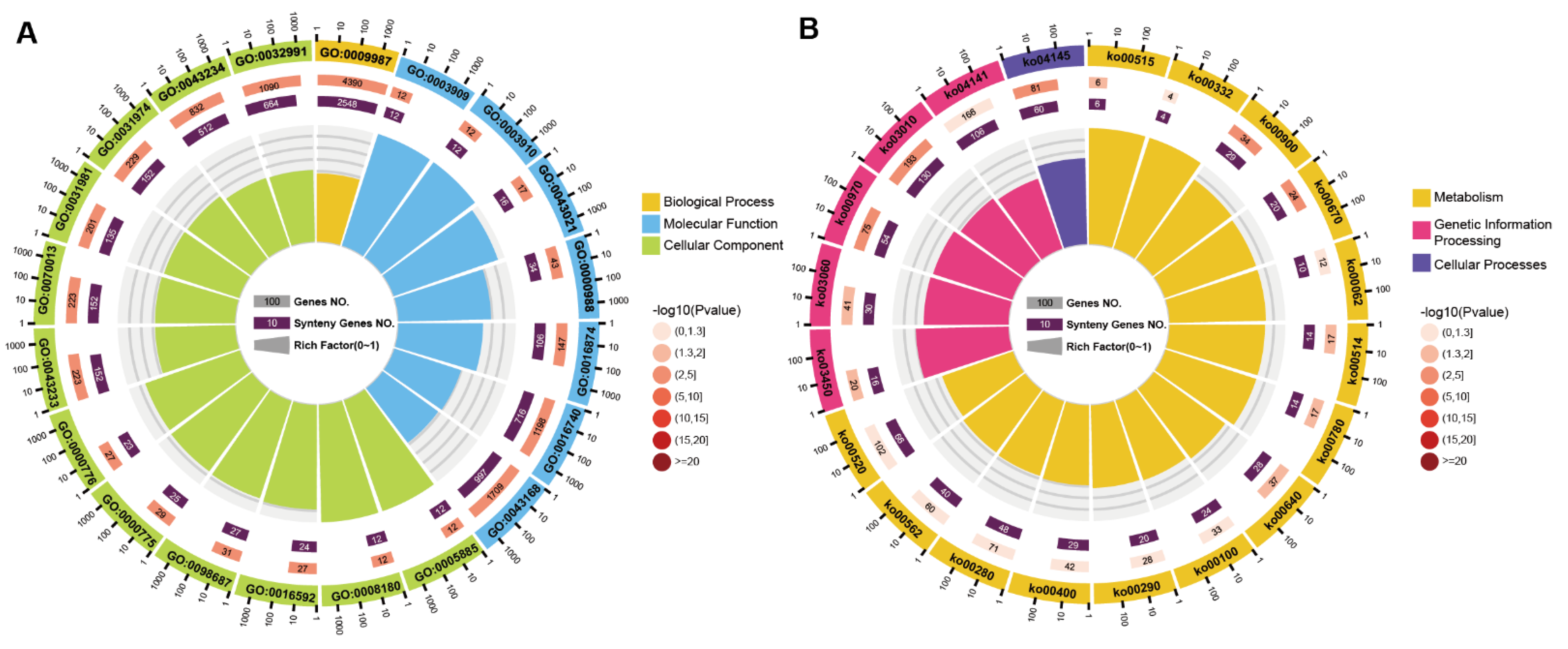
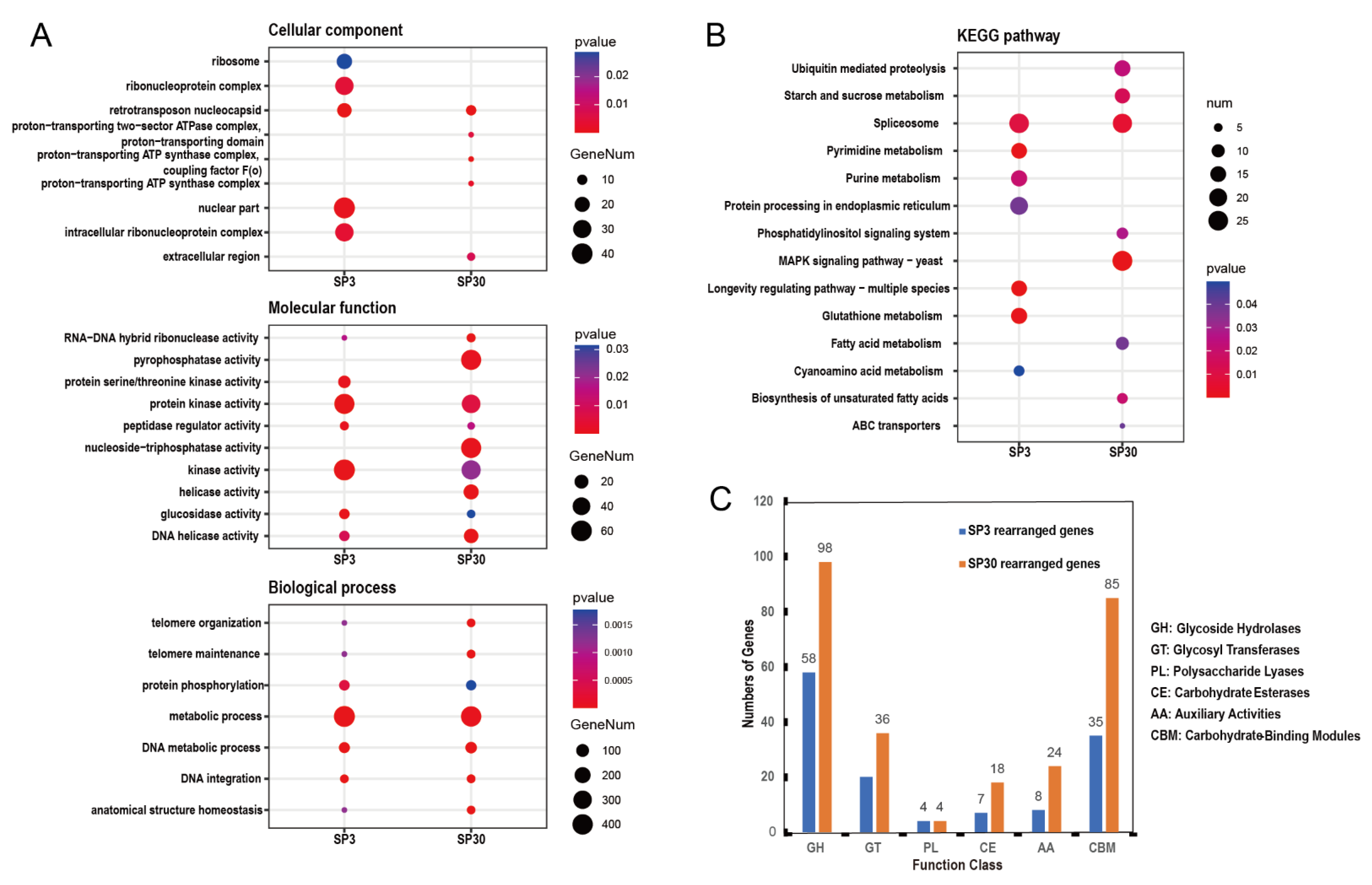
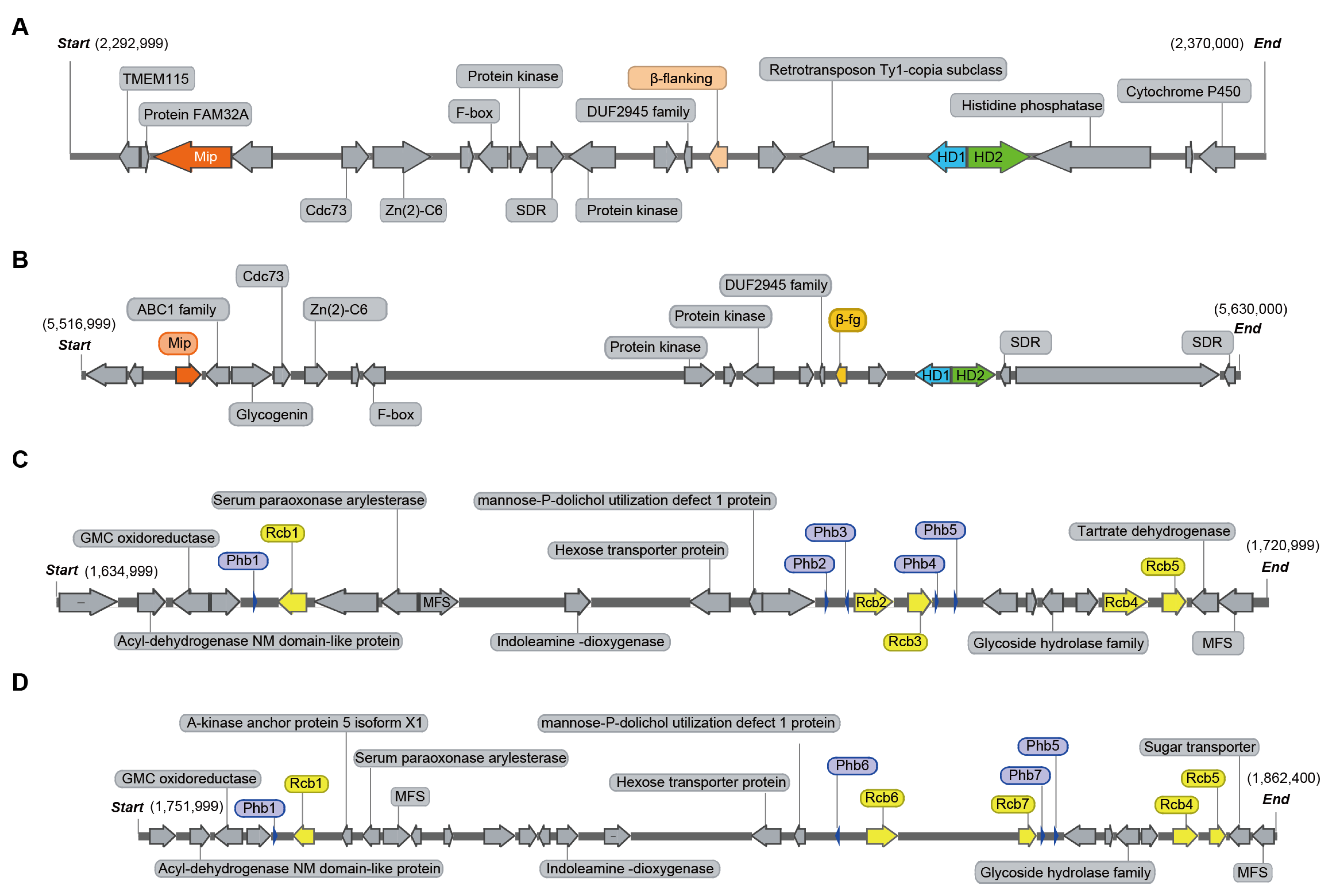
3.3. Comparative Analysis of SP3 and SP30
3.4. Analysis of Mating-Type Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, D.; Gao, Q.; Rong, C.; Liu, Y.; Song, S.; Yu, Q.; Zhou, K.; Liao, Y. Comparative transcriptome analysis of abnormal cap and healthy fruiting bodies of the edible mushroom Lentinula edodes. Fungal Genet. Biol. 2021, 156, 103614. [Google Scholar] [CrossRef]
- Yan, D.; Liu, Y.; Rong, C.; Song, S.; Zhao, S.; Qin, L.; Wang, S.; Gao, Q. Characterization of brown film formed by Lentinula edodes. Fungal Biol. 2020, 124, 135–143. [Google Scholar] [CrossRef]
- Zhao, S.; Gao, Q.; Rong, C.; Wang, S.; Zhao, Z.; Liu, Y.; Xu, J. Immunomodulatory effects of edible and medicinal mushrooms and their bioactive immunoregulatory products. J. Fungi 2020, 6, 269. [Google Scholar] [CrossRef]
- Chang, S.; Miles, P. Historical record of the early cultivation of Lentinus in China. Mushroom J. Trop. 1987, 7, 31–37. [Google Scholar]
- Zhang, J.; Shen, N.; Li, C.; Xiang, X.; Liu, G.; Gui, Y.; Patev, S.; Hibbett, D.S.; Barry, K.; Andreopoulos, W.; et al. Population genomics provides insights into the genetic basis of adaptive evolution in the mushroom-forming fungus Lentinula edodes. J. Adv. Res. 2021, in press. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, X.; Liu, W. De novo transcriptomic analysis during Lentinula edodes fruiting body growth. Gene 2018, 641, 326–334. [Google Scholar] [CrossRef]
- Wang, G.; Chen, L.; Tang, W.; Wang, Y.; Zhang, Q.; Wang, H.; Zhou, X.; Wu, H.; Guo, L.; Dou, M.; et al. Identifying a melanogenesis-related candidate gene by a high-quality genome assembly and population diversity analysis in Hypsizygus marmoreus. J. Genet. Genom. 2021, 48, 75–87. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Shim, D.; Park, S.-G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.-S.; Ro, H.-S.; Kim, M.; Ryoo, R.; Rhee, S.-K.; et al. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Zhou, Q.; Tang, D.; Huang, W.; Yang, Z.; Zhang, Y.; Hamilton, J.P.; Visser, R.G.F.; Bachem, C.W.B.; Robin Buell, C.; Zhang, Z.; et al. Haplotype-resolved genome analyses of a heterozygous diploid potato. Nat. Genet. 2020, 52, 1018–1023. [Google Scholar] [CrossRef]
- Gao, Q.; Yan, D.; Wang, D.; Gao, S.; Zhao, S.; Wang, S.; Liu, Y. Variations in nuclear number and size in vegetative hyphae of the edible mushroom Lentinula edodes. Front. Microbiol. 2019, 10, 1987. [Google Scholar] [CrossRef]
- Vollger, M.R.; Logsdon, G.A.; Audano, P.A.; Sulovari, A.; Porubsky, D.; Peluso, P.; Wenger, A.M.; Concepcion, G.T.; Kronenberg, Z.N.; Munson, K.M.; et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet. 2020, 84, 125–140. [Google Scholar] [CrossRef]
- Zhao, J.; Chang, S.-T. Monokaryotization by protoplasting heterothallic species of edible mushrooms. World J. Microbiol. Biotechnol. 1993, 9, 538–543. [Google Scholar] [CrossRef]
- Manicom, B.; Bar-Joseph, M.; Rosner, A.; Vigodsky-Haas, H.; Kotze, J. Potential applications of random DNA probes and restriction fragment length polymorphisms in the taxonomy of the Fusaria. Phytopathology 1987, 77, 669–672. [Google Scholar] [CrossRef]
- Yang, X.; Liu, D.; Liu, F.; Wu, J.; Zou, J.; Xiao, X.; Zhao, F.; Zhu, B. HTQC: A fast quality control toolkit for Illumina sequencing data. BMC Bioinform. 2013, 14, 33. [Google Scholar] [CrossRef]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Chen, N. Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Jurka, J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000, 16, 418–420. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. 