
Heme Oxygenase-1 (HMX1) Loss of Function Increases the In-Host Fitness of the Saccharomyces ‘boulardii’ Probiotic Yeast in a Mouse Fungemia Model

 ,
,  , and
, and
Abstract
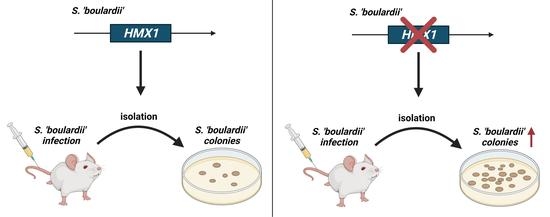
:
1. Introduction
1.1. Probiotic Yeast as a Cause of Fungemia
1.2. The Role of Iron during Pathogenicity
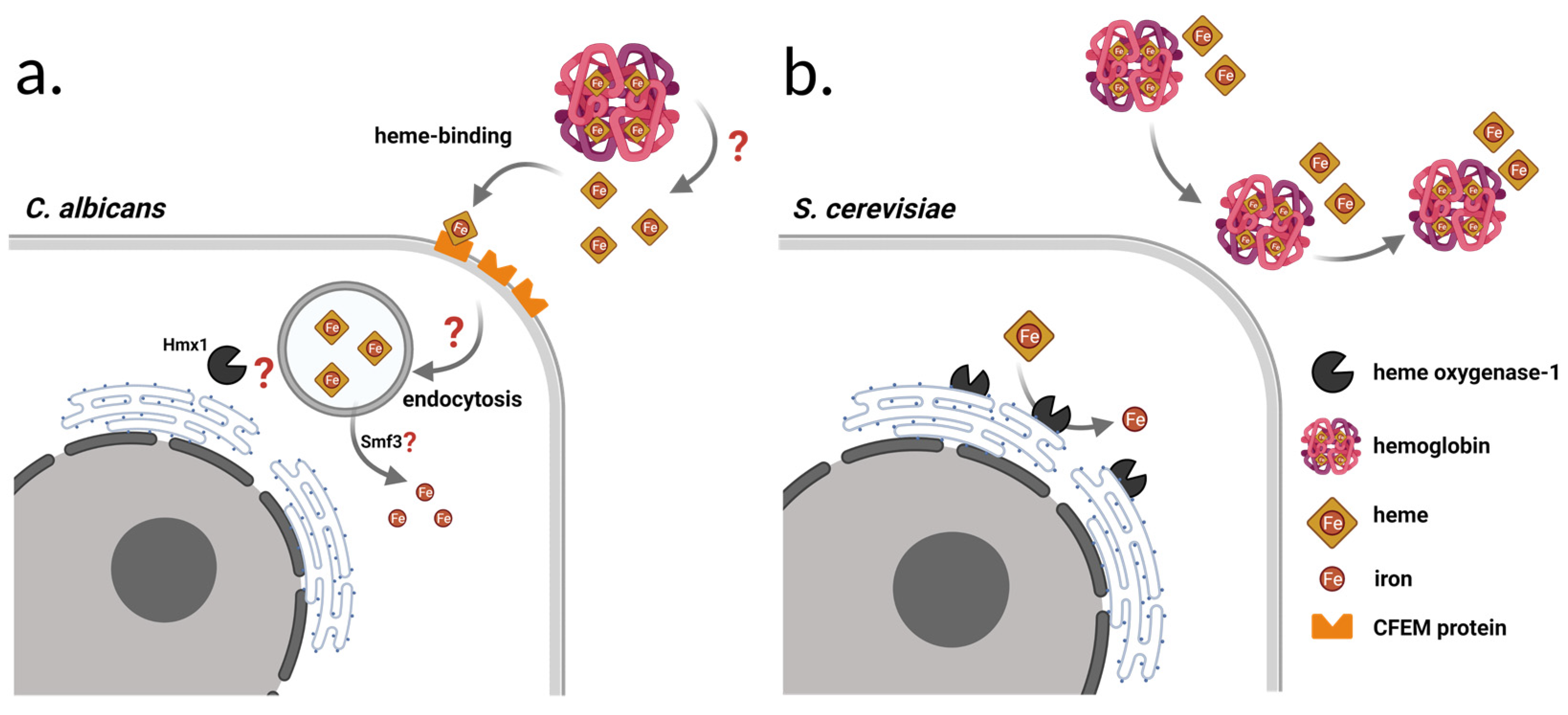
1.3. Heme-Iron Utilization by the Enzyme Heme Oxygenase-1 (Hmx1)
1.4. Hemolytic Activity of S. cerevisiae
1.5. A potential Role of HMX1 in the Pathogenicity of S. ‘boulardii’
2. Materials and Methods
2.1. Isolates, Mutant Strains, and Patient Data
2.2. Genome Sequencing and Assembly
2.3. Comparative Genomic Analysis
2.4. Phylogenomics
2.5. Plasmids and Constructs for HMX1 Deletion
2.6. Yeast Transformations
2.7. Iron Starvation, Hemolytic Activity
2.8. Yeast Growth in Immunosuppressed BALB/c Mice
2.9. Statistics and Data Visualization
3. Results
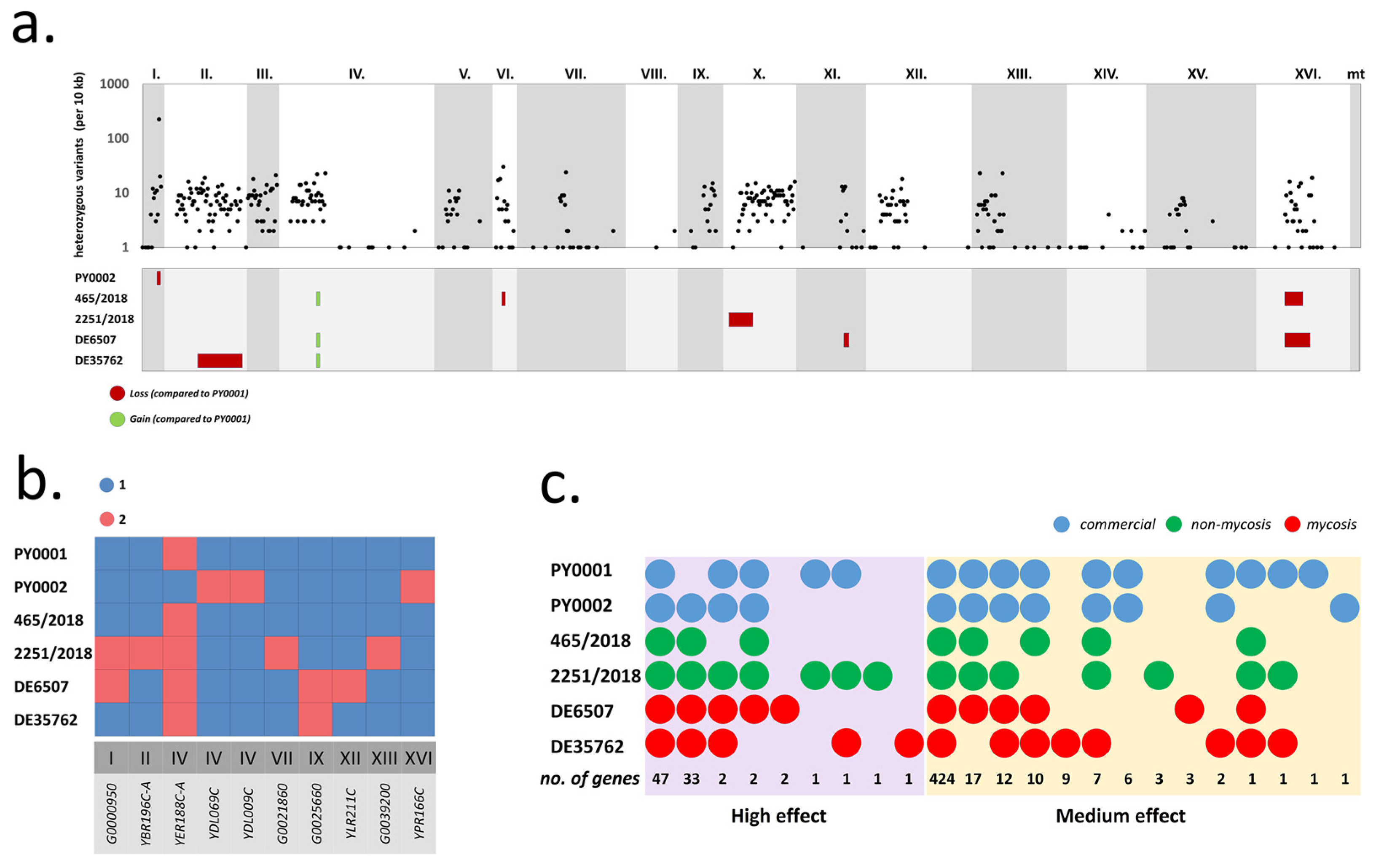
3.1. Comparison of the Genetic Backgrounds of S. ‘boulardii’ Yeasts Used in This Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Identifier | Gene Name | Description | Interaction Type | Mutation Effect | Position (on Chromosome) | Mutation Type | PY0001 | PY0002 | 465/2018 | 2251/2018 | DE6507 | DE35762 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YBR274W | CHK1 | Serine/threonine kinase and DNA damage checkpoint effector | Genetic | Moderate | 729,433 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | homoz. |
| YJR017C | ESS1 | Peptidylprolyl-cis/trans-isomerase (PPIase) | Genetic | Moderate | 471,176 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| Moderate | 471,227 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| YDL171C | GLT1 | NAD+-dependent glutamate synthase (GOGAT) | Genetic | High | 156,574 | Frameshift | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| High | 156,577 | Frameshift | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| Moderate | 160,753 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| YJR094C | IME1 | Master regulator of meiosis that is active only during meiotic events | Genetic | Moderate | 597,121 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| Moderate | 597,137 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| Moderate | 597,835 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| YJR054W | KCH1 | Potassium transporter that mediates K+ influx | Genetic | Moderate | 529,412 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| Moderate | 530,009 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| Moderate | 530,230 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| YPL055C | LGE1 | Protein involved in histone H2B ubiquitination | Genetic | Moderate | 441,438 | Missense | het. z. | het. z. | het. z. | het. z. | homoz. | het. z. |
| Moderate | 441,813 | Missense | het. z. | het. z. | het. z. | het. z. | homoz. | het. z. | ||||
| YER042W | MXR1 | Methionine-S-sulfoxide reductase | Genetic | Moderate | 231,971 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| YLR014C | PPR1 | Zinc finger transcription factor | Genetic | Moderate | 158,716 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| YDL013W | SLX5 | Subunit of the Slx5-Slx8 SUMO-targeted Ub ligase (STUbL) complex | Genetic | High | 436,686 | Frameshift | Not present | homoz. | homoz. | homoz. | homoz. | homoz. |
| YFL002C | SPB4 | Putative ATP-dependent RNA helicase | Genetic | Moderate | 121,701 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| Moderate | 122,057 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| YIL036W | CST6 | Basic leucine zipper (bZIP) transcription factor from ATF/CREB family involved in stress-responsive regulatory network | Transcription regulator | Moderate | 264,026 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
| Moderate | 264,770 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. | ||||
| Moderate | 264,849 | Missense | het. z. | het. z. | het. z. | het. z. | het. z. | het. z. |
3.2. Successful CRISPR/Cas9-Based Gene Deletion of HMX1
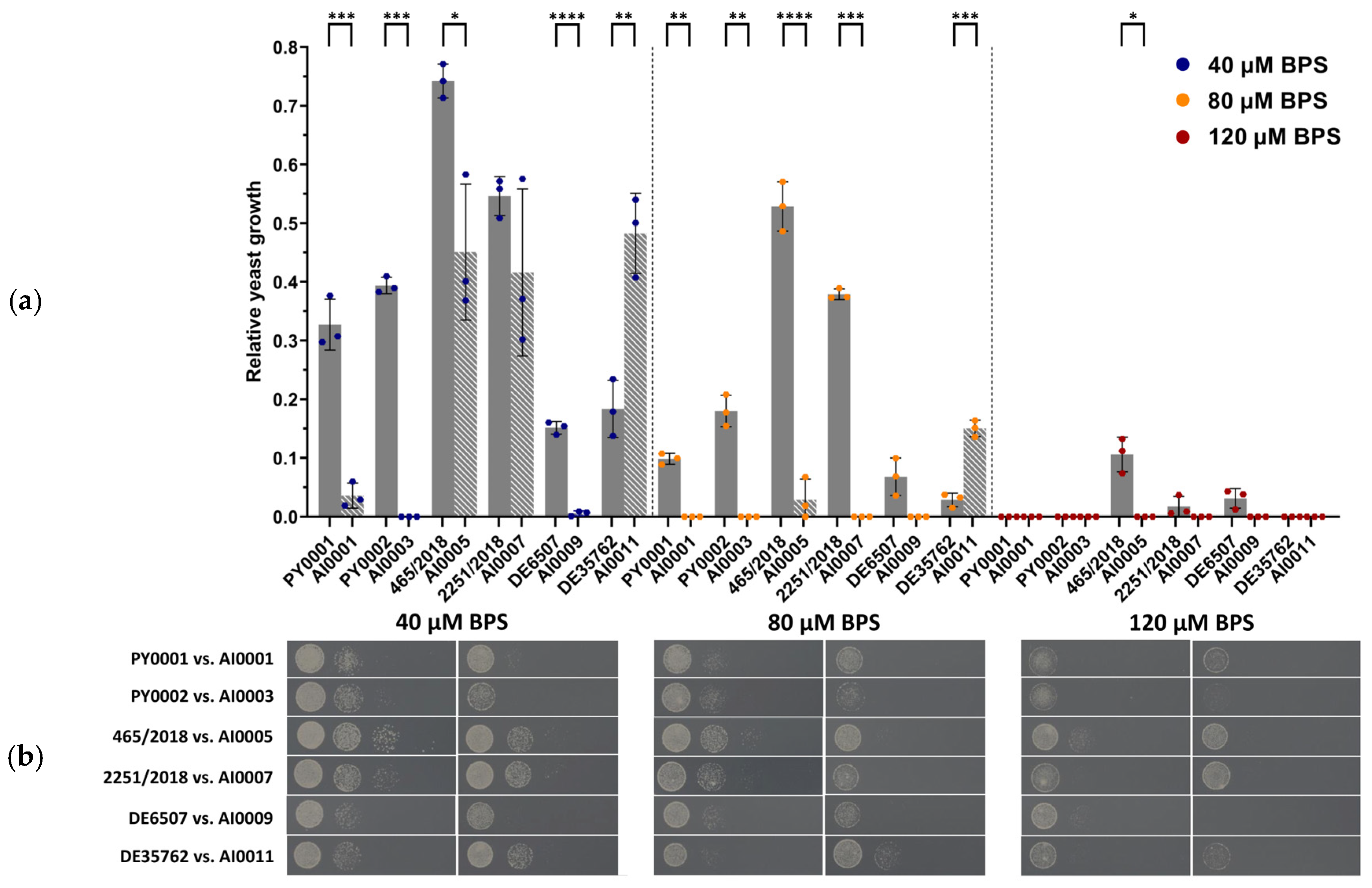
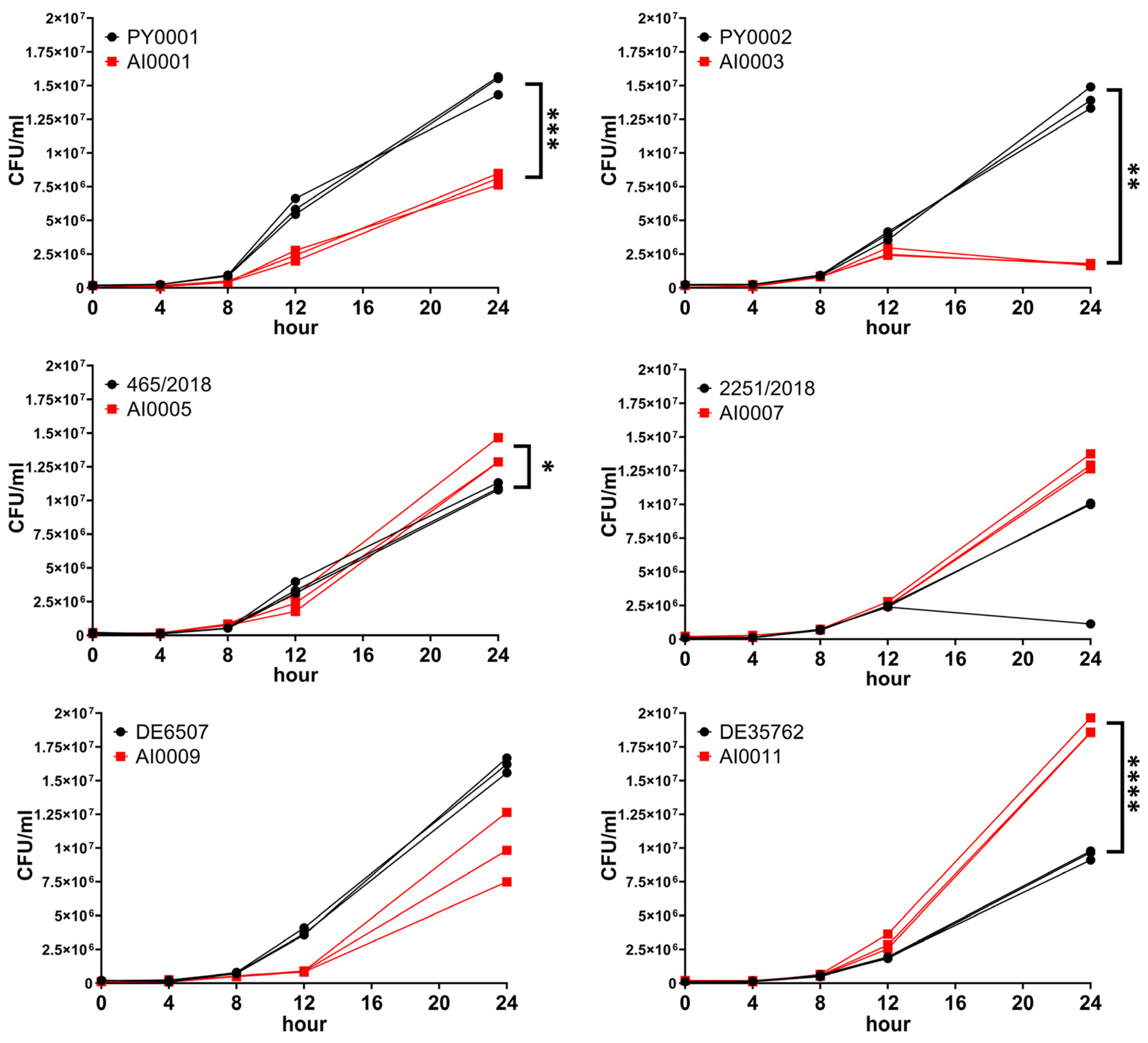
3.3. Deletion of HMX1 Resulted in Decreased Growth under Iron Deprivation
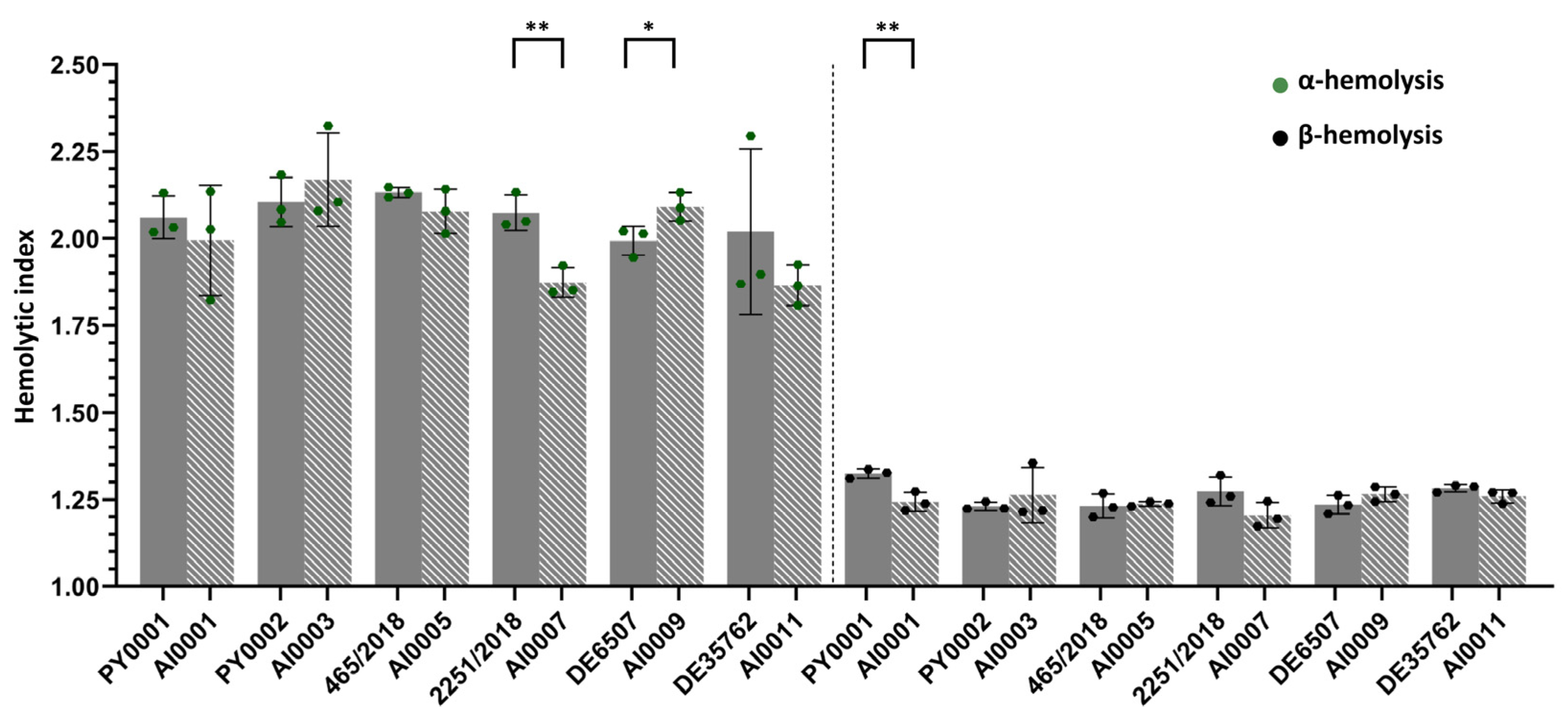
3.4. Hemolytic Activity Was Not Affected by HMX1 Deletion
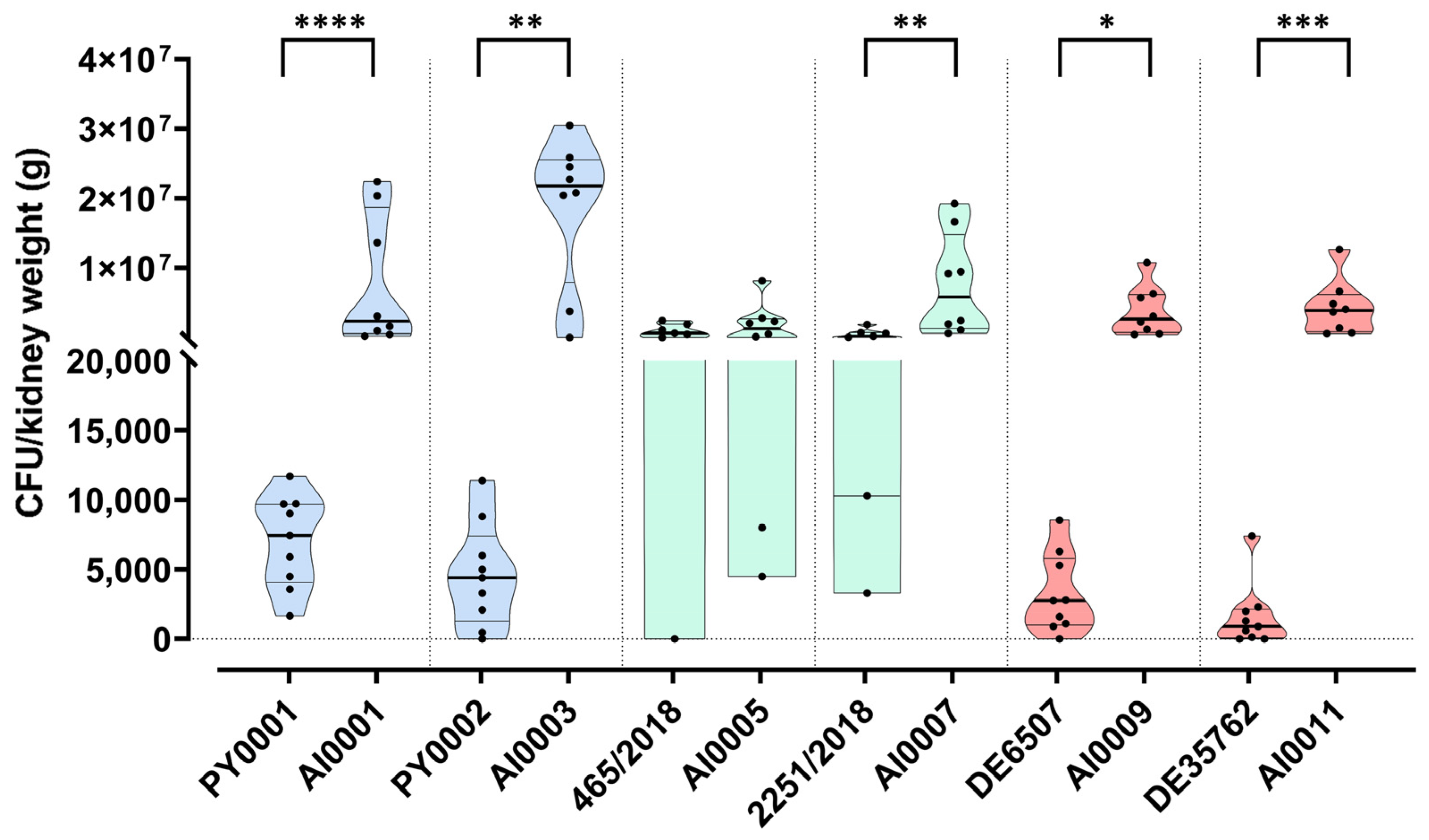