1), i351–i358. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35 (Suppl. 2), W265–W268. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34 (Suppl. 2), W435–W439. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33 (Suppl. 2), W465–W467. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32 (Suppl. 2), W309–W312. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.-R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2007, 36 (Suppl. 1), D281–D288. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chiaromonte, F.; Yap, V.; Miller, W. Scoring pairwise genomic sequence alignments. In Biocomputing 2002; World Scientific: Singapore, 2001; pp. 115–126. [Google Scholar]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Ayudhya, S.P.N.; Sugimura, S.; Ozaki, Y.; Aimi, T.; Shimomura, N. Three-dimensional analysis of ectomycorrhizas formed in Pinus thunbergii roots inoculated with Rhizopogon roseolus. Mushroom Sci. Biotechnol. 2019, 27, 93–97. [Google Scholar]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Wei, L.; Jiang, T.; Xuan, J. A New Mushroom Variety Liaofu No. 4; Breeding Technical Report; 2014; pp. 172–178. [Google Scholar]
- Ha, B.; Kim, S.; Kim, M.; Moon, Y.J.; Song, Y.; Ryu, J.-S.; Ryu, H.; Ro, H.-S. Diversity of A mating type in Lentinula edodes and mating type preference in the cultivated strains. J. Microbiol. 2018, 56, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J. Co-evolution of plant LTR-retrotransposons and their host genomes. Protein Cell 2013, 4, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Ongaro, L.; Scliar, M.O.; Flores, R.; Raveane, A.; Marnetto, D.; Sarno, S.; Gnecchi-Ruscone, G.A.; Alarcón-Riquelme, M.E.; Patin, E.; Wangkumhang, P.; et al. The genomic impact of european colonization of the Americas. Curr. Biol. CB 2019, 29, 3974–3986.e4. [Google Scholar] [CrossRef] [PubMed]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef]