3.5. Deletion of HMX1 Caused a Significantly Higher Kidney Burden in Immunosuppressed BALB/c Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muñoz, P.; Bouza, E.; Cuenca-Estrella, M.; Marı, J.; Jesu, M.; Sa, M.; Rinco, C.; Hortal, J.; Pela, T.; Maran, G. Saccharomyces cerevisiae fungemia: An emerging infectious disease. Clin. Infect. Dis. 2005, 40, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Enache-Angoulvant, A.; Hennequin, C. Invasive Saccharomyces infection: A comprehensive review. Clin. Infect. Dis. 2005, 41, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Chitasombat, M.N.; Kofteridis, D.P.; Jiang, Y.; Tarrand, J.; Lewis, R.E.; Kontoyiannis, D.P. Rare opportunistic (non-Candida, non-Cryptococcus) yeast bloodstream infections in patients with cancer. J. Infect. 2012, 64, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, D.; Nguyen, D.; English, A.M. Ctt1 catalase activity potentiates antifungal azoles in the emerging opportunistic pathogen Saccharomyces cerevisiae. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergstrom, A.; Sigwalt, A.; Barré, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Imre, A.; Rácz, H.V.; Antunovics, Z.; Rádai, Z.; Kovács, R.; Lopandic, K.; Pócsi, I.; Pfliegler, W.P. A new, rapid multiplex PCR method identifies frequent probiotic origin among clinical Saccharomyces isolates. Microbiol. Res. 2019, 227, 126298. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Ruiz-Ojeda, F.J.; Gil-Campos, M.; Gil, A. Mechanisms of action of probiotics. Adv. Nutr. 2019, 10, S49–S66. [Google Scholar]
- Suzuki, H.; Perencevich, E.; Diekema, D.; Livorsi, D.; Nair, R.; Kralovic, S.; Roselle, G.; Goto, M. Temporal trends of candidemia incidence rates and potential contributions of infection control initiatives over 18 years within the United States Veterans Health Administration System: A joinpoint time-series analysis. Clin. Infect. Dis. 2021, 73, 689–696. [Google Scholar] [CrossRef]
- Wombwell, E.; Bransteitter, B.; Gillen, L. Incidence of Saccharomyces cerevisiae fungemia in hospitalized patients administered Saccharomyces boulardii probiotic. Mycoses 2021, 64, 1521–1526. [Google Scholar] [CrossRef]
- Imre, A.; Kovács, R.; Pázmándi, K.; Nemes, D.; Jakab, Á.; Fekete, T.; Rácz, H.V.; Dóczi, I.; Bácskay, I.; Gácser, A.; et al. Virulence factors and in-host selection on phenotypes in infectious probiotic yeast isolates (Saccharomyces ’boulardii’). J. Fungi 2021, 7, 746. [Google Scholar] [CrossRef]
- Dlouhy, A.C.; Outten, C.E. The iron metallome in eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241. [Google Scholar] [CrossRef] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nairz, M.; Weiss, G. Iron in infection and immunity. Mol. Aspects Med. 2020, 75, 100864. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, D.; Oski, F.; Ginsburg, D.; Orkin, S.; Look, A. Nathan and Oski’s Hematology and Oncology of Infancy and Childhood; Elsevier/Saunders: Philadelphia, PA, USA, 2003. [Google Scholar]
- Peyssonnaux, C.; Zinkernagel, A.; Datta, V.; Lauth, X.; Johnson, R.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Prentice, A. Hepcidin and the iron-infection axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammella, E.; Correnti, M.; Cairo, G.; Recalcati, S. Iron availability in tissue microenvironment: The key role of ferroportin. Int. J. Mol. Sci. 2021, 22, 2986. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Sonnweber, T.; Weiss, G. The struggle for iron—A metal at the host-pathogen interface. Cell. Microbiol. 2010, 12, 1691–1702. [Google Scholar] [CrossRef]
- Weiss, G. Modification of iron regulation by the inflammatory response. Best Pract. Res. Clin. Haematol. 2005, 18, 183–201. [Google Scholar] [CrossRef]
- Masson, P.; Heremans, J.; Schonne, E. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J. Exp. Med. 1969, 130, 643–658. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Alonso, L.; Romero, A.; Martínez-Pastor, M.; Puig, S. Iron regulatory mechanisms in Saccharomyces cerevisiae. Front. Microbiol. 2020, 11, 582830. [Google Scholar] [CrossRef]
- Kim, D.; Yukl, E.T.; Moënne-Loccoz, P.; Ortiz De Montellano, P.R. Fungal heme oxygenases: Functional expression and characterization of Hmx1 from Saccharomyces cerevisiae and CaHmx1 from Candida albicans. Biochemistry 2006, 45, 14772–14780. [Google Scholar] [CrossRef] [PubMed]
- Protchenko, O.; Philpott, C.C. Regulation of intracellular heme levels by HMX1, a homologue of heme oxygenase, in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 36582–36587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, T.; Tóth, Z.; Tóth, R.; Vágvölgyi, C.; Gácser, A. Iron metabolism, pseudohypha production, and biofilm formation through a multicopper oxidase in the human-pathogenic fungus Candida parapsilosis. mSphere 2020, 5, e00227-20. [Google Scholar] [CrossRef] [PubMed]