- Gehrmann, T.; Pelkmans, J.F.; Ohm, R.A.; Vos, A.M.; Sonnenberg, A.S.M.; Baars, J.J.P.; Wösten, H.A.B.; Reinders, M.J.T.; Abeel, T. Nucleus-specific expression in the multinuclear mushroom-forming fungus Agaricus bisporus reveals different nuclear regulatory programs. Proc. Natl. Acad. Sci. USA 2018, 115, 4429–4434. [Google Scholar] [CrossRef]
- Sonnenberg, A.S.M.; Gao, W.; Lavrijssen, B.; Hendrickx, P.; Sedaghat-Tellgerd, N.; Foulongne-Oriol, M.; Kong, W.-S.; Schijlen, E.G.W.M.; Baars, J.J.P.; Visser, R.G.F. A detailed analysis of the recombination landscape of the button mushroom Agaricus bisporus var. bisporus. Fungal Genet. Biol. 2016, 93, 35–45. [Google Scholar] [CrossRef]
- Vidal-Diez de Ulzurrun, G.; Lee, Y.-Y.; Stajich, J.E.; Schwarz, E.M.; Hsueh, Y.-P. Genomic analyses of two Italian oyster mushroom Pleurotus pulmonarius strains. G3 Genes Genomes Genet. 2020, 11, jkaa007. [Google Scholar] [CrossRef]
- Sun, S.; Xu, J. Chromosomal rearrangements between serotype A and D strains in Cryptococcus neoformans. PLoS ONE 2009, 4, e5524. [Google Scholar] [CrossRef]
- Ma, X.-L.; Wang, Z.-Y.; Nie, L.-M.; Wang, H.-L.; Zhang, Q.; Wang, J.-P. Comparative genomics studies of homoeologous chromosomes pairs concerted evolution in grass genome. J. Hebei United Univ. 2014. [Google Scholar]
- Bartnicki-Garcia, S. Fundamental Aspects of Hyphal Morphogenesis, Microbe Differentiation 23rd Symposium of the Society of General Microbiology; Cambridge University Press: Cambridge, UK, 1973; pp. 245–267. [Google Scholar]
- Riquelme, M.; Roberson, R.W.; Sánchez-León, E. 3 Hyphal tip growth in filamentous fungi. In Growth, Differentiation and Sexuality; Wendland, J., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 47–66. [Google Scholar]