- Gerwien, F.; Safyan, A.; Wisgott, S.; Hille, F.; Kaemmer, P.; Linde, J.; Brunke, S.; Kasper, L.; Hube, B. A novel hybrid iron regulation network combines features from pathogenic and nonpathogenic yeasts. MBio 2016, 7, e01782-16. [Google Scholar] [CrossRef] [Green Version]
- Skrzypek, M.S.; Binkley, J.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database (CGD): Incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017, 45, D592. [Google Scholar] [CrossRef] [Green Version]
- Navarathna, D.H.M.L.P.; Roberts, D.D. Candida albicans heme oxygenase and its product CO contribute to pathogenesis of candidemia and alter systemic chemokine and cytokine expression. Free Radic. Biol. Med. 2010, 49, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Weissman, Z.; Pinsky, M.; Donegan, R.K.; Reddi, A.R.; Kornitzer, D. Using genetically encoded heme sensors to probe the mechanisms of heme uptake and homeostasis in Candida albicans. Cell. Microbiol. 2021, 23, e13282. [Google Scholar] [CrossRef]
- Roy, U.; Kornitzer, D. Heme-iron acquisition in fungi. Curr. Opin. Microbiol. 2019, 52, 77–83. [Google Scholar] [CrossRef]
- Fourie, R.; Kuloyo, O.O.; Mochochoko, B.M.; Albertyn, J.; Pohl, C.H. Iron at the centre of Candida albicans interactions. Front. Cell. Infect. Microbiol. 2018, 1, 185. [Google Scholar] [CrossRef] [Green Version]
- Weissman, Z.; Kornitzer, D. A family of Candida cell surface haem-binding proteins involved in haemin and haemoglobin-iron utilization. Mol. Microbiol. 2004, 53, 1209–1220. [Google Scholar] [CrossRef]
- Manns, J.M.; Mosser, D.M.; Buckley, H.R. Production of a hemolytic factor by Candida albicans. Infect. Immun. 1994, 62, 5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcok, H.K.; Aktas, E.; Ayyildiz, A.; Yigit, N.; Yazgi, H. Hemolytic activities of the Candida species in liquid medium. Eurasian J. Med. 2009, 41, 95. [Google Scholar] [PubMed]
- Tsang, C.S.P.; Chu, F.C.S.; Leung, W.K.; Jin, L.J.; Samaranayake, L.P.; Siu, S.C. Phospholipase, proteinase and haemolytic activities of Candida albicans isolated from oral cavities of patients with type 2 diabetes mellitus. J. Med. Microbiol. 2007, 56, 1393–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuster, A.; Osherov, N.; Rosenberg, M. Alcohol-mediated haemolysis in yeast. Yeast 2004, 21, 1335–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuster, A.; Korem, M.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Rosenberg, M. Microbial alcohol-conferred hemolysis is a late response to alcohol stress. FEMS Yeast Res. 2011, 11, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Weissman, Z.; Shemer, R.; Conibear, E.; Kornitzer, D. An endocytic mechanism for haemoglobin-iron acquisition in Candida albicans. Mol. Microbiol. 2008, 69, 201–217. [Google Scholar] [CrossRef]
- Pereira, R.; Jadhav, R.; Baghela, A.; Barretto, D. In Vitro Assessment of probiotic potential of Saccharomyces cerevisiae DABRP5 isolated from bollo batter, a traditional Goan fermented food. Probiotics Antimicrob. Proteins 2021, 13, 796–808. [Google Scholar] [CrossRef]
- Fernández-Pacheco, P.; Monge, I.M.R.; Fernández-González, M.; Colado, J.M.P.; Arévalo-Villena, M. Safety evaluation of yeasts with probiotic potential. Front. Nutr. 2021, 8, 659328. [Google Scholar] [CrossRef]
- Rannikko, J.; Holmberg, V.; Karppelin, M.; Arvola, P.; Huttunen, R.; Mattila, E.; Kerttula, N.; Puhto, T.; Tamm, Ü.; Koivula, I.; et al. Fungemia and other fungal infections associated with use of Saccharomyces boulardii probiotic supplements. Emerg. Infect. Dis. 2021, 27, 2103–2109. [Google Scholar] [CrossRef]
- Poncelet, A.; Ruelle, L.; Konopnicki, D.; Miendje Deyi, V.Y.; Dauby, N. Saccharomyces cerevisiae fungemia: Risk factors, outcome and links with S. boulardii-containing probiotic administration. Infect. Dis. Now 2021, 51, 293–295. [Google Scholar] [CrossRef]
- Gupta, P.; Singh, Y.P.; Taneja, A. Saccharomyces: A friend or foe in ICU (A case report with solution). Indian J. Crit. Care Med. 2019, 23, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Wibawa, T.; Praseno; Aman, A.T. Virulence of Candida albicans isolated from HIV infected and non infected individuals. Springerplus 2015, 4, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rácz, H.V.; Mukhtar, F.; Imre, A.; Rádai, Z.; Gombert, A.K.; Rátonyi, T.; Nagy, J.; Pócsi, I.; Pfliegler, W.P. How