- Roncero, C.; Vázquez de Aldana, C.R. Glucanases and Chitinases. In The Fungal Cell Wall: An Armour and a Weapon for Human Fungal Pathogens; Latgé, J.-P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 131–166. [Google Scholar]
- Coelho, M.A.; Bakkeren, G.; Sun, S.; Hood, M.E.; Giraud, T.; Heitman, J.; Gow, N.A.R. Fungal sex: The Basidiomycota. Microbiol. Spectr. 2017, 5, 5-3. [Google Scholar] [CrossRef] [PubMed]
- Day, P.R. The structure of the a mating type locus in Coprinus Lagopus. Genetics 1960, 45, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Lukens, L.; Yicun, H.; May, G. Correlation of genetic and physical maps at the A mating-type locus of Coprinus cinereus. Genetics 1996, 144, 1471–1477. [Google Scholar] [CrossRef]
- Bao, D. Research progress on the mating-typing locus structures of basidiomycete mushrooms. Mycosystema 2019, 38, 2061–2077. [Google Scholar]
- Van Peer, A.F.; Park, S.-Y.; Shin, P.-G.; Jang, K.-Y.; Yoo, Y.-B.; Park, Y.-J.; Lee, B.-M.; Sung, G.-H.; James, T.Y.; Kong, W.-S. Comparative genomics of the mating-type loci of the mushroom Flammulina velutipes reveals widespread synteny and recent inversions. PLoS ONE 2011, 6, e22249. [Google Scholar] [CrossRef]
- Wang, W.; Lian, L.; Xu, P.; Chou, T.; Mukhtar, I.; Osakina, A.; Waqas, M.; Chen, B.; Liu, X.; Liu, F.; et al. Advances in understanding mating type gene organization in the mushroom-forming fungus Flammulina velutipes. G3 Genes Genomes Genet. 2016, 6, 3635–3645. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.C.; Hu, D.M. Exploring Genotype-Environment Interactions for Mushroom Breeding. Acta Edulis Fungi 2018, 25, 1–22. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | SP3 | SP30 |
|---|---|---|
| Numbers of Scaffold | 116 | 82 |
| Total Length (bp) | 50,830,139 | 49,800,313 |
| Scaffold N50 (bp) | 5,320,140 | 5,235,283 |
| Contig Num | 124 | 90 |
| Contigs N50 (bp) | 3,399,954 | 2,637,355 |
| Pseudo chromosome | 10 | 10 |
| Pseudo-chromosome total length (bp) | 47,651,827 | 46,345,524 |
| Pseudo-chromosome N50 length (bp) | 5,320,140 | 5,235,283 |
| Chromosome anchoring rate for contigs (%) | 93.74 | 93.05 |
| Chromosome anchoring rate for contigs longer than 100 Kb (%) | 99.63 | 98.91 |
| Term | SP3 | SP30 | ||
|---|---|---|---|---|
| BUSCO Number | Proportion (%) | BUSCO Number | Proportion (%) | |
| Complete BUSCOs | 1287 | 96.4 | 1285 | 96.2 |
| Complete and single-copy BUSCOs | 1257 | 94.2 | 1262 | 94.5 |
| Complete and duplicated BUSCOs | 30 | 2.2 | 23 | 1.7 |
| Fragmented BUSCOs | 27 | 2 | 27 | 2 |
| Missing BUSCOs | 21 | 1.6 | 23 | 1.8 |
| Total BUSCO groups searched | 1335 | 100 | 1335 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Q.; Yan, D.; Song, S.; Fan, Y.; Wang, S.; Liu, Y.; Huang, Y.; Rong, C.; Guo, Y.; Zhao, S.; et al. Haplotype-Resolved Genome Analyses Reveal Genetically Distinct Nuclei within a Commercial Cultivar of Lentinula edodes. J. Fungi 2022, 8, 167. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020167
Gao Q, Yan D, Song S, Fan Y, Wang S, Liu Y, Huang Y, Rong C, Guo Y, Zhao S, et al. Haplotype-Resolved Genome Analyses Reveal Genetically Distinct Nuclei within a Commercial Cultivar of Lentinula edodes. Journal of Fungi. 2022; 8(2):167. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020167
Chicago/Turabian StyleGao, Qi, Dong Yan, Shuang Song, Yangyang Fan, Shouxian Wang, Yu Liu, Yu Huang, Chengbo Rong, Yuan Guo, Shuang Zhao, and et al. 2022. "Haplotype-Resolved Genome Analyses Reveal Genetically Distinct Nuclei within a Commercial Cultivar of Lentinula edodes" Journal of Fungi 8, no. 2: 167. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020167