to characterize a strain? Clonal heterogeneity in industrial Saccharomyces influences both phenotypes and heterogeneity in phenotypes. Yeast 2021, 38, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.; Xiao, W. Isolation of nucleic acids. Methods Mol. Biol. 2006, 313, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Yue, J.-X.; Liti, G. Long-read sequencing data analysis for yeasts. Nat. Protoc. 2018, 13, 1213–1231. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Zhu, Y.O.; Sherlock, G.; Petrov, D.A. Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 2016, 6, 2421–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, S.R.; Wong, E.D.; Nash, R.S.; Aleksander, S.; Alexander, M.; Douglass, E.; Karra, K.; Miyasato, S.R.; Simison, M.; Skrzypek, M.S.; et al. New Data and collaborations at the Saccharomyces Genome Database: Updated reference genome, alleles, and the Alliance of Genome Resources. Genetics 2021, 220, iyab224. [Google Scholar] [CrossRef] [PubMed]
- HMX1 Interactions|SGD. Available online: https://www.yeastgenome.org/locus/S000004195/interaction (accessed on 7 March 2022).
- Ortiz, E.M. vcf2phylip v2.0: Convert a VCF Matrix into Several Matrix Formats for Phylogenetic Analysis. 2019. Available online: https://zenodo.org/record/2540861#.YoSha1RBzIV (accessed on 9 March 2022). [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Benchling. Available online: https://benchling.com (accessed on 1 May 2022).
- Lee, M.E.; Deloache, W.C.; Cervantes, B.; Dueber, J.E. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 2015, 4, 975–986. [Google Scholar] [CrossRef]
- Akhmetov, A.; Laurent, J.M.; Gollihar, J.; Gardner, E.C.; Garge, R.K.; Ellington, A.D.; Kachroo, A.H.; Marcotte, E.M. Single-step precision genome editing in yeast using CRISPR-Cas9. Bio-Protocol 2018, 8, e2765. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef]
- Lowry, R. VassarStats. Available online: http://vassarstats.net/index.html (accessed on 1 May 2022).
- Complex Online Web Statistical Calculators. Available online: https://astatsa.com/ (accessed on 21 October 2021).
- Statistics Online—Checks Assumptions, Interprets Results. Available online: https://www.statskingdom.com/index.html (accessed on 21 October 2021).
- Petropavlovskiy, A.; Tauro, M.; Lajoie, P.; Duennwald, M. A Quantitative imaging-based protocol for yeast growth and survival on agar plates. STAR Protoc. 2020, 1, 100182. [Google Scholar] [CrossRef] [PubMed]
- Pfliegler, W.P.; Boros, E.; Pázmándi, K.; Jakab, Á.; Zsuga, I.; Kovács, R.; Urbán, E.; Antunovics, Z.; Bácsi, A.; Sipiczki, M.; et al. Commercial strain-derived clinical Saccharomyces cerevisiae can evolve new phenotypes without higher pathogenicity. Mol. Nutr. Food Res. 2017, 61, 1601099. [Google Scholar] [CrossRef] [Green Version]
- Ventoulis, I.; Sarmourli, T.; Amoiridou, P.; Mantzana, P.; Exindari, M.; Gioula, G.; Vyzantiadis, T.-A. Bloodstream infection by Saccharomyces cerevisiae in two COVID-19 patients after receiving supplementation of Saccharomyces in the ICU. J. Fungi 2020, 6, 98. [Google Scholar] [CrossRef]
- McCullough, M.J.; Clemons, K.V.; Mccusker, J.H.; Stevens, D.A. Species identification and virulence attributes of Saccharomyces boulardii (nom. inval.). J. Clin. Microbiol. 1998, 36, 2613–2617. [Google Scholar] [CrossRef] [Green Version]
- Caudal, E.; Friedrich, A.; Jallet, A.; Garin, M.; Hou, J.; Schacherer, J. Population-level survey of loss-of-function mutations revealed that background dependent fitness genes are rare and functionally related in yeast. bioRxiv 2021. [Google Scholar] [CrossRef]
- Coutinho, J.; Peixoto, T.; de Menezes, G.; Carvalho, C.; Ogaki, M.; Gomes, E.; Rosa, C.; Rosa, L.; Arantes, R.; Nicoli, J.; et al. In vitro and in vivo evaluation of the probiotic potential of Antarctic yeasts. Probiotics Antimicrob. Proteins 2021, 13, 1338–1354. [Google Scholar] [CrossRef]
- Siscar-Lewin, S.; Hube, B.; Brunke, S. Antivirulence and avirulence genes in human pathogenic fungi. Virulence 2019, 10, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, R.T.; Kupiec, M.; Magnelli, P.; Abeijon, C.; Fink, G.R. A Saccharomyces cerevisiae mutant with increased virulence. Proc. Natl. Acad. Sci. USA 2003, 100, 2766–2770. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Mizobuchi, A.; Kato, T.; Kasahara, E.; Ito, C.; Watanabe, H.; Kanzaki, K.; Kitagawa, S.; Tachibana, T.; Azuma, M. Isolation of diploid baker’s yeast capable of strongly activating immune cells and analyses of the cell wall structure. Biosci. Biotechnol. Biochem. 2014, 78, 911–915. [Google Scholar] [CrossRef] [PubMed]





| ID | Isolate | ΔΔHMX1 Mutant | Type | Formulation | Component | Place of Acquisition | Date of Acquisition | Country of Manufacturing | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PY0001 | AI0001 | Commercial probiotic | Active dry | Single component | Debrecen, Hungary | March 2015 | France | [6] | ||
| 2 | PY0002 | AI0003 | Commercial probiotic | Active dry | Single component | Debrecen, Hungary | November 2017 | France | [6] | ||
| ID | Isolate | ΔΔHMX1 Mutant | Type | Age (yr) at Sampling | Sex | Medical Condition during Saccharomyces isolation | Mycosis Case | Anatomical Origin/Sample Type | Date of Sampling | Geographic Origin | Reference |
| 3 | 465/2018 | AI0005 | Non-mycosis isolate (probiotic-derived) | 41 | ♀ | Amenorrhea | no | Vagina | 3 January 2018 | Szeged, University Clinic | - |
| 4 | 2251/2018 | AI0007 | Non-mycosis isolate (probiotic-derived) | 17 | ♂ | Ulcerative colitis | no | Feces | 8 January 2018 | Szeged, University Clinic | - |
| 5 | DE6507 | AI0009 | Mycosis isolate (probiotic-derived) | 63 | ♂ | Pneumonia | yes | Hemoculture | 18 February 2017 | Debrecen, University Clinic | [6] |
| 6 | DE35762 | AI0011 | Mycosis isolate (probiotic-derived) | 66 | ♀ | Respiratory failure | yes | Hemoculture | 5 November 2015 | Debrecen, University Clinic | [6] |
| Oligos for Repair DNA: | Forward: 5′-ATGGAGGACAGTAGCAATACAATCATACCCTCACCCACTGACGTGGGGGCGCTAGCAAACcactaactaactaagcgtcg-3′ Reverse: 5′-TTTTGATATTATTTCATGTATATATTATGTTTGTATTTAGACTTTTTTTTTTATACGTTAcgacgcttagttagttagtg-3′ | |
| Oligos for gRNA: | Forward: 5′-gactttCATCCACGAAAACATACACA-3′ Reverse: 5′-aaacTGTGTATGTTTTCGTGGATGaa-3′ | |
| Verification Primers | Primers to Verify Gene Deletion | Forward: 5′-TGTTATTGTACCCATAGAGG-3′ Reverse: 5′-AAATATAGTGATGCAACTCG-3′ |
| Primers to Verify Locus of Insertion | Forward: 5′-TGTTATTGTACCCATAGAGG-3′ Reverse: 5′-CGACGCTTAGTTAGTTAGTG-3′ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imre, A.; Kovács, R.; Tóth, Z.; Majoros, L.; Benkő, Z.; Pfliegler, W.P.; Pócsi, I. Heme Oxygenase-1 (HMX1) Loss of Function Increases the In-Host Fitness of the Saccharomyces ‘boulardii’ Probiotic Yeast in a Mouse Fungemia Model. J. Fungi 2022, 8, 522. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050522
Imre A, Kovács R, Tóth Z, Majoros L, Benkő Z, Pfliegler WP, Pócsi I. Heme Oxygenase-1 (HMX1) Loss of Function Increases the In-Host Fitness of the Saccharomyces ‘boulardii’ Probiotic Yeast in a Mouse Fungemia Model. Journal of Fungi. 2022; 8(5):522. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050522
Chicago/Turabian StyleImre, Alexandra, Renátó Kovács, Zoltán Tóth, László Majoros, Zsigmond Benkő, Walter P. Pfliegler, and István Pócsi. 2022. "Heme Oxygenase-1 (HMX1) Loss of Function Increases the In-Host Fitness of the Saccharomyces ‘boulardii’ Probiotic Yeast in a Mouse Fungemia Model" Journal of Fungi 8, no. 5: 522. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